

Abstract
Background & Aims
Fibroblast growth factor receptor (FGFR) 4 controls bile acid metabolism and protects the liver from fibrosis, but the roles of FGFR1 and FGFR2 in the adult liver are largely unknown. We investigated the functions and mechanisms of action of these receptors in liver homeostasis, regeneration, and fibrosis.
Methods
We generated mice with hepatocytes that lack FGFR1 and FGFR2 and subjected them to acute and chronic carbon tetrachloride-induced liver injury and partial hepatectomy; mice were also injected with FGF7. We performed histology, histomorphometry, real-time reverse transcription PCR, and immunoblot analyses.
Results
In hepatocytes, loss of FGFR1 and FGFR2 eliminated responsiveness to FGF7 and related FGF family members, but did not affect toxin-induced liver injury and fibrosis. However, mortality after partial hepatectomy increased because of severe hepatocyte necrosis. These effects appeared to be mediated by a failure of hepatocyes to induce the expression of the transcriptional regulators Dbp and Tef upon liver surgery; this affected expression of their target genes, which encode detoxifying cytochrome P450 enzymes. We found that Dbp and Tef expression was directly controlled by FGFR signalling in hepatocytes. As a consequence of the reduced expression of genes that control detoxification, the liver tissue that remained after partial hepatectomy failed to efficiently metabolize endogenous compounds and the drugs applied for anaesthesia/analgesia.
Conclusions
We identified a new, cytoprotective effect of FGFR1 and FGFR2 in the regenerating liver and suggest the use of recombinant FGF7 to increase survival of patients after surgical resection of large amounts of liver tissue.
Keywords: liver disease, cirrhosis, drug toxicity, cytoprotection
Introduction
The liver is the only organ in mammals that can fully regenerate after injury. This potential is required after surgery, toxin-induced necrosis, or viral infections to restore liver integrity. Hepatic tissue loss initiates a well-defined program, which finally results in restoration of the initial liver mass and re-establishment of the liver’s essential functions in metabolic regulation and compound detoxification 1. A commonly used model to study liver regeneration in rodents is partial hepatectomy (PH), where 70% of the liver is surgically removed. In response to PH, hepatocytes enter the cell cycle and proliferate to restore the original liver mass 2,3.
Unfortunately, the regenerative capacity of the liver is frequently inadequate, in particular after chronic injury by toxins or viral infections. This causes replacement of functional epithelial tissue by connective tissue, resulting in fibrosis and cirrhosis 4,5. A suitable animal model to study toxin-induced liver injury is application of carbon tetrachloride (CCl4), which initially causes centrilobular necrosis and inflammation, and - upon long-term application – liver fibrosis 6.
We recently showed a role of FGFs in liver regeneration 7, although the functions and mechanisms of action of individual FGFs and their receptors are largely unclear. FGFs consist of a family of 22 polypeptides, which regulate migration, proliferation, differentiation, and survival of different cell types 8,9. They activate four transmembrane tyrosine kinase receptors, designated FGFR1-FGFR4. Further diversity is generated by alternative splicing in the third immunoglobulin-like domains of FGFR1-3, resulting in IIIb and IIIc variants with different ligand binding specificities. The IIIb variants are predominantly expressed in epithelial cells, whereas mesenchymal cells express mainly the IIIc variants 8.
FGFs are essential regulators of liver development 10,11, and they are also involved in liver regeneration 7, 12. We previously demonstrated that transgenic mice expressing a dominant-negative FGFR2-IIIb mutant in hepatocytes showed impaired hepatocyte proliferation after PH 7. Since the dominant-negative mutant inhibits the action of all FGF receptors in response to common FGF ligands 13, the role of individual FGFs and FGFRs in liver regeneration remains to be determined.
FGFR4 is strongly expressed by hepatocytes and is a regulator of bile acid homeostasis 14. FGFR4-deficient mice showed normal regeneration after PH 14, but enhanced fibrosis after chronic CCl4 application 15. On the other hand, mice lacking FGF1 and FGF2 showed less fibrosis in this model, suggesting that these FGFs accelerate liver fibrosis through activation of other FGFRs 16. Since FGFR2 and FGFR1 are also expressed by hepatocytes 7,14,17 (Fig. 1), we determined the consequences of their loss in hepatocytes for liver homeostasis, regeneration and fibrosis.
Fig. 1. Verification of the loss of FGFR1 and FGFR2 in hepatocytes.

(A) RNAs from total liver or primary hepatocytes of Alb-R1/R2 (ko) and control mice (ctrl) were analyzed for Fgfr or Gapdh expression using qRT-PCR. Expression levels in control mice were arbitrarily set as 1. RNA from mouse epidermis was used as a positive control for FGFR3 expression. (B) Lysates from total liver of Alb-R2-IIIb and control mice were analyzed by western blotting for expression of FGFR2. Differently glycosylated forms of FGFR2 are indicated. (C) Sections from the liver of Alb-R2-IIIb and control mice were analyzed by immunohistochemistry for expression of FGFR2. (D) Primary hepatocytes from control and Alb-R1/R2 mice were serum-starved, treated with 10 ng/ml FGF7, harvested at the indicated time points and analyzed by western blotting for phosphorylated FRS2α and GAPDH. Cells incubated for 60 min in the absence of FGF7 were used as control (60c).
Materials and Methods
Animals
Animal maintenance and experiments were approved by the local veterinary authorities of Zurich, Switzerland. The animals were free of pathogens, including mouse hepatitis virus.
CCl4-induced acute liver injury
Mice were injected intraperitoneally (i.p.) with a single dose of CCl4 (0.4mg/g body weight in mineral oil (Sigma, Buchs, Switzerland)) or mineral oil alone. They were sacrificed at different time points after injury.
CCl4–induced liver fibrosis
Mice were injected i.p.15 times every third day with CCl4 (0.2mg/g body weight in olive oil) or vehicle alone. They were sacrificed 72h after the last injection.
PH
8–10-week-old male mice, which had received food and water ad libitum prior surgery, were anaesthetized by i.p. injection of ketamine (0.1mg/g body weight)/xylazine (5μg/g body weight) in isotonic saline or by inhalation of isoflurane (2%). PH was previously described 7. Three liver lobes including the previously emptied gall bladder were removed. After surgery mice were injected with buprenorphine (Temgesic, Essec Chemie AG, Switzerland; 0.1 μg/g body weight). They were sacrificed by CO2 inhalation and the remaining livers were harvested at different time points after PH. For sham surgery the mice were anaesthetized, the abdomen was opened, and the liver lobes were briefly pulled out from the abdominal cavity. In most experiments the liver removed in the surgical process served as control. Livers from non-operated or sham-operated mice served as additional controls in some experiments. All experiments were performed in the morning between 8 and 11 am.
Culture of primary hepatocytes and analysis of the FGF response
Murine hepatocytes were isolated as described 18, plated on collagen R (Serva, Heidelberg, Germany; 0.2 mg/ml)-coated dishes in RPMI medium (Invitrogen) at a density of 5×105/cm2, and left to adhere for 3h. Subsequently, the medium was changed to remove non-attached cells. After 30 min 10 ng/ml FGF7 (Palifermin, Amgen, Thousank Oaks, CA) was added. At different time points after FGF addition cells were lysed and analyzed by western blotting for phosphorylated FRS2α and GAPDH.
Histology and histomorphometry
Liver samples were fixed in 4% paraformaldehyde in PBS and embedded in paraffin. Sections (3.5μm) were stained with hematoxylin/eosin, photographed (3–5 pictures per animal), and the necrotic area was determined morphometrically. Fibrotic area was measured in sections stained with Sirius Red or with an antibody against fibronectin.
Identification of proliferating cells
Proliferating cells were identified by 5-bromo-2′ deoxyuridine (BrdU) labeling as previously described 19 and counted in four independent microscopic fields (200× magnification) per animal.
Serum collection and analysis
Mice were euthanized by CO2 inhalation and blood was taken by heart punctuation. Following coagulation, it was snap frozen and stored at −80°C until serology tests were performed using standard procedures.
Statistical analysis
Statistical analysis was performed using the Prism4 software. Quantitative data are expressed as mean +/− SEM. Significance was calculated using the Mann-Whitney U test. *P<0.05, **P<0.01, and ***P<0.001. For quantitative Real-Time RT-PCR (qRT-PCR) experiments RNA from single mice was used, and at least four mice were analyzed per condition and genotype.
Results
Generation of mice lacking FGFR1 and FGFR2 in hepatocytes
Mice with floxed fgfr1 20 and fgfr2 alleles 21 were mated with albumin-Cre transgenic mice, which express Cre in hepatocytes postnatally 22. Albumin-Cre mice with wild-type fgfr alleles as well as mice with floxed fgfr alleles but without the Cre transgene were used as controls, and they never revealed phenotypic abnormalities. The knockout mice were designated Alb-R1, Alb-R2, Alb-R2-IIIb and Alb-R1/R2. Since hepatocytes express predominantly or even exclusively the IIIb variant of FGFR2 (Fig. 1A), Alb-R2 and Alb-R2-IIIb mice have the same defect.
Loss of fgfr1 and fgfr2 expression was verified by qRT-PCR. Reduced levels of fgfr1 and fgfr2 mRNAs were observed in total liver and particularly in isolated primary hepatocytes of Alb-R1/R2 mice compared to control mice (Fig. 1A). Using primers specific for the IIIb and IIIc splice variants, we found an almost complete loss of FGFR1-IIIb and FGFR2-IIIb in the liver. This is consistent with the expression of FGFR2-IIIb in hepatocytes but not in other liver cells 23. By contrast, only a slight and non-significant reduction was seen for the (stromal) IIIc variants (Fig. 1A). Expression of FGFR4 was not affected, and FGFR3 was not expressed in the liver (Fig. 1A). Loss of FGFR2 in hepatocytes was verified by western blotting of total liver lysates (Fig. 1B). The residual expression most likely results from the expression in non-parenchymal cells, e.g. endothelial cells, as determined by immunohistochemistry (Fig. 1C). Treatment of serum-starved, cultured primary hepatocytes with FGF7 led to increased phosphorylation of FGF receptor substrate 2α (FRS2α) in cells from control mice but not from Alb-R1/R2 mice (Fig. 1D), demonstrating loss of FGF7-induced signalling. Under the same conditions, hepatocytes from mice of both genotypes responded appropriately to epidermal growth factor (EGF) (Suppl. Fig. S1A).
Loss of FGFR1 and FGFR2 in hepatocytes does not affect liver function
Alb-R1/R2 mice were healthy and macroscopically normal. Their liver weight to body weight ratio was unaltered, and no histological abnormalities were seen in the liver (Suppl. Fig. S1B). Serum analysis for albumin and alkaline phosphatase did not reveal alterations in the mutant mice. There was no sign of liver damage as revealed by the normal serum levels of aspartate aminotransferase (AST), and alanine aminotransferase (ALT) (Suppl. Fig. S1C).
Loss of FGFR1 and FGFR2 in hepatocytes reduces CCl4-induced inflammation
A single injection of CCl4 caused severe liver necrosis in mice of both genotypes. There was only a slight increase in the area of necrosis in Alb-R1/R2 mice compared to control mice 24h after CCl4 injection (Fig. 2A), and apoptosis was not altered as determined by TUNEL staining (data not shown). Serum levels of ALT and AST were only slightly, but non-significantly increased in Alb-R1/R2 mice (Fig. 2B). The speed of repair was even slightly higher in Alb-R1/R2 animals as indicated by the reduction in necrotic area between 24h and 48h after CCl4 injection and by the increased rate of liver cell proliferation in some knockout mice at 36h (Fig. 2A,C). Surprisingly, the inflammatory response was less pronounced in Alb-R1/R2 mice compared to controls as demonstrated by significantly lower numbers of neutrophils and macrophages (Fig. 2D) and by the strongly reduced induction of several pro-inflammatory cytokines and chemokines (Fig. 2E).
Fig. 2. Loss of FGFR1 and FGFR2 in hepatocytes protects from CCl4-induced inflammation.

(A–E) Mice were injected once with CCl4 in mineral oil and sacrificed at different time points. (A) Representative hematoxylin/eosin-stained sections are shown. Necrotic area can be distinguished from normal liver tissue by the brighter colour and the inflammatory cell infiltrates (encircled areas). It was determined by measuring 4–5 independent microscopic fields (N≥5 per genotype) and is indicated as percent of total liver area. Bar: 100μm. (B) AST and ALT levels were determined in the serum 48h after CCl4 injection. N≥5 per genotype. (C) Liver sections from BrdU-injected animals were stained with an antibody against BrdU. Representative sections from Alb-R1/R2 mice and controls 36h after CCl4 injection are shown. The percentage of BrdU-positive cells was determined by counting 4–5 independent microscopic fields. N≥5 mice per time point. Bar: 50μm. (D) Liver sections were stained with antibodies against neutrophils (Ly6G) or macrophages (ER-MP23). Representative pictures are shown. Inflammatory cells were counted in 4–5 independent microscopic fields (200 × magnification, N≥5 per genotype). Bar: 50μm. (E) RNAs from the liver of control and Alb-R1/R2 mice at different time points after CCl4 injection were analyzed by qRT-PCR for the levels of IL-1β, TNF-α, S100A8, S100A9, MIP-1α, or MIP-1β mRNAs. Rps29 was used for normalization.
Histopathological analysis of liver sections after long-term treatment with CCl4 did not reveal a difference in the extent of fibrosis. Cell proliferation, liver weight to body weight ratio, AST and ALT levels in the serum, and inflammatory cell infiltration were also not affected (Suppl. Fig. S2A–F).
These data demonstrate that FGFR1 and FGFR2, unlike FGFR4, neither protect the liver from acute toxin-induced injury nor from chronic injury and fibrosis, but rather enhance the initial inflammatory response.
FGFR1 and FGFR2 cooperate to enhance survival of hepatocytes after PH
We next determined the roles of FGFR1 and FGFR2 in the regeneration process after PH. We first analyzed the expression of putative ligands of FGFR1-IIIb and FGFR2-IIIb 24,25 in the regenerating liver of wild-type mice. We also analyzed the expression of FGFs in the spleen, since FGF7 is expressed in this organ and reaches the liver via the portal vein 26. RNase protection assay revealed expression of FGF1 in normal and injured liver. FGF1, FGF2 and FGF7 were expressed in the spleen, and their expression increased after PH (Suppl. Fig. S3A,B). Low levels of FGF22 and FGF3 mRNAs were also detected in normal and injured liver, whereas FGF10 was neither expressed in the non-injured adult liver nor within 6h and 7d after PH (data not shown). These results suggest that FGF1 derived from liver and spleen, as well as spleen-derived FGF7 are the major ligands of FGFR1-IIIb and FGFR2-IIIb in the regenerating liver. Since there was no compensatory upregulation of the IIIc variants (Fig. 1A), it seems unlikely that other FGFs, which can only bind to FGFR1-IIIc and/or FGFR2-IIIc, affect liver regeneration via FGFR1 or FGFR2 on hepatocytes.
When control and Alb-R1/R2 mice were subjected to PH, more than 40% of the mutant mice died within 24h–48h (Fig. 3A), whereas the mortality rate of control or single knockout mice was below 5%. The fact that most of the knockout mice survived the first 24h indicates a defect in the onset of regeneration and/or metabolic abnormalities as the cause of death, rather than an acute toxicity exerted by the operation. This hypothesis is supported by the normal survival rate of sham-operated animals.
Fig. 3. FGFR1 and FGFR2 cooperate to enhance survival of mice and to prevent hepatocyte necrosis after PH.

Mice were subjected to PH. (A) The percentage of surviving animals at different time points after PH is indicated. Numbers above the bars indicate the number of animals observed at each time point per genotype. (B) AST and ALT levels in the serum were determined 6h after PH. N≥5 per genotype. (C,D) Macroscopic (C) and histological analysis (D) revealed the presence of necrotic lesions in the liver of Alb-R1/R2 mice 24h after PH (N≥12 per genotype). Bar: 50μm.
Serum levels of AST were significantly higher in Alb-R1/R2 mice compared to control animals 6h after PH, and ALT levels were also increased (Fig. 3B). In line with this finding necrotic lesions were observed macroscopically 24h after PH in Alb-R1/R2 mice but not in control mice (Fig. 3C) or sham-operated Alb-R1/R2 mice. This was confirmed histologically, and strong centrilobular necrosis was seen in Alb-R1/R2 mice (Fig. 3D). Since necrosis occurred only upon removal of liver tissue, the functional activity of the remaining tissue is rate limiting. The rate of apoptosis was equally low in mice of both genotypes (data not shown).
Hepatocyte proliferation is enhanced in surviving Alb-R1/R2 mice after PH
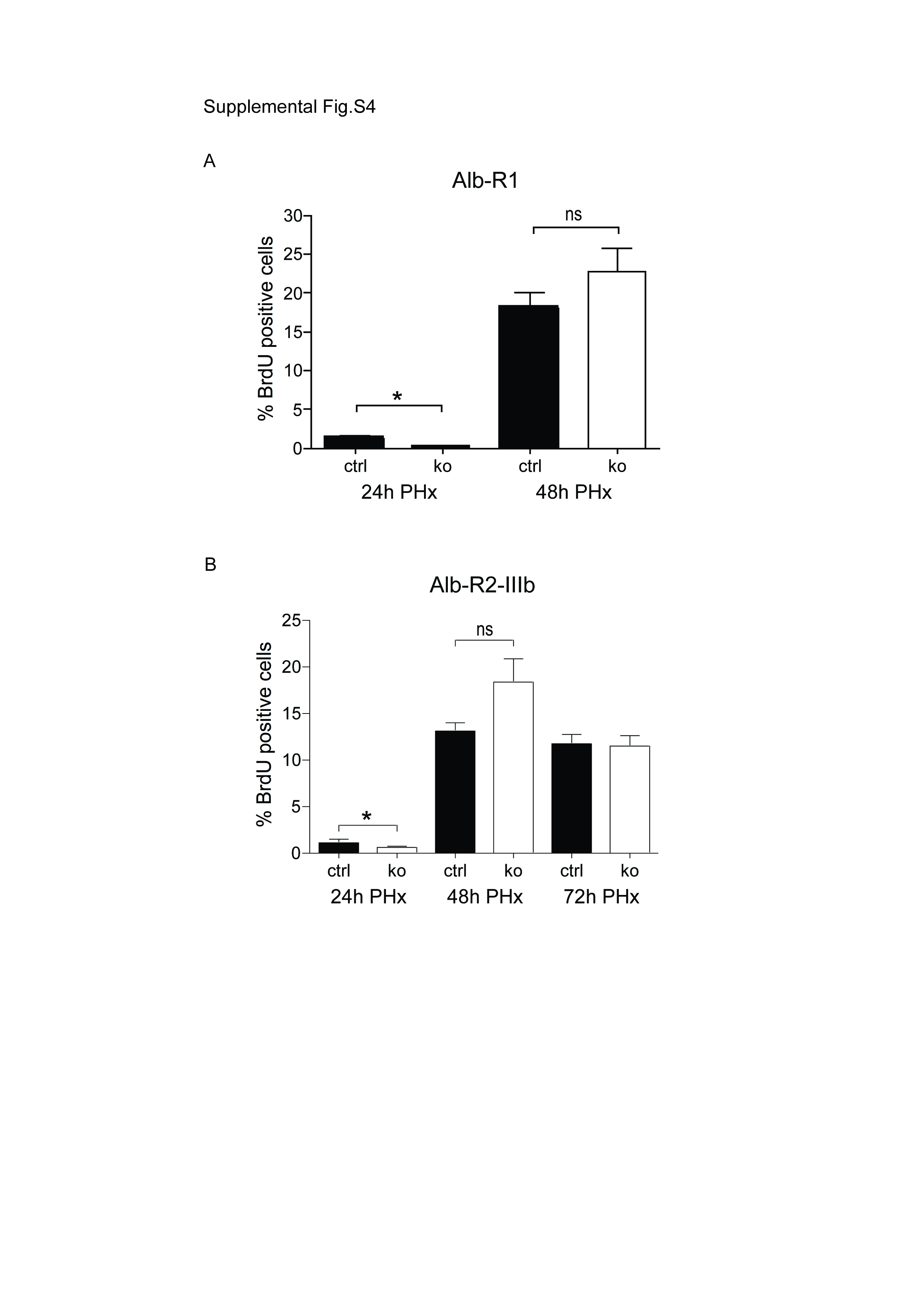
24h after surgery, only few proliferating cells were detected in mice of both genotypes as determined by BrdU labelling. 24h later, strong proliferation was seen in control mice, and the proliferation rate was even significantly increased in the surviving Alb-R1/R2 mice (Fig. 4A,B). This hyperproliferation most likely reflects the necessity to repair the necrotic area in addition to the surgically removed tissue. The onset of hepatocyte proliferation was delayed in the single knockout mice, but a slightly increased proliferation rate was observed at 48h (Suppl. Fig. S4). Proliferation subsequently declined in mice of all genotypes, and was completed 7d after injury (Fig. 4B). At this time point the liver to body weight ratio was similar in control and Alb-R1/R2 mice (data not shown), demonstrating that the surviving double knockout mice can completely regenerate. Increased hepatocyte proliferation was not observed in either control or Alb-R1/R2 mice after sham injury (shown for the 48h time point in Fig. 4A).
Fig. 4. Delayed activation of major signalling pathways and enhanced proliferation of hepatocytes after PH in surviving Alb-R1/R2 mice.

(A) Cell proliferation in the liver at 48h after PH was assessed by BrdU incorporation. Representative sections from injured liver (48h after PH) are shown. Bar: 50μm. (B) The percentage of proliferating cells was determined by counting 4–5 independent microscopic fields/per liver at 200× magnification, N>6 per genotype and time point. (C) Liver lysates from control and Alb-R1/R2 mice before and at different time points after PH were analyzed by western blotting for the levels of total and phosphorylated ERK1/2, p38, Akt, JNK1/2, STAT3 and GAPDH. Each lysate was obtained from pooled livers of three animals per time point and genotype. All results shown on this figure were obtained with the same lysates. Results were reproduced in an independent experiment using lysates from different animals.
The enhanced hepatocyte proliferation in Alb-R1/R2 mice at the 48h time point was also reflected by the enhanced phosphorylation (activation) of Erk1/2 and p38 as determined by Westernblot analysis of total liver lysates (137% or 35% increase, respectively). By contrast, the activation of these signalling proteins and of STAT3 was strongly reduced 6h after PH (45% reduction of P-Erk1/2, 63% reduction of P-p38 and 55% reduction of P-STAT3). Activation of Akt and JNK was not affected (Fig. 4C). These results indicate a defect in the priming phase, which may result in impaired activation of genes required for cell survival.
FGFR1 and FGFR2 control compound detoxification in the regenerating liver
We next tested if the enhanced mortality of the mutant mice results from impaired metabolism of the drugs used for anaesthesia (ketamine/xylazine) and/or analgesia (buprenorphine). When ketamine/xylazine was replaced by the less toxic inhalent anaesthetic isofluorane 27, the survival rate was indeed increased to 94% (Fig. 5A). Necrosis was only rarely observed, and the rate of hepatocyte proliferation was similar in control and Alb-R1/R2 mice at 48h, reflecting the fact that compensatory hyperproliferation is not required under these conditions (Fig. 5B). Since buprenorphine is hepatotoxic 28, we next omitted it after ketamine/xylazine anaesthesia and PH. Under these conditions we observed a survival rate of 85% (Fig. 5A). Nevertheless, there was still necrosis in the mutant mice of all treatment groups (data not shown). This was not unexpected, since the narcotic drugs as well as endogenous compounds still need to be detoxified, and this may well be impaired in the Alb-R1/R2 mice.
Fig. 5. FGFR1 and FGFR2 signaling is important for compound detoxification in the regenerating liver.

(A) Mice were subjected to PH. One group was anaesthetized with ketamine/xylazine followed by buprenorphine treatment (K/X/B), in the second group ketamine/xylazine was replaced by isoflurane (Iso/B), and in the third group buprenorphine was omitted after ketamine/xylazine anaesthesia (K/X). The percentage of surviving animals at different time points after PH is indicated. Numbers above the bars indicate the number of animals observed at each time point per genotype and treatment group. (B) Cell proliferation was assessed in mice of the Iso/B group 48h after PH using BrdU incorporation. N>6 per genotype and treatment group. (C) Expression of Dbp, Tef, Cyp2a5, and Cyp2c38 was analyzed by qRT-PCR using RNAs from livers of untreated Alb-R1/R2 mice and control animals. RNAs from the liver tissue, which was removed upon PH (0h PH), or from the remaining liver 24h after PH were analyzed for comparison. (D) Levels of Dbp and Tef mRNAs were analyzed by qRT-PCR in non-injured Alb-R1/R2 and control mice at different day times. Rps29 was used for normalization.
FGFR1 and FGFR2 control the hepatectomy-induced expression of circadian transcription factors involved in compound detoxification
Detoxification of buprenorphine and ketamine/xylazine is performed by cytochrome P450 (Cyp) enzymes 28,29, which are regulated by the PAR-domain basic leucine zipper transcription factors Dbp (D site albumin promoter binding protein), Tef (thyrotrophic embryonic factor), and Hlf (hepatic leukemia factor) 30. Therefore, we analyzed the expression of these genes in control and mutant mice. Liver samples were obtained from untreated mice, from the liver tissue that was removed during the PH surgery (10 min after ketamine/xylazine injection and approximately 5 min after beginning of the surgery) as well as liver tissue obtained 24h after PH.
Whereas expression of Hlf was not affected by the surgery, we found a strong increase in the expression the Dbp and Tef in the liver tissue of control mice that was removed during the surgery (0h time point) compared to liver tissue from untreated mice (Fig. 5C). Expression declined to basal levels 24h after PH. The upregulation was independent of the type of anaesthesia (Suppl. Fig. S5A). Ketamine/xylazine alone was not sufficient to upregulate Dbp and Tef expression, whereas isoflurane caused a mild increase in Dbp expression in mice of both genotypes (Suppl. Fig. S5B). In particular, the subsequent manipulation of the animals, which included opening of the abdominal cavity and ligation of the liver lobes, strongly induced the expression of both transcription factors in control but not in Alb-R1/R2 mice. Consistent with the lack of Dbp and Tef induction in the mutant animals, expression of the Dbp and Tef targets Cyp2c38 and Cyp2a5 was strongly affected (Fig. 5C). Expression of these Cyp enzymes was also induced by ketamine/xylazine anaesthesia alone within 10 min, whereas no increase occurred in response to isoflurane within this time frame (Suppl. Fig. S5B).
Dbp and Tef show a strong circadian regulation in the liver 30,31, but this was not affected by the loss of FGFR1 and FGFR2 (Fig. 5D). Thus, only the strong increase that occurs during the surgical procedure was dependent on FGFR signalling.
FGFs are direct regulators of Dbp and Tef expression in hepatocytes
Since hepatocytes rapidly loose expression of Cyp enzymes upon culturing 32 we studied their response to FGFs in vivo. When FGF7 was injected intraperitoneally into control mice, levels of phosphorylated FRS2α, Erk1/2 and to a lesser extent p38 increased in comparison to saline-injected mice, whereas phosphorylation of Akt was not affected. The FGF7-induced activation of FRS2α and p38 was not seen in Alb-R1/R2 mice, and the basal levels of phosphorylated p38 form were already much lower in the knockout animals. Only a minor and non-significant Erk1/2 activation was observed in Alb-R1/R2 mice (Fig. 6A and Suppl. Fig. S6). Most importantly, FGF7 caused a strong upregulation of Dbp and Tef expression in wild-type mice, but not in Alb-R1/R2 mice, demonstrating that the increase is dependent on FGFR signalling in hepatocytes (Fig. 6B,C). Consistent with the increase in Dbp and Tef expression, the mRNA levels of their targets Cyp2a5 and Cyp2c38 increased strongly in control mice but not in Alb-R1/R2 mice 30 min after FGF7 injection (Fig. 6D).
Fig. 6. FGF7 activates a cytoprotective response in the liver.

(A) Liver lysates were prepared from control and Alb-R1/R2 mice 10 and 30 min after injection of saline or 5 μg FGF7 and analyzed by western blotting for the levels of phosphorylated FRS2α and total and phosphorylated ERK1/2, p38, Akt and GAPDH. Each lysate was obtained from a single mouse. For statistical analysis see Suppl. Fig. S6. (B) Control mice were i.p. injected with 0.5 or 5 μg FGF7 or vehicle, sacrificed 10 min after injection, and RNAs from the liver were analyzed by qRT-PCR for expression of Dbp and Tef. (C,D) Alb-R1/R2 mice and control littermates were injected i.p. with saline or 5 μg FGF7. They were sacrificed 10 or 30 min after injection, and RNAs from the liver were analyzed by qRT-PCR for expression of Dbp and Tef (C) or Cyp2a5 and Cyp2c38 (D). Rps29 was used for normalization.
These studies demonstrate that FGFs control the expression of Dbp and Tef and their Cyp targets and strongly suggest that the loss of this regulation in Alb-R1/R2 mice causes impaired compound detoxification and thus increased injury in the regenerating liver.
Discussion
In this study we identified essential cytoprotective functions of FGFR1 and FGFR2 after PH, whereas CCl4-induced liver injury and fibrosis were not affected. Interestingly, the opposite finding was obtained for FGFR4 knockout mice 14, demonstrating different functions of these receptors in the injured liver. Hepatocyte proliferation after PH was neither affected in FGFR4 knockout mice nor in Alb-R1/R2 mice, whereas this process was impaired in mice expressing a dominant-negative FGFR2-IIIb mutant in hepatocytes 7. Since this mutant receptor inhibits signalling through all FGFR variants in response to common ligands, it seems likely that signalling by a single type of FGFR is sufficient for the proliferative response.
The most dramatic phenotype of the Alb-R1/R2 mice was the reduced survival after PH, which resulted from impaired compound metabolism as revealed by the almost complete rescue of the lethality upon omission of buprenorphine or by replacement of ketamine/xylazine by isofluorane. Increased mortality after PH was also observed in mice lacking the hepatocyte growth factor receptor or the EGF receptor in hepatocytes 33–35. Although various other abnormalities were found in these mice, it will be interesting to determine if expression of detoxifying enzymes is also impaired in these animals.
The impaired compound detoxification seen in Alb-R1/R2 mice most likely results from the loss of the strong surgery-induced expression of Dbp and Tef. Although FGFR signalling may also regulate Cyp expression independent of Dbp and Tef, the inability of Alb-R1/R2 mice to upregulate these transcription factors in response to PH is likely to contribute strongly to the reduced induction of Cyp enzymes. As a consequence, the capacity to appropriately metabolize endogenous and exogenous compounds is reduced, providing a likely explanation for the liver necrosis and death of the animals. Individual differences in metabolism/clearance of these drugs or minor differences in the amount of drugs applied may explain why some of the mice survived after PH. Consistent with our hypothesis, mice lacking Dbp, Tef and Hlf in all cells had increased serum transaminase levels, and this was also observed after PH in Alb-R1/R2 mice. In addition, the loss of Dbp, Tef and Hlf in the liver impaired the clearance of pentobarbital and enhanced the toxicity of anticancer drugs 30.
Although the omission of buprenorphine or the use of isofluorane strongly improved the survival rate, hepatic necrosis was not completely abolished. This is likely due to impaired detoxification of the drug used for anaesthesia as well as of endogenous compounds during the regeneration process. In addition, the remaining liver tissue may no longer fulfill further metabolic functions required for efficient regeneration and survival. In particular, triglyceride metabolism appears to be impaired, since we found prolonged hepatic steatosis in Alb-R1/R2 mice after PH (our unpublished data).
Since ketamine/xylazine and buprenorphine treatment of non-injured liver did not cause necrosis, the detoxification capacity of the non-injured liver of Alb/R1/R2 mice is obviously sufficient. This is consistent with the unaltered basal expression levels of Dbp and Tef. By contrast, liver injury results in upregulation of Dbp, Tef and their Cyp targets in control mice, and this is most likely required to increase the detoxification capacity of the remaining liver tissue, which is otherwise rate-limiting.
The strong and rapid upregulation of Dbp and Tef expression upon liver surgery in control but not in Alb-R1/R2 mice demonstrates that FGFR signalling controls the expression of these genes in hepatocytes. This control is obviously direct, since a similar upregulation of Dbp and Tef was observed upon injection of FGF7 into wild-type but not into Alb-R1/R2 mice. Therefore, it seems likely that the surgical procedure normally results in activation of FGFR signalling and subsequent induction of Dbp/Tef expression. The extremely rapid increase after surgery may result from mobilization of FGFs from the matrix upon manipulation of the liver. However, ligand-independent activation of FGFR signalling cannot be fully excluded, and this needs to be further explored.
Our results provide first evidence for a role of Dbp, Tef and their Cyp targets in liver regeneration and reveal a crucial role of FGFs in their regulation. These results suggest that therapeutic application of FGFs may be a promising strategy to prevent liver necrosis and to improve the survival rate of patients after surgical resection of large amounts of liver tissue, e.g. in patients with liver metastases. FGF7 may be most suitable for this purpose, since it activates FGFR2-IIIb on hepatocytes and it is already clinically approved for the treatment of mucositis in cancer patients 36. Future studies will reveal if the clinical use of FGFs 9 can be extended to the regenerating liver.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Grant support: ETH Zurich (TH 41/04-2) and the Swiss National Science Foundation (3100A0-109340) (to S.W.), the German Research Association (He 2458/14-1) and the Medical Faculty of the University of Regensburg (ReForM) (to CH), and the National Institutes of Health (grant HD04908 to D.M.O.).
We thank Dr. W. Xu, A. Kälin, O. Antsiferova, N. Hallschmid, and C. Born-Berclaz for valuable experimental help, Drs. P. Bugnon and T. Ramadan for performing PH, Drs. W. Kovacs and R. Grose for helpful suggestions and for providing primers, and Dr. A. Wendel for helpful suggestions with the manuscript. F.B. was a member of the Zurich graduate program in Molecular Life Sciences, Zurich.
Abbreviations
- BrdU
5-bromo-2′-deoxyuridine
- Cyp
Cytochrome P450 enzyme
- Dbp
D site albumin promoter binding protein
- FGF
Fibroblast growth factor
- FGFR
Fibroblast growth factor receptor
- FRS2
Fibroblast growth factor receptor substrate 2
- Hlf
hepatic leukemia factor
- MIP
Macrophage inflammatory protein
- PH
Partial hepatectomy
- RT-PCR
Reverse transcription polymerase chain reaction
- Tef
Thyrotrophic embryonic factor
Footnotes
Disclosures: The authors have no conflict of interest
Author contributions:
FB, TS, CH: data acquisition; FB, TS, CH and SW analyzed and interpreted data; CD, JP and DMO provided knockout mice.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–47. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 2.Fausto N. Involvement of the innate immune system in liver regeneration and injury. J Hepatol. 2006;45:347–9. doi: 10.1016/j.jhep.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diehl AM. Liver regeneration. Front Biosci. 2002;7:e301–14. doi: 10.2741/A925. [DOI] [PubMed] [Google Scholar]
- 5.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–50. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 6.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–36. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- 7.Steiling H, Wustefeld T, Bugnon P, et al. Fibroblast growth factor receptor signalling is crucial for liver homeostasis and regeneration. Oncogene. 2003;22:4380–8. doi: 10.1038/sj.onc.1206499. [DOI] [PubMed] [Google Scholar]
- 8.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:REVIEWS3005. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–53. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung J, Zheng M, Goldfarb M, et al. Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science. 1999;284:1998–2003. doi: 10.1126/science.284.5422.1998. [DOI] [PubMed] [Google Scholar]
- 11.Berg T, Rountree CB, Lee L, et al. Fibroblast growth factor 10 is critical for liver growth during embryogenesis and controls hepatoblast survival via beta-catenin activation. Hepatology. 2007;46:1187–97. doi: 10.1002/hep.21814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kan NG, Junghans D, Izpisua Belmonte JC. Compensatory growth mechanisms regulated by BMP and FGF signaling mediate liver regeneration in zebrafish after partial hepatectomy. Faseb J. 2009 doi: 10.1096/fj.09-131730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ueno H, Gunn M, Dell K, et al. A truncated form of fibroblast growth factor receptor 1 inhibits signal transduction by multiple types of fibroblast growth factor receptor. J Biol Chem. 1992;267:1470–6. [PubMed] [Google Scholar]
- 14.Yu C, Wang F, Kan M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275:15482–9. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 15.Yu C, Wang F, Jin C, et al. Increased carbon tetrachloride-induced liver injury and fibrosis in FGFR4-deficient mice. Am J Pathol. 2002;161:2003–10. doi: 10.1016/S0002-9440(10)64478-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C, Wang F, Jin C, et al. Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis. Am J Pathol. 2003;163:1653–62. doi: 10.1016/S0002-9440(10)63522-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Z, Evarts RP, Fujio K, et al. Expression of fibroblast growth factor receptors flg and bek during hepatic ontogenesis and regeneration in the rat. Cell Growth Differ. 1995;6:1019–25. [PubMed] [Google Scholar]
- 18.Latta M, Kunstle G, Leist M, et al. Metabolic depletion of ATP by fructose inversely controls CD95- and tumor necrosis factor receptor 1-mediated hepatic apoptosis. J Exp Med. 2000;191:1975–85. doi: 10.1084/jem.191.11.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beyer TA, Xu W, Teupser D, et al. Impaired liver regeneration in Nrf2 knockout mice: role of ROS-mediated insulin/IGF-1 resistance. Embo J. 2008;27:212–23. doi: 10.1038/sj.emboj.7601950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trokovic R, Trokovic N, Hernesniemi S, et al. FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. Embo J. 2003;22:1811–23. doi: 10.1093/emboj/cdg169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu K, Xu J, Liu Z, et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–74. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- 22.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–50. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 23.Steiling H, Muhlbauer M, Bataille F, et al. Activated hepatic stellate cells express keratinocyte growth factor in chronic liver disease. Am J Pathol. 2004;165:1233–41. doi: 10.1016/S0002-9440(10)63383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Ibrahimi OA, Olsen SK, et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ornitz DM, Xu J, Colvin JS, et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–7. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki M, Itoh T, Osada H, et al. Spleen-derived growth factor, SDGF-3, is identified as keratinocyte growth factor (KGF) FEBS Lett. 1993;328:17–20. doi: 10.1016/0014-5793(93)80956-u. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc. 2008;3:1167–70. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 28.Berson A, Fau D, Fornacciari R, et al. Mechanisms for experimental buprenorphine hepatotoxicity: major role of mitochondrial dysfunction versus metabolic activation. J Hepatol. 2001;34:261–9. doi: 10.1016/s0168-8278(00)00050-7. [DOI] [PubMed] [Google Scholar]
- 29.Capponi L, Schmitz A, Thormann W, et al. In vitro evaluation of differences in phase 1 metabolism of ketamine and other analgesics among humans, horses, and dogs. Am J Vet Res. 2009;70:777–86. doi: 10.2460/ajvr.70.6.777. [DOI] [PubMed] [Google Scholar]
- 30.Gachon F, Olela FF, Schaad O, et al. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab. 2006;4:25–36. doi: 10.1016/j.cmet.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 31.Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part II: The C/EBPs and D site-binding protein in cell cycle control, carcinogenesis, circadian gene regulation, liver regeneration, apoptosis, and liver-specific gene regulation. Pharmacol Rev. 2004;56:291–330. doi: 10.1124/pr.56.2.5. [DOI] [PubMed] [Google Scholar]
- 32.Maslansky CJ, Williams GM. Primary cultures and the levels of cytochrome P450 in hepatocytes from mouse, rat, hamster, and rabbit liver. In Vitro. 1982;18:683–93. doi: 10.1007/BF02796423. [DOI] [PubMed] [Google Scholar]
- 33.Borowiak M, Garratt AN, Wustefeld T, et al. Met provides essential signals for liver regeneration. Proc Natl Acad Sci U S A. 2004;101:10608–13. doi: 10.1073/pnas.0403412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huh CG, Factor VM, Sanchez A, et al. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101:4477–82. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A. 2007;104:17081–6. doi: 10.1073/pnas.0704126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finch PW, Rubin JS. Keratinocyte growth factor expression and activity in cancer: implications for use in patients with solid tumors. J Natl Cancer Inst. 2006;98:812–24. doi: 10.1093/jnci/djj228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.