

Abstract
The mammalian immune response is mediated by a heterotetrameric transcriptional control complex, called RFX, that regulates expression of the major histocompatibility complex class II (MHCII) genes. RFX comprises four proteins: RFX5 (two copies), RFXAP, and RFXB, and mutations and deletions that prevent assembly of the RFX complex have been linked to a severe immunodeficiency disorder. Two RFX5 and one RFXAP molecules assemble in the cytoplasm prior to nuclear localization, a process mediated by an N-terminal “dimerization domain” of RFX5 (RFX5N) and a C-terminal domain of RFXAP (RFXAPC). We previously presented evidence that RFXAPC is unstructured in the absence of RFX5N but adopts a regular structure in the RFX5N2·RFXAPC complex and that the RFX5N2ÄR FXAPC complex binds RFXB with high affinity. We now report the structure of the RFX5N2ÄR FXAPC complex, determined in solution by 15N- and 13C-edited NMR spectroscopy. RFX5N consists of a long central helix flanked by two shorter helices. The central helices of the two RFX5N molecules form an anti-parallel coiled coil, and the flanking helices pack at the ends of the long helices in a perpendicular arrangement such that the RFX5N dimer is shaped like a staple. RFXAPC consists of two α-helices that form a V-shaped structure that packs within the RFX5N2 staple. Leucine residues in the leucine-rich region of RFX5N (62-LYLYLQL-68) that are critical for MHCII gene expression in vivo contribute to both the dimer (Leu 64 and Leu 68) and RFX5N:RFXAPC interfaces (Leu 62 and Leu 66). The clustering of hydrophobic residues from different regions of RFXAPC suggests a potential binding site for RFXB.
Introduction
Major Histocompatibility complex class II (MHCII) molecules are essential for the initiation and regulation of the mammalian immune response. MHCII molecules present peptides that are derived from invading species on the surface of antigen-presenting cells. The MHCII-antigen complex is then bound by T-cell receptors on the surface of helper T-cells, which initiates a cascade of events leading to the adaptive immune response 1,2.
Expression of the MHCII gene products is tightly regulated by a cell-specific multi-protein complex 3. The core DNA binding component of this complex is the RFX complex, which serves to nucleate assembly of the other components on the MHCII promoters 4-10. The RFX complex is comprised of three proteins: RFX5, RFXAP, and RFXB. Deletions or point mutations in the RFX proteins that prevent formation of the complex, DNA binding, or recruitment of other components of the multi-protein complex have been shown to lead to a rare severe immunodeficiency disorder termed Bare lymphocyte syndrome (BLS). Although BLS affects few people around the world, patients with this disease lead to the discovery of a prototypical “disease of gene regulation” in the immune response pathway 6. Since the aberrant expression of any of the MHCII regulatory proteins results in a severe autosomal recessive disease 6, it is essential to have a better structural understanding of how the RFX proteins assemble to illustrate a generic view of how the human immune response is regulated.
RFX5 has four distinct domains: the oligomerization domain, the RFX DNA binding domain, the dimerization domain, and the transactivation domain 11. The oligomerization domain (RFX5N) is named due to its ability to form a tetramer when isolated, but is dimeric when domains C-terminal to it are present or when in a complex with RFXAP and RFXB 12. It also contains a leucine-rich region that has been shown to be essential for MHCII gene expression 13,14. RFX5 contains a second dimerization domain that, together with the oligomerization domain, flanks the RFX DNA binding domain and inhibit its ability to bind to DNA in the absence of RFXAP and RFXB 10. The transactivation domain of RFX5 is involved in recruiting other components of the multi-protein complex required to initiate and regulate MHCII gene expression 11,15. RFXAP is a 272 amino acid protein that contains a C-terminal domain (RFXAPC) that is necessary and sufficient for formation of the RFX complex 11,12,16-21. RFXB is a 260 amino acid protein that interacts with RFX5 and RFXAP through a series of ankyrin repeats 6,11,22. A dimer of RFX5 is proposed to form a complex with a monomer of RFXAP in the cytoplasm of antigen presenting cells through contacts made between the oligomerization domain of RFX5 and the C-terminal domain of RFXAP 11,12,16,21,23. The RFX52·RFXAP complex then translocates to the nucleus and binds to RFXB 23. RFXB simultaneously interacts with a region of RFX5 that comprises the RFX DNA binding domain and the dimerization domain, and the C-terminal domain of RFXAP 12. The RFX52·RFXAP·RFXB complex then binds to the MHCII promoter and recruits other components of the MHCII gene specific transcription complex 7,8,22,24-29.
We have previously shown that the C-terminal domain of RFXAP is unstructured but becomes structured upon binding to the oligomerization domain of RFX5 21. We have now solved the solution structure of the oligomerization domain of RFX5 bound to the C-terminal domain of RFXAP. The structure reveals why the C-terminal domain of RFXAP is unfolded in the absence of RFX5, and how RFX5 stabilizes its tertiary structure. The complex reveals the importance of a glutamine-rich region within the C-terminal domain of RFXAP, and a leucine-rich region in RFX5. The structure also suggests a potential interaction site for RFXB binding to RFXAP that would explain why the RFX5N2·RFXAPC complex has a higher affinity for RFXB than RFXAP alone.
RESULTS
Overview of the solution structure of the RFX5(25-90)2·RFXAP(214-272) complex
A stereo view of the top 20 structures of the RFX5(25-90)2·RFXAP(214-272) comple with the lowest target functions, along with a cartoon representation, is depicted in Figure 1. The x structures show good convergence for each of the α-helices and afforded pairwise backbone r.m.s deviations of 0.87 ± 0.14 Å. To differentiate between the two RFX5 oligomerization domain monomers in the structure, one monomer will be denoted with the identifier “A” and the second monomer with the identifier “B”. Each monomer of RFX5(25-90) forms three α-helices – Helix 1 (residues 30 – 36), Helix 2 (residues 40 – 55), and Helix 3 (residues 58 – 67) – that are connected by short loops (Figure 1B). The orientation of the three α-helices resemble a “C” shape, with the axes of Helix-1 and Helix-3 running approximately anti-parallel to each other and perpendicular to Helix-2. It deviates from the “C” shape by Helix-2 being below the plane formed by the axes from Helix-1 and Helix-2. In the homodimer, Helix-A2 and Helix-B2 from each monomer pack against each other in an anti-parallel manner with an approximately 40° angle between the axes of the two helices. Helix-A1 from one monomer and Helix-B3 from the other monomer pack against each other in a parallel manner. Helix-A3 and Helix-B1 from each monomer interact in a similar fashion. The C-terminal domain of RFXAP forms two α-helices – Helix-C1 (residues 226 – 238), and Helix-C2 (residues 242 – 261) – that are connected by a short loop. The two helices pack against each other close to the C-terminal end of Helix-C1 and at the N-terminal end of Helix-C2, with the helical axes making an approximately 50° angle to each other. In the complex, the two helices of RFXAPC pack against a hydrophobic surface formed by Helix-2A and Helix-3A from one monomer, and Helix-B2 and Helix-B3 from the other monomer of RFX5.
Figure 1.
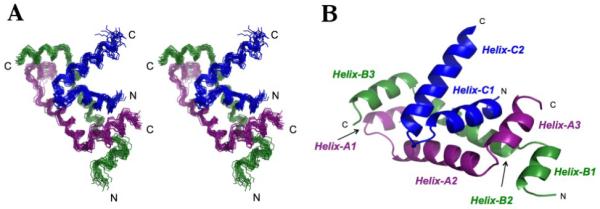
Overview of the complex between the N-terminal domain of RFX5 and the C-terminal domain of RFXAP (A) Stereo view of best-fit superposition of the backbone heavy atoms for the 20 refined complex structures for RFX5 residues 28-68 and RFXAP residues 225-260. (B) Representative ribbon diagram of the RFX5N2·RFXAPC complex. RFX5N monomer A is colored purple, monomer B colored green, and RFXAPC is colored blue. This figure and subsequent figures were generated using Pymol46.
Structure of the RFX5 oligomerization domain monomer
The details for the structure of each monomer of RFX5N are essentially identical and only one of them will be discussed here. Each monomer of RFX5N contains three α-helices that are connected by short β-extended “hinge regions” (Figure 2). The first helix spans Leu 30A through Gly 36A. Helix-A1 is initiated by an N-terminal helix cap formed by a hydrogen bond between the side-chain hydroxyl of Thr 29A and the backbone amide proton of Gln 32A. Helix-A1 terminates with a C-terminal helix cap hydrogen bond between the amide proton of Thr 37A and the backbone carbonyl of Leu 34A. The two helices are bridged by contacts made by the loop residue Ile 38A, which exhibits long range intra-molecular NOEs to residues in both Helix-A1 and Helix-A2. Specifically, the side chain protons of Ile 38A exhibit NOEs to Leu 34A in helix-A1, and strong NOEs to Val 42A and Gln 43A methyl protons in Helix-A2. The NOEs observed for this hinge region are characteristic of long-range hydrophobic interactions that stabilize backbone electrostatic helix capping 30. The multiple NOEs observed for Ile 38A orient Helix-A1 and Helix-A2 in an orthogonal manner. We have denoted the interactions between Helix-A1 and Helix-A2, comprised of residues Leu 34A, Ile 38A, and Val 42A, as “Hinge-1A” (Figure 2B).
Figure 2.
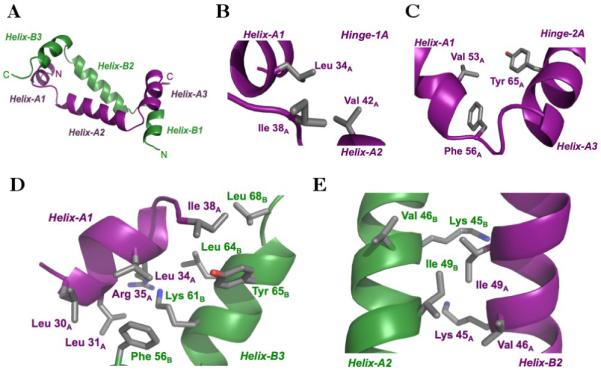
Structure of the RFX5 oligomerization dimer. (A) Ribbon representation of the RFX5N dimer. (B) Arrangement of hinge-1A residues in monomer A. (C) Arrangement of hinge-2A residues in monomer A. (D) Interactions between Helix-A1 and Hinge-1A with Hinge-2B and Helix-B3, referred to as “region 1” in the text. (E) Interactions between Helix-A2 and Helix-B2, referred to as “region 2” in the text. Side chains are represented as sticks with the carbon, oxygen, and nitrogen atoms colored gray, red, and blue, respectively.
Helix-A2 extends from Lys 40A to Lys 55A and is the longest helix in the RFX5 monomer. This helix has an N-terminal capping box formed by a hydrogen bond between the side-chain of Ser 39A and the backbone amide proton of Val 42A. Typical amide-amide NOEs between Ile 38A and Gln 43A, support this N-terminal helix cap. Interestingly, Phe 56A in the loop connecting Helix-A2 and Helix-A3 is the C-terminal helix-capping residue for Helix-A2. The carbonyl oxygen of Val 53A accepts the amide proton of Phe 56A to form a uniquely stabilized C-terminal aromatic helix cap. Characteristically, protons within an aromatic ring plane are up-field shifted and those on the edge of the ring are downfield shifted 31. The aromatic face of Phe 56A packs against the Hα and methyl protons of Val 53A, thereby up-field shifting these proton chemical shifts. These hydrophobic interactions further stabilize the helix-A2 C-terminal helix cap. In addition to exhibiting NOEs to Val 53A, Phe 56A also exhibits NOEs to Tyr 65A from Helix-A3, resulting in orienting these two helices in an orthogonal manner. We have denoted the region connecting Helix-A2 and Helix-A3 as “Hinge-2A”, where it is comprised of Val 53A, Phe 56A, and Tyr 65A (Figure 2C).
Helix-A3 spans residues Asp 58A to Gln 67A. An N-terminal helix cap leading into Helix-A3 is formed by a hydrogen bond between the side-chain of Ser 57A and the backbone amide proton of Asp 60A. The end of Helix-A3 terminates with a C-terminal helix cap hydrogen bond between the carbonyl oxygen of Tyr 65A and the backbone amide proton of Leu 68A. The aromatic face of Tyr 65A further stabilizes the Helix-A3 C-terminal helix cap through hydrophobic packing against the Leu 68A methyl protons. The Leu 68A side-chain methyl protons exhibit an up-field chemical shift corroborating a side-chain interaction with the aromatic face of Tyr 65A.
Structure of the RFX5 oligomerization domain dimer
The oligomerization domain dimer is stabilized by interactions between the monomers in three distinct regions: Region 1 is comprised of Helix-A1 and Hinge-1A packing against Hinge-2B and Helix-B3, Region 2 is comprised of Helix-A2 packing against Helix-B2, and Region 3 is comprised of Hinge-2A and Helix-A3 packing against Hinge-1B and Helix-B1. The first and third regions are related by the two-fold symmetry of the oligomerization domain dimer and are essentially identical. As a result only Region 1 and 2 will be discussed in detail. In Region 1, Ile 38A from Hinge-1A and Phe 56B from Hinge-2B have a key role at the protein-protein interface Figure 2D). Ile 38A packs against the side chains of Tyr 65B and Leu 68B from Helix-B3. The position of Ile 38A is supported by the side chain protons exhibiting an up-field chemical shift as a result of packing against the aromatic face of Tyr 65B. Phe 56B packs against a hydrophobic surface formed by Leu 30A, Leu 31A, and Leu 34A from Helix-A1. This binding mode is supported by NOEs between the side chain protons of Leu 30A and Leu 31A with the aromatic protons of Phe 56B. In addition to these “Hinge residue – Helix” interactions, there are several interactions between residues in Helix-A1 and Helix-B3. Leu 31A, and potentially Leu 34A, are involved in hydrophobic interactions with the aliphatic region of Lys 61B, whose extended position is supported by a direct NOE between Leu 31A and Lys 61B. In addition, the methyl groups of Leu 64B pack tightly against the main chain nitrogen and α-carbon of Arg 35A. This interaction correlates with unambiguous NOEs between the Leu 64A methyl group hydrogens and side chain hydrogens of Arg35B. Considering the close packing of Leu 64B against the main chain atoms of Arg 35A, this interaction clearly has an important role in positioning Helix-A1 against Helix-B3. The second region of interaction occurs at the crossover point of Helix-A2 and Helix-B2 (Figure 2E). The side chain of Val 49A packs against the plane of the peptide bond between Lys 45B and Val 46B, in addition to making hydrophobic interactions with the side chains. The symmetry-related residues, Val 49B, Lys 45A and Val 46A, make similar interactions.
Structure of the C-terminal domain of RFXAP
RFXAPC forms two α-helices, Helix-C1 and Helix-C2, that are connected together by a short loop (Figure 1B). Helix-C1 extends from Leu 226 through Leu 238. A traditional N-terminal helix cap stabilizes this helix via a hydrogen bond between the side chain of Thr 225 and the backbone amide proton of Glu 228. There were no observed NOE cross peaks indicative of a C-terminal helix cap for Helix-C1. NOE derived backbone distance restraints were obtained for Helix-C2 extending from Pro 242 to Glu 261. The N-terminus of Helix-C2 is protected by a hydrogen bond between the side-chain of Ser 241 and the Val 245 backbone amide proton. Long range side-chain NOEs for the C-terminal portion of RFXAPC beyond Lys 250 were ambiguously assigned due to a considerable amount of solvent exposure and signal overlap of glutamine residues.
Helix-C1 and Helix-C2 interact with each other through a small network of hydrophobic contacts between Leu 236 and Ser 237 from Helix-C1 and Val 245 and Leu 248 from Helix-C2 (Figure 3). The positions of the side chains was supported by a series of NOEs between the side-chain protons of Leu 236 and Ser 237 to the side-chain methyls of Val 245. Signal degeneracy prevented unambiguous assignment of side-chain to side-chain NOEs between a series of glutamines at the middle and N-terminal end of Helix-C1 and at the C-terminal end of Helix-C2 that faced each other. Molecular dynamic simulations for the RFX5(25-90)2·RFXAP(214-272) complex, using the Amber-10 Generalized Born solvation model, demonstrated that a polar zipper 32 comprised of glutamines could mediate interactions between RFXAP Helix-C1 and Helix-C2. An analysis of the average hydrogen bond pattern for the side-chains of Gln 229 and Gln 232 in Helix-C1 demonstrated that in 60% of the structures calculated Gln 229 formed at least one hydrogen bond with Gln 249, Gln 252, or Gln 253, and in 70% of structures Gln 232 formed van der Waals interactions with Gln 249 in addition to accepting and donating hydrogen bonds.
Figure 3.
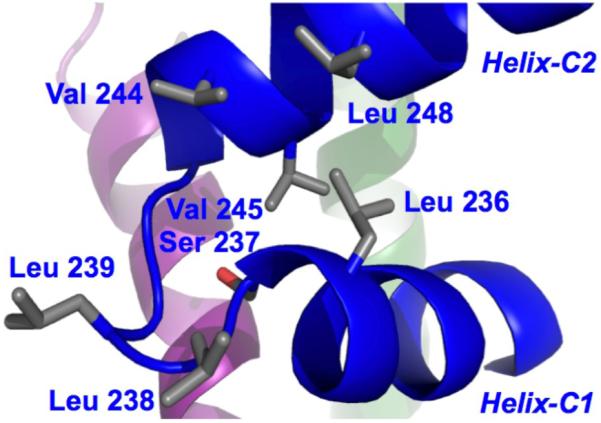
Intra-molecular interactions in the C-terminal domain of RFXAP.
Description of the RFX5N2·RFXAPC complex interactions
The distribution of amino acids on the surface of the RFX5 oligomerization domain dimer results in one face being comprised primarily of hydrophilic residues, and the other face, which interacts with RFXAP, being comprised primarily of hydrophobic residues (Figure 4). Contacts are made from both monomers of RFX5N to residues along Helix-C1, the loop connecting Helix-C1 and Helix-C2, and Helix-C2 of RFXAP (Figure 5). Helix-C1 from RFXAPC is stabilized by a series of hydrophobic interactions along the entire length of the helix with residues primarily from Helix-A2, but also from Helix-A3, Hinge-B1 and Helix-B2 of RFX5N (Figure 5A and 5B). At the N-terminus of Helix-C1, Leu 227 packs against a hydrophobic pocket formed by Ile 38B from Hinge-B1, and Leu 68A, and Tyr 65A from Helix-A3. Tyr 65A also has a key role in the packing of the central portion of Helix-C1 against Helix-A3. The position of Tyr 65A was defined by unambiguous NOEs to both Val 230 and Leu 231 side chain methyl groups as well as spin diffused signals to their backbone amide protons. The Tyr 65A aromatic protons have a single NOE to the Hγ2 of Val 230 and to the Hδ1 of L231, placing the aromatic edge of Tyr 65A between these two residues. This observation is supported by the up-field shifted proton chemical shift of the L231 Hδ1 methyl group. Together with Leu 66A, Tyr 65A packs against Leu 231 at the center of Helix-C1. Val 230 bridges interactions from both monomers of RFX5, packing against Val 46B from Helix-B2 and Val 53A from Helix-A2. At the C-terminal end of Helix-C3, NOEs between Lys 234 and Val 53A position the lysine side chain to pack against Val 50A and Val 53A from Helix-A3 (Figure 5B). Similarly, NOEs between Arg 235 and Leu 62A from Helix-A3 position the β- and γ-carbons of the arginine side chain in close proximity to the leucine side chain.
Figure 4.
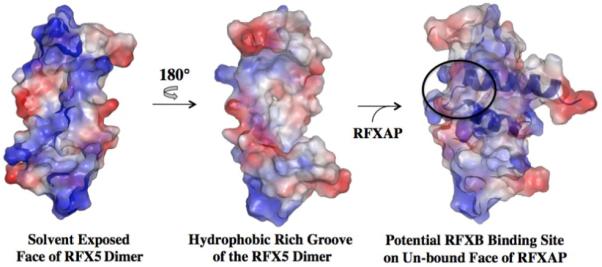
Surface electrostatic potential representation of the RFX5N dimer in the absence and presence of RFXAPC. Positively charged, negatively charged, and neutral regions are represented by blue, red, and white coloring. The potential binding site for RFXB on the complex is indicated by the black oval. The orientation of the RFX5N2·RFXAPC complex is similar to that shown in Figure 3.
Figure 5.
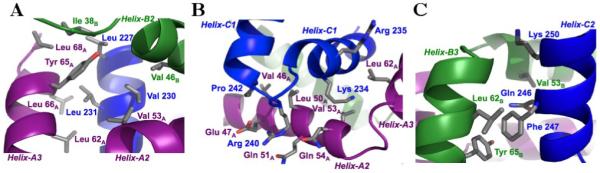
Interactions between the oligomerization domain of RFX5 and C-terminal domain of RFXAP. (A) Interaction between N-terminal residues of Helix-C1 of RFXAPC with RFX5N. B) Interaction between C-terminal residues of Helix-C1 and the loop region between Helix-C1 and Helix-C2 of RFXAPC and RFX5N. C) Interaction between Helix-C2 of RFXAPC with Helix-3B of RFX5N.
NOEs between Leu 50A in Helix-2A with Arg 240 and Pro 242 showed that the loop region between Helix-C1 and Helix-C2 is anchored against Helix-A2 (Figure 5B). The aliphatic portion of Arg 240 and Pro 242 pack against the side chains of Leu 50A and Val 46A, respectively. The positively charged guanidinium group of Arg 240 can also potentially form a salt bridge with the acidic carboxyl group of Glu 47A and/or hydrogen bonds with Gln 51A and Gln 54A.
The majority of contacts between Helix-C2 of RFXAPC and RFX5N are centered on a network of hydrophobic interactions formed by Phe 247 and residues from Helix-3B (Figure 5C). The edge of the Phe 247 aromatic ring inserts perpendicular to Tyr 65B for a T-stacking or edge-face aromatic interaction, which is supported by an NOE between Tyr 65B(Hε) and Phe 247(Hζ). The aromatic face of Phe 247 packs against the main chain and side chain of Leu 62B of Helix-B3, causing the Hα of Leu 62B to exhibit an up-field proton chemical shift. In addition to the contacts made by Phe 247, NOEs between Val 53B of Helix-B2 and Lys 250 position Val 53B to pack against Gln 246 and the aliphatic side chain of Lys 250.
RFX5N dimer asymmetry leads to peak doubling in the NMR data
The symmetry of the RFX5N dimer is disrupted to differing degrees at the RFXAP binding interface. We found that a majority of the NOEs for residues at the RFX5N2·RFXAPC complex interface directly overlapped in the 4D data even though they exhibited distinct HN peak doubling in the HSQC and in the triple resonance experiments. The observed peak doubling aided in the determination of residues located at both the dimer and complex interface. A region of the HSQC spectrum illustrating peak doubling of residues at the RFX5 dimer and RFXAP complex interface are highlighted in Figure 6A. The difference in signal resonance is dependent upon where the residue is located in the structure. For residues like Val 46 (Figure 2E), which are almost completely buried in the hydrophobic core of the RFX5 dimer, there is only a small shift in signal resulting from the asymmetry introduced upon RFXAP binding. In contrast, the local environment of residues directly involved at both the dimer and complex interface experience a greater change in their local environment, as illustrated by amide signal differences of Tyr 65 in Figure 6A. We also observed peak doubling in the 3D-HNCA, which was inconsistent throughout the backbone assignments. Small stretches of residues exhibiting doubling of signal occurred at various places in RFX5N and then would converge as illustrated in Figure 6B for residues Asn 44 – Gly 48. Since the signal separation only occurred in small stretches and in small differences in magnitude, we were unable to differentiate between monomers of RFX5N in the 3D experiments. Although the chemical shifts of residues in the HSQC and 3D experiments exhibited peak doubling, the equivalent residues in each monomer of RFX5N were indistinguishable in all the experiments. Interestingly, long-range NOEs between each monomer of RFX5 and NOEs between RFX5N and RFXAPC exhibited signal overlap in the 4D datasets. An example of 4D data signal overlap for a residue that had peak doubling in the HSQC is illustrated in the CCNOE plane for the Hε aromatic protons of Tyr 65 in Figure 6C. This residue is critical to both RFX5N dimerization and complex formation with RFXAP. The dimerization interactions are evident in the redundant NOEs with Ile 38B methyl protons. Interestingly, the 4D-CCNOE plane for the Tyr 65 Hε aromatic protons revealed sources of contact for Tyr 65A and Tyr 65B to RFXAPC helices C1 and C2, respectively (Figure 6C). The Tyr 65 Hε CCNOE plane demonstrates that Tyr 65A-Hε (monomer A) has an NOE to Val 230 and Leu 231 methyl protons (RFXAPC Helix C1), and an aromatic-aromatic NOE between Tyr 65B (monomer B) and Phe 247 Hδ (RFXAPC Helix C2). While there were no distinguishing NOEs between Tyr 65A and Tyr 65B in the 4D data, we propose that the HSQC peak on the left (Figure 6A) most likely corresponds to Tyr 65B based on its down-field shifted amide resonance potentially induced by the aromatic edge of Phe 247. The 4D data for the Tyr 65 aromatic protons was essential to determining the tertiary fold of the RFX5N2·RFXAPC binary complex.
Figure 6.

RFX5 NMR data asymmetry (A) Enlarged section of the HSQC highlighting the observed peak doubling. (B) HNCA strips for a stretch of amino acids showing peak doubling. Dotted lines indicate doubling of signal for the proton chemical shifts. (C) 4D CCNOE plane for Tyr 65 Hε to show overlapping signals that correspond to Tyr 65A and Tyr 65B in one plane. Specifically, Tyr 65A Hε (monomer A) has an NOE to Val 230 and L231 methyl protons (RFXAP helix-C1), and an aromatic-aromatic signal between Tyr 65B and F247 Hε (RFXAP helix-C2). *Indicates spin diffusion peaks.
DISCUSSION
The solution structure supports the presence of a dimer of RFX5 in the RFX complex
There are currently two models for the composition of the RFX complex. In the first model there is only one copy of RFX5, RFXAP and RFXB in the RFX complex 33. In our model there are two copies of RFX5 and one copy of RFXAP and RFXB 12. The structure of the RFX5(25-90)2·RFXAP(214-272) complex presented here clearly shows that residues from both monomers of RFX5N contacts RFXAPC and that a dimer would be required to form a stable complex between RFX5 and RFXAP (Figure 5). In addition, the contacts made by RFX5N to RFXAPC give rise to an equilibrium binding constant in the low nM range 21, supporting its biological relevance. The structure therefore supports the presence of a dimer of RFX5 in the RFX complex.
The key functional and structural regions of RFX5Nare contained within residues 30 – 68
Previous studies of RFX5 have shown that the first 19 residues can be removed without affecting the function of RFX515. Further deletion to residue 39 (RFX5ΔN39) still allowed formation of the RFX complex, although there was evidence that the protein or protein complex was unstable. The instability is likely due to the loss of Helix 1 in RFX5N, which is involved in dimerization of the oligomerization domain of RFX5 (Figure 2D). RFX5ΔN39 still contains the regions of RFX5N that interact with RFXAP (Figure 5), and can still dimerize through the second dimerization domain of RFX5. Taken together with the ability of RFXB to bridge interactions between RFX5 and RFXAP, it is clear how the RFX complex can still form with the RFX5ΔN39 construct. The last 20 residues of the oligomerization domain of RFX5 do not adopt a well defined structure and therefore likely serves as a flexible linker between the oligomerization domain and the RFX DNA binding domain. It is therefore difficult to make any assertions as to how the RFX DNA binding domain of RFX5 is positioned relative to the oligomerization domain. Further structural and/or biophysical studies will be required to understand how RFXAP and RFXB promote DNA binding of the RFX domain of RFX5.
RFXAPC cannot form a stable tertiary structure in the absence of RFX5
RFXAP is a member of a growing family of proteins that contain domains that are unfolded or only transiently folded in the absence of a partner protein 34,35. In our previous studies we showed that the C-terminal domain of RFXAP was unfolded in the absence of RFX5, but could adopt come degree of helical structure 21. The structure of the RFX5(25-90)2·RFXAP(214-272) complex presented here supports these observations. In the structure, RFXAPC forms two α-helices connected by a short loop. The two helices interact with each other through a small hydrophobic network formed by residues near the C-terminus of the first helix and at the N-terminus of the second helix. In addition, glutamines in each helix can potentially interact to form a glutamine polar zipper 32,36. In the absence of RFX5, it is highly unlikely that these contacts are sufficient to stabilize the two helices of the interaction domain of RFXAP relative to each other into a well-defined tertiary structure. RFX5N is able to stabilize this folded state of RFXAPC through numerous, primarily hydrophobic, contacts along almost the entire length of RFXAPC.
Effects of deletions and mutations on Helix-C2 of RFXAPC
Previous deletion and mutational studies have sought to define the regions of RFXAP that are important for function 11,16,18-20,22,37. Deletion of residues 267 – 272 of RFXAP did not have an effect on complex formation or MHCII gene expression, which is supported by the last 12 residues of RFXAP being exposed to solvent. Deletion of residues 246 – 272, however, prevented formation of the complex and expression of MHCII genes. This deletion removes most of Helix-C2, including residues that are involved at the interface between RFX5 and RFXAP, such as Phe 247 (Figure 5C). This deletion would therefore significantly destabilize formation of the RFX complex. The second helix of RFXAPC also encompasses a large proportion of the glutamine-rich region, which is contained within residues 246 – 266 (11 glutamines are located in this region in total). Mutation of the glutamines to alanine has been shown to have no effect on MHCII gene expression 20. In the complex, the majority of the glutamines are exposed to the solvent, which supports the observation that they serve a non-essential role in complex formation. Gln 246 is the only residue that is not exposed to the solvent, and is potentially involved in a Van der Waals interaction with the side chain of Val 53B. Since mutation of Gln 246 to alanine has no effect on MHCII gene expression it is unlikely that this interaction contributes to the stability of the complex. In addition to mutation of the glutamines, the study also analyzed the effect of mutating hydrophobic residues within the glutamine-rich region using the following mutations: RFXAP(F247A, L248A) and RFXAP(L254A, L255A, V269A, L260A). The RFXAP(L254A, L255A, V269A, L260A) mutant did not show any effect on MHCII gene expression, which is supported by these residues being exposed to solvent in the complex structure. In contrast, the RFXAP(F247A, L248A) mutation had a significant effect on MHCII gene expression. This is likely a result of the loss of Phe 247, which packs against Leu 62B in the complex (Figure 5C). Loss of this contact would destabilize formation of the RFX complex.
The leucine-rich region of RFX5N contributes to dimer formation and interaction with RFXAP
Analysis of cell lines that were defective in MHCII gene expression identified a single point mutation in RFX5 that prevented DNA binding and transactivation 13,14. The point mutation, RFX5(L66H), was located in a distinct leucine-rich region, which had the sequence 62–LYLYLQL–68. Mutation of any of the other leucines in this region produced the same phenotype as RFX5(L66H). In the complex structure, Leu 62 and Leu 66 are located adjacent to each other on one face of Helix-3 and are involved with interactions with RFXAP. Leu 62A and Leu 66A from one monomer of RFX5 form part of the hydrophobic interface that Leu 231 of RFXAP packs against (Figure 5A). In the other RFX5 monomer, Leu 66B is exposed to solvent, but Leu 62B packs closely against Phe 247 of RFXAP. Mutation of either Leu 62 or Leu 66 of RFX5 would therefore be expected to destabilize the RFX complex. Previous studies of RFX5(L66A) also suggested that this mutant may interfere with dimerization of the oligomerization domain 12-14. Leu 66 is not directly involved at the monomer-monomer interface and it is therefore not clear from the complex structure why its mutation would affect dimerization. Leu 64 and Leu 68 are also located adjacent to each other on another face of Helix-3 and appear to be particularly important for the positioning of Helix-1 against Helix-3 in RFX5 (Figure 2D). Mutation of these residues would be predicted to have derogatory consequences for the ability for the oligomerization domain of RFX5 to dimerize, and therefore form the appropriate surface for interaction with RFXAP. In support of these observation, crosslinking experiments of RFX5(L64A) and RFX5(L68A) reveal they form monomers in solution (unpublished data). In addition, Leu 68 is involved at the RFX5N2:RFXAPC interface through contacting Leu 227 of RFXAP.
A hydrophobic cluster on the surface of RFXAPC suggests a potential binding site for RFXB
In our previous studies we showed that RFXB forms a weak interaction with RFXAP(214-272), which dissociates at μM concentrations 12,21. In contrast, RFXB binds to the RFX5(25-90)2·RFXAP(214-272) complex with a Kd of 76 nM to form a highly stable complex. Since RFXB does not interact with the isolated oligomerization domain of RFX5, the interactions between RFXB and the complex are likely to be primarily mediated by RFXAP. Analysis of the accessible surface area of RFXAPC in the solution structure of the RFX5(25-90)2·RFXAP(214-272) complex revealed two clusters of exposed hydrophobic residues that could be involved in the interaction with RFXB. The first cluster is at the C-terminal end of Helix-C2 and comprises Leu 254, Leu 255, and Val 259. These are unlikely to have a major role in the interaction since their combined mutation to alanines had little effect on MHCII gene expression 19,20. The second cluster is comprised of Leu 236 from Helix-C1, Leu 238 and Leu 239 from the loop, and Val 244 and Leu 248 from Helix-C2 (Figure 3). This cluster is a particularly good candidate for the RFXB binding surface as it requires the positioning of the two helices of RFXAP(214-272) in a particular manner by RFX5 in order to form the hydrophobic surface. We therefore propose that RFX5 increases the affinity of RFXAP for RFXB by initially reducing the entropic cost of stabilizing the flexible C-terminal domain of RFXAP into a single conformation in the RFX52·RFXAP complex. The loss in entropy is presumably outweighed by the numerous contacts made to Helix-C1, the loop region, and Helix-C2 of RFXAPC by RFX5. The contacts made to RFXAPC by RFX5N promotes positioning of residues to form the RFXB binding site. In the absence of RFX5, RFXB may not be able to make sufficient contacts to RFXAP that would overcome the entropic cost associated with stabilizing the C-terminal domain of RFXAP into one conformation.
MATERIALS AND METHODS
Production and purification of the RFX5(25-90)2·RFXAP(214-272) complex
A polycistronic expression vector was constructed that expressed RFXAP(214-272) with a chitin binding domain (CBD) affinity tag and RFX5(25-90) with a polyhisitidine affinity tag. Both affinity tags contained TEV protease cleavage sites between the affinity tag and the protein in order to remove the affinity tag. The vector was constructed in an identical manner to a similar vector described previously 21. The polycistronic vector was sequenced at the University of Maryland Biotechnology Institute, Center for Biosystems Research DNA Sequencing Facility (College Park, MD). The 15N-labeled protein was expressed in E coli in an identical manner as described previously 21. The double-labeled sample was prepared in a similar manner, but supplementing with 13C-glucose as the sole carbon source.
The cell pellets were resuspended in 50 mM Citrate pH 5.5, 50 mM NaCl, 1 mM EDTA, and 1 mM DTT. A pH of 5.5 was used to protonate the histidines to facilitate binding to a SP sepharose cation exchange column (GE Healthcare). The protein was eluted from the SP sepharose column using an increasing gradient of NaCl. The eluted complex was dialyzed into 50 mM Tris pH 8.0, 500 mM NaCl, and 20 mM Imidizole and then passed over a Ni charged HisTrap column (GE Healthcare). The complex was eluted with an increasing concentration of imidazole and the affinity tags were cleaved with TEV protease for 4hrs at room temperature. After dialyzing overnight to remove the excess imidazole, the polyhistidine affinity tag, and the polyhisitidine-tagged TEV protease, was removed by passing back over the Ni HisTrap column. Size exclusion chromatography using a Superdex 75 16/60 column (GE Healthcare) was used to remove the cleaved chitin binding domain from the complex and as a final purification step. The purified 15N-labeled or 15N,13C-labeled RFX5(25-90)2·RFXAP(214-272) samples were dialyzed into 20mM Phosphate pH 7.0, 150 mM NaCl, 1mM DTT, and 1mM EDTA. Upon completion of each labeled RFX5(25-90)2·RFXAP(214-272) sample, 100 μL of 100 μM protein was sent to David King at the University of California, Berkeley to elucidate isotope incorporation using electrospray ionization (ESI) mass spectrometry and potential sample degradation. All of the RFX5(25-90)2·RFXAP(214-272) samples were stable and had between 96-99% 15N and or 13C isotope incorporation.
Multidimensional protein NMR spectroscopy
All NMR data of the RFX5(25-90)2·RFXAP(214-272) complex was collected at the Howard Hughes Medical Institute at UMBC using Bruker DRX 800MHz and/or DMX 600 MHz NMRs equipped with z-gradient cryoprobes at 35°C (308 K) using TopSpin acquisition software (Bruker). In all experiments, quadrature detection was acquired using the States-TPPI method 38. All NMR data was processed using NMRPipe 39 and analyzed using NMRVIEWJ 40. A combined set of standard 2D, 3D and 4D NOESY experiments were collected using the 15N and 15N,13C-edited RFX5(25-90)2·RFXAP(214-272) at 35° C using approximately 1mM sample concentrations. Protein backbone assignments were carried out following standard triple resonance methods using a 15N-edited HNCA and 15N-edited HNCOCA 31,41,42. Side-chain and long-range NOE assignments were made from 3D 15N-edited CBCA(CO)NH 42, 3D 15N-edited NOESY-HSQC (τmix= 150 ms) 43, 4D 15N, 13C-edited NOESY (τmix= 100 ms), and 4D 13C, 13C-edited NOESY (τmix = 100 ms collected in 100% D2O) datasets 43.
NOE cross peak assignments were assigned upper interproton distance restraints of 2.7 Å for strong, 3.3 Å for medium, and 5.0 Å for weak intensity signals 41. Additional distances were added to for NOEs with methyl groups (0.5 Å), methylene protons (0.8 Å), pseudo methyls (1.5 Å), and pseudo-aromatic protons (2.3 Å) 44,45. Backbone upper and lower limits for O-H bonds were set to 2.20 and 1.80 respectively. Similarly, the upper and lower limits were set to 3.20 Å and 2.70 Å for O-N backbone interactions. Only functional restraints were incorporated into structure calculations, which exclude restrictive side-chain NOEs that limit flexibility of particularly extended charged residues such as Arg or Lys. Six intra-residue NOEs were included to restrain the orientation capping residues and to preserve the symmetry along the Helix-A2 and Helix-B2 axis of the models according to supporting evidence in the data. Structure calculations and convergence statistics were carried out using CYANA 3.0 through simulated annealing and torsion angle refinement 45. Dimer NOEs between each monomer of RFX5 were manually tabulated to give a symmetrical unit. Statistical information is provided in Table 1 and summarizes the distance geometry restraints and structural statistics for the RFX52N·RFXAPC complex. The top 20 structures generated from 620 starting models with the lowest target functions were evaluated with PROCHECK-NMR 46.
Table 1.
| RFX52N·RFXAPC | |
|---|---|
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | 782 |
| Intra-residue | 6 |
| Inter-residue | |
| Sequential (|i − j| = 1) | 378 |
| Medium and long range (|i − j| > 1) | 292 |
| Intermolecular | |
| RFX5 dimer | 64 |
| RFX52-RFXAP | 42 |
| Backbone H-bonds (4/ H-bond) | 158 |
| Side-chain H-bonds (4/H-bond) | 16 |
| Target function (Å2) | |
| Mean (SD) | 0.11 (1.35E −02) |
| Minimum | 8.15E −02 |
| Maximum | 0.13 |
| Restraint violations (Å) | |
| Distance constraints (SD) | 0 |
| Van der Waals (SD) | 0.10 (0.03) |
| PROCHECK statistics (%) | |
| Residues in most favored regions | 91.0 |
| Residues in additional allowed regions | 6.0 |
| Residues in generously allowed regions | 1.0 |
| Residues in disallowed regions | 2.0 |
| Structure convergence (Å2) | |
| Average Heavy atom (SD) | 0.87 (0.14) |
| Average Backbone (SD) | 1.26 (0.14) |
Pairwise r.m.s. deviation was calculated among 20 refined structures.
Energy minimization refinement calculations of the RFX5N2·RFXAPC complex were carried out using AMBER-10 program package 48. The related force field library was generated using LEaP 48 and the protein complex subjected to restrained molecular dynamics at 25°C (1 ps). Upper limit distance restraints defining intermolecular NOEs corresponding to strong (2.7Å), medium (3.3Å), and weak (5.0Å) were employed.
To solve the structure of homodimers such as RFX5N2 the use of asymmetric labeling is often required. This was not the case with the RFX5N2·RFXAPC complex as the RFX5N dimer symmetry is disrupted by RFXAP and we were able to clearly differentiate between each monomer by NOEs resulting from interactions with RFXAP packing against the helices in RFX5N. For example, NOEs between Leu 231-Hδ and Tyr 65A-Hε clearly indicate that helix C1 of RFXAP packs against helix-A3 of RFX5, whereas NOEs between Phe 247-Hδ and Leu 62B Hδ are indicative of packing between helix C2 of RFXAP and Helix-B3 of the second RFX5 monomer. Likewise, NOEs observed between the side chain of RFXAP Arg 240 with the Hγ of RFX5 Glu 47A, confirmed that RFXAP indeed packs within the hydrophobic groove of the RFX5 dimer. The packing of the RFXAP C1-C2 junction within the middle of helix-A2 also breaks the symmetry of the RFX5 A2-B2 dimer interface, therefore verifying the NOEs observed for the helix A3-C1 and helix B3-C2 RFX5N2·RFXAPC complex interactions, respectively.
Molecular dynamic simulations for the complex were carried out under the Generalized Born (GB) implicit solvation model using Amber-10 48 to predict formation of hydrogen bonds between glutamines in Helix-C1 and Helix-C2 in RFXAPC. Briefly, the GB solvation model is used in molecular dynamic calculations to represent the solvent surrounding a macromolecule as a continuous medium and can be used to approximate the average behavior of solvent exposed surfaces 48,49. The dynamic simulations were carried out for 20 structures at a temperature range of 0 to 25 K over 600,000 steps using NOE derived distance constraints obtained from the NMR data.
Atomic Coordinates
The atomic coordinates for the RFX5N2·RFXAPC complex have been deposited in the Protein Data Bank under the accession code 2KW3
ACKNOWLEDGEMENTS
The authors of this work would like to thank Robert Edwards, Chen Yu, and the members of the Summers lab in the Department of Chemistry and Biochemistry at UMBC for help with NMR data collection, processing of NMR data and technical support. The authors also thank David Shaw King for mass spectrometry analysis of labeled protein samples. This work was supported by University of Maryland Baltimore County internal research grants and the NIGMS Initiative for Minority Student Development; Grant Number R25-GM55036.
Abbreviations used
- MHCII
major histocompatibilty complex class II
- RFX
regulatory factor X
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- TEV
Tobacco etch virus
- HTT
6 histidine affinity tag with TEV cleavage site
- HSQC
heternonuclear single quantum coherence
- CCNOE
carbon-carbon nuclear overhauser effect
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu. Rev. Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 2.Ting JP, Zhu XS. Class II MHC genes: a model gene regulatory system with great biologic consequences. Microbes Infect. 1999;1:855–861. [PubMed] [Google Scholar]
- 3.Boss JM, Jensen PE. Transcriptional regulation of the MHC class II antigen presentation pathway. Curr. Opin. Immunol. 2003;15:105–111. doi: 10.1016/s0952-7915(02)00015-8. [DOI] [PubMed] [Google Scholar]
- 4.Emery P, Durand B, Mach B, Reith W. RFX proteins, a novel family of DNA binding proteins conserved in the eukaryotic kingdom. Nucleic Acids Res. 1996;24:803–807. doi: 10.1093/nar/24.5.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hake SB, Masternak K, Kammerbauer C, Janzen C, Reith W, Steimle V. CIITA leucine-rich repeats control nuclear localization, in vivo recruitment to the major histocompatibility complex (MHC) class II enhanceosome, and MHC class II gene transactivation. Mol. Cell Biol. 2000;20:7716–7725. doi: 10.1128/mcb.20.20.7716-7725.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu. Rev. Immunol. 2001;19:331–373. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- 7.Durand B, Kobr M, Reith W, Mach B. Functional complementation of major histocompatibility complex class II regulatory mutants by the purified X-box-binding protein RFX. Mol. Cell. Biol. 1994;14:6839–6847. doi: 10.1128/mcb.14.10.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasegawa SL, Boss JM. Two B cell factors bind the HLA-DRA X box region and recognize different subsets of HLA class II promoters. Nucleic Acids Res. 1991;19:6269–6276. doi: 10.1093/nar/19.22.6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, Aebersold R, et al. A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat. Genet. 1998;20:273–277. doi: 10.1038/3081. [DOI] [PubMed] [Google Scholar]
- 10.Garvie CW, Boss JM. Assembly of the RFX complex on the MHCII promoter: role of RFXAP and RFXB in relieving autoinhibition of RFX5. Biochim. Biophys. Acta. 2008;1779:797–804. doi: 10.1016/j.bbagrm.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 11.DeSandro AM, Nagarajan UM, Boss JM. Associations and interactions between bare lymphocyte syndrome factors. Mol. Cell. Biol. 2000;20:6587–6599. doi: 10.1128/mcb.20.17.6587-6599.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garvie CW, Stagno JR, Reid S, Singh A, Harrington E, Boss JM. Characterization of the RFX complex and the RFX5(L66A) mutant: implications for the regulation of MHC class II gene expression. Biochemistry. 2007;46:1597–1611. doi: 10.1021/bi6023868. [DOI] [PubMed] [Google Scholar]
- 13.Brickey WJ, Wright KL, Zhu XS, Ting JP. Analysis of the defect in IFN-gamma induction of MHC class II genes in G1B cells: identification of a novel and functionally critical leucine-rich motif (62-LYLYLQL-68) in the regulatory factor X 5 transcription factor. J. Immunol. 1999;163:6622–6630. [PubMed] [Google Scholar]
- 14.Jabrane-Ferrat N, Nekrep N, Tosi G, Esserman LJ, Peterlin BM. Major histocompatibility complex class II transcriptional platform: assembly of nuclear factor Y and regulatory factor X (RFX) on DNA requires RFX5 dimers. Mol. Cell Biol. 2002;22:5616–5625. doi: 10.1128/MCB.22.15.5616-5625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villard J, Peretti M, Masternak K, Barras E, Caretti G, Mantovani R, Reith W. A functionally essential domain of RFX5 mediates activation of major histocompatibility complex class II promoters by promoting cooperative binding between RFX and NF-Y. Mol. Cell Biol. 2000;20:3364–3376. doi: 10.1128/mcb.20.10.3364-3376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durand B, Sperisen P, Emery P, Barras E, Zufferey M, Mach B, Reith W. RFXAP, a novel subunit of the RFX DNA binding complex is mutated in MHC class II deficiency. Embo. J. 1997;16:1045–1055. doi: 10.1093/emboj/16.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caretti G, Cocchiarella F, Sidoli C, Villard J, Peretti M, Reith W, Mantovani R. Dissection of functional NF-Y-RFX cooperative interactions on the MHC class II Ea promoter. J. Mol. Biol. 2000;302:539–552. doi: 10.1006/jmbi.2000.4028. [DOI] [PubMed] [Google Scholar]
- 18.Nekrep N, Jabrane-Ferrat N, Peterlin BM. Mutations in the bare lymphocyte syndrome define critical steps in the assembly of the regulatory factor X complex. Mol. Cell Biol. 2000;20:4455–4461. doi: 10.1128/mcb.20.12.4455-4461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peretti M, Villard J, Barras E, Zufferey M, Reith W. Expression of the three human major histocompatibility complex class II isotypes exhibits a differential dependence on the transcription factor RFXAP. Mol Cell Biol. 2001;21:5699–5709. doi: 10.1128/MCB.21.17.5699-5709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long AB, Ferguson AM, Majumder P, Nagarajan UM, Boss JM. Conserved residues of the bare lymphocyte syndrome transcription factor RFXAP determine coordinate MHC class II expression. Mol. Immunol. 2006;43:395–409. doi: 10.1016/j.molimm.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 21.Briggs L, Laird K, Boss JM, Garvie CW. Formation of the RFX gene regulatory complex induces folding of the interaction domain of RFXAP. Proteins. 2009;76:655–664. doi: 10.1002/prot.22379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nekrep N, Geyer M, Jabrane-Ferrat N, Peterlin BM. Analysis of ankyrin repeats reveals how a single point mutation in RFXANK results in bare lymphocyte syndrome. Mol. Cell Biol. 2001;21:5566–5576. doi: 10.1128/MCB.21.16.5566-5576.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagarajan UM, Long AB, Harreman MT, Corbett AH, Boss JM. A hierarchy of nuclear localization signals governs the import of the regulatory factor X complex subunits and MHC class II expression. J. Immunol. 2004;173:410–419. doi: 10.4049/jimmunol.173.1.410. [DOI] [PubMed] [Google Scholar]
- 24.Reith W, Satola S, Sanchez CH, Amaldi I, Lisowska-Grospierre B, Griscelli C, et al. Congenital immunodeficiency with a regulatory defect in MHC class II gene expression lacks a specific HLA-DR promoter binding protein, RF-X. Cell. 1988;53:897–906. doi: 10.1016/s0092-8674(88)90389-3. [DOI] [PubMed] [Google Scholar]
- 25.Jabrane-Ferrat N, Fontes JD, Boss JM, Peterlin BM. Complex architecture of major histocompatibility complex class II promoters: reiterated motifs and conserved protein-protein interactions. Mol. Cell Biol. 1996;16:4683–4690. doi: 10.1128/mcb.16.9.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mudhasani R, Fontes JD. Multiple interactions between BRG1 and MHC class II promoter binding proteins. Mol. Immunol. 2005;42:673–682. doi: 10.1016/j.molimm.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 27.Masternak K, Muhlethaler-Mottet A, Villard J, Zufferey M, Steimle V, Reith W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14:1156–1166. [PMC free article] [PubMed] [Google Scholar]
- 28.Scholl T, Mahanta SK, Strominger JL. Specific complex formation between the type II bare lymphocyte syndrome-associated transactivators CIITA and RFX5. Proc. Natl. Acad. Sci. USA. 1997;94:6330–6334. doi: 10.1073/pnas.94.12.6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu XS, Linhoff MW, Li G, Chin KC, Maity SN, Ting JP. Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB to cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol. Cell Biol. 2000;20:6051–6061. doi: 10.1128/mcb.20.16.6051-6061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aurora R, Rose GD. Helix capping. Protein. Sci. 1998;7:21–38. doi: 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; New York: 1986. [Google Scholar]
- 32.Perutz M. Polar zippers: their role in human disease. Protein Sci. 1994;3:1629–1637. doi: 10.1002/pro.5560031002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burd AL, Ingraham RH, Goldrick SE, Kroe RR, Crute JJ, Grygon CA. Assembly of major histocompatability complex (MHC) class II transcription factors: association and promoter recognition of RFX proteins. Biochemistry. 2004;43:12750–12760. doi: 10.1021/bi030262o. [DOI] [PubMed] [Google Scholar]
- 34.Wright PE, Dyson HJ. Linking folding and binding. Curr. Opin. Struct. Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 36.Guo L, Han A, Bates DL, Cao J, Chen L. Crystal structure of a conserved N-terminal domain of histone deacetylase 4 reveals functional insights into glutamine-rich domains. Proc. Natl. Acad. Sci. USA. 2007;104:4297–4302. doi: 10.1073/pnas.0608041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westerheide SD, Boss JM. Orientation and positional mapping of the subunits of the multicomponent transcription factors RFX and X2BP to the major histocompatibility complex class II transcriptional enhancer. Nucleic Acids Res. 1999;27:1635–1641. doi: 10.1093/nar/27.7.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A, Gronenborn AM, Clore GM. Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: application to interleukin 1 beta. Biochemistry. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 39.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 40.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 41.Clore GM, Gronenborn AM. Structures of larger proteins in solution: three- and four-dimensional heteronuclear NMR spectroscopy. Science. 1991;252:1390–1399. doi: 10.1126/science.2047852. [DOI] [PubMed] [Google Scholar]
- 42.Grzesiek S, Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 43.Kay LE, Clore GM, Bax A, Gronenborn AM. Four-dimensional heteronuclear triple-resonance NMR spectroscopy of interleukin-1 beta in solution. Science. 1990;249:411–414. doi: 10.1126/science.2377896. [DOI] [PubMed] [Google Scholar]
- 44.Clore GM, Gronenborn AM, Nilges M, Ryan CA. Three-dimensional structure of potato carboxypeptidase inhibitor in solution. A study using nuclear magnetic resonance, distance geometry, and restrained molecular dynamics. Biochemistry. 1987;26:8012–8023. doi: 10.1021/bi00398a069. [DOI] [PubMed] [Google Scholar]
- 45.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 46.Laskowski RJA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 47.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2008. [Google Scholar]
- 48.Walker RC, Crowley MF, Case DA. The implementation of a fast and accurate QM/MM potential method in Amber. J. Comput. Chem. 2008;29:1019–1031. doi: 10.1002/jcc.20857. [DOI] [PubMed] [Google Scholar]
- 49.Onufriev A, Case DA, Bashford D. Effective Born radii in the generalized Born approximation: the importance of being perfect. J. Comput. Chem. 2002;23:1297–1304. doi: 10.1002/jcc.10126. [DOI] [PubMed] [Google Scholar]