

Abstract
Background and aim
Friedreich's ataxia (FRDA) and ataxia with oculomotor apraxia type 2 (AOA2) are the two most frequent forms of autosomal recessive cerebellar ataxias. However, brain metabolism in these disorders is poorly characterized and biomarkers of the disease progression are lacking. We aimed at assessing the neurochemical profile of the pons, the cerebellar hemisphere and the vermis in patients with FRDA and AOA2 to identify potential biomarkers of these diseases.
Methods
Short-echo, single voxel proton (1H) magnetic resonance spectroscopy data were acquired from 8 volunteers with FRDA, 9 volunteers with AOA2, and 38 control volunteers at 4T. Disease severity was assessed by the Friedreich's Ataxia Rating Scale (FARS).
Results
Neuronal loss/dysfunction was indicated in the cerebellar vermis and hemispheres in both diseases by lower total N-acetylaspartate levels than controls. The putative gliosis marker myo-inositol was higher than controls in the vermis and pons in AOA2 and in the vermis in FRDA. Total creatine, another potential gliosis marker, was higher in the cerebellar hemispheres in FRDA relative to controls. Higher glutamine in FRDA and lower glutamate in AOA2 than controls were observed in the vermis, indicating different mechanisms possibly leading to altered glutamatergic neurotransmission. In AOA2, total N-acetylaspartate levels in the cerebellum strongly correlated with the FARS score (p < 0.01).
Conclusion
Distinct neurochemical patterns were observed in the two patient populations, warranting further studies with larger patient populations to determine if the alterations in metabolite levels observed here may be utilized to monitor disease progression and treatment.
Keywords: MRS, Friedreich's, AOA2, ataxia, brain, metabolites
1. Introduction
Autosomal recessive cerebellar ataxias (ARCAs) are a genetically and clinically heterogeneous group of neurodegenerative diseases. The most common ARCA in the Indo-European population is Friedreich's ataxia (FRDA) (Anheim et al. 2009a, Campuzano et al. 1996), and recent epidemiological studies indicate that ataxia with oculomotor apraxia type 2 (AOA2) is the second most frequent ARCA identified so far (Anheim et al. 2009a, Le Ber et al. 2004). Clinically, AOA2 bears some resemblance to FRDA (age of onset, cerebellar signs, peripheral neuropathy), leading to misdiagnosis in AOA2; however, these diseases belong to different categories of ataxias based on the function of the affected protein. Friedreich's ataxia is considered a progressive, metabolic ataxia, whereas AOA2 belongs to the group of ataxias that result from defects in the DNA repair machinery (Palau et al. 2006).
In FRDA, the gene coding for frataxin is affected (Campuzano et al. 1996). Although its exact function is still unknown, frataxin is involved in iron homeostasis (Calabrese et al. 2005, Lodi et al. 2006, Pandolfo et al. 2009). In patients, a GAA repeat expansion results in decreased protein transcription (Campuzano et al. 1996), which leads to excessive reactive oxygen species production, altered mitochondrial respiratory chain function and iron deposition in cardiac, muscular, spinal and cerebral tissue (Bradley et al. 2000, Calabrese et al. 2005, Koeppen et al. 2007). In particular, the increase in reactive oxygen and nitrogen species and the altered energy balance due to mitochondrial dysfunction are thought to lead to neuronal death (Calabrese et al. 2005, Lodi et al. 2006, Voncken et al. 2004). In the central nervous system, FRDA is characterized by the degeneration of large sensory neurons in the dorsal root ganglia, followed by degeneration of sensory posterior columns, spinocerebellar tracts, corticospinal motor tracts and atrophy of the large sensory fibers in peripheral nerves. In the brain, the dentate nucleus is largely affected and exhibits grumose degeneration and neuronal loss (Koeppen et al. 2007). Imaging studies reveal mild vermian atrophy in advanced stages of the disease (Ormerod et al. 1994). Olivopontocerebellar structures seem unaffected (Anheim et al. 2009a, Mascalchi et al. 2002, Ormerod et al. 1994), although alterations of the pons, medulla and inferomedial portions of the cerebellar hemispheres have been observed in some cases (Della Nave et al. 2004, Della Nave et al. 2008, Guerrini et al. 2009, Oppenheimer 1979, Pagani et al. 2010).
AOA2 was recently shown to result from mutations in the senataxin gene (Asaka et al. 2006, Duquette et al. 2005, Moreira et al. 2004), likely causing a loss of function of the senataxin protein. Senataxin is thought to be involved in double strand break DNA repair and response to oxidative stress but also in transcription regulation via exon splicing (Airoldi et al. 2009, Suraweera et al. 2007, Suraweera et al. 2009). Patients with AOA2 have elevated α-fetoprotein (AFP) levels, a tumor marker (Anheim et al. 2009b, Le Ber et al. 2005), but without increased occurrence of cancer as observed in ataxiatelangiectesia. Oculomotor apraxia is not a systematic finding and appears to be present in about 50% of patients (Anheim et al. 2009b). Peripheral neuropathy has also been described in this disease (Asaka et al. 2006, Criscuolo et al. 2006, Gazulla et al. 2009, Le Ber et al. 2004, Le Ber et al. 2005, Tazir et al. 2009). In the brain, imaging studies revealed extensive vermian atrophy (Anheim et al. 2009a, Bernard et al. 2008, Le Ber et al. 2004, Nicolaou et al. 2008, Schols et al. 2008) which is stable shortly after the onset of the disease (Anheim et al. 2009a). Criscuolo et al. reported extensive Purkinje cell loss and mild fibrous gliosis in the cerebellar cortex, more severely in the vermis compared to the hemispheres (Criscuolo et al. 2006). The same authors reported a slight reduction in the size of the brain stem and spinal cord, and a lower number of neurons in the dentate nuclei.
Little is known about the metabolic status of the brain in both FRDA and AOA2. High field 1H magnetic resonance spectroscopy (1H-MRS) is a powerful tool for the non-invasive characterization of biochemical alterations in the brain because it enables the measurement of neurochemical profiles of 10-15 metabolites in localized brain regions in humans (Öz et al. 2010a, Tkáč et al. 2009). Few MRS studies of FRDA have been published to date (Franca et al. 2009, Guerrini et al. 2009, Mascalchi et al. 2002, Viau et al. 2005), reporting metabolite ratios (NAA/creatine, choline/creatine, myo-inositol/creatine, glutamate+glutamine/creatine), while AOA2 has not been studied by MRS so far. Neurochemical levels measured by MRS in ARCAs can provide insights into the disease processes and could potentially provide non-invasive biomarkers of disease progression. Our goal was therefore to measure the neurochemical profiles of the pons, cerebellar hemisphere (encompassing the dentate nucleus), and vermis in patients with FRDA or AOA2 in order to identify disease biomarkers. We used single-voxel, short-echo time MRS at 4T to measure metabolite levels in the brain and investigated their correlation with scores on a validated ataxia rating scale (Fahey et al. 2007, Subramony et al. 2005). In addition, cerebrospinal fluid (CSF) was collected from a subset of the volunteers and F2-isoprostanes were measured as an independent marker of oxidative stress (Greco et al. 2000).
2. Results
Patient demographics and CSF data
Table 1 shows the patient demographics. The FARS score ranged from 34 to 109 in patients with FRDA, and from 44 to 75 in patients with AOA2. Mean FARS scores of the two patient groups were comparable and significantly higher than controls as expected. F2-isoprostane levels in CSF were comparable between patients and controls. In both groups of patients, CSF glucose concentration was higher than their respective control groups, while lactate levels were comparable between the groups. In controls, lactate concentration correlated with age (r2=0.8, p=0.00001).
Table 1.
Group demographics and CSF and serum measures. FRDA, patients with Friedreich's ataxia; AOA2, patients with ataxia with oculomotor apraxia type 2; CTRL FRDA and CTRL AOA2, control populations age matched for the FRDA and AOA2 groups, respectively. All data are presented as means ± standard deviations.
| FRDA (N = 8) | CTRL FRDA (N = 29) | AOA2 (N = 9) | CTRL AOA2 (N = 33) | |
|---|---|---|---|---|
| Gender (F/M) | 5/3 | 15/14 | 5/4 | 14/19 |
| Age (years) | 27 ± 9 | 26 ± 6 | 37 ± 9 | 33 ± 12 |
| Age of onset (years) | 12 ± 6 | n/a | 16 ± 6 | n/a |
| Disease duration (years) | 14 ± 9 | n/a | 21 ± 7 | n/a |
| # of GAA triplet repeats (short allele) | 611 ± 237 (range: 120 - 970) | n/a | n/a | n/a |
| CSF glucose (mg/dL) | 61 ± 2* (N=3) | 51 ± 5 (N = 20) | 65 ± 5**** (N = 7) | 52 ± 6 (N = 23) |
| CSF lactate (mmol/L) | 2.3 ± 1.2 (N=3) | 1.7 ± 0.2 (N=9) | 1.9 ± 0.5 (N=5) | 1.7 ± 0.2 (N=9) |
| CSF F2-isoprostanes (pg/mL) | 31 ± 10 (N=3) | 29 ± 8 (N=20) | 23 ± 5 (N=7) | 31 ± 9 (N=23) |
| CSF vermis (%) | 18 ± 5*** | 11 ± 4 | 50 ± 13**** | 11 ± 4 |
| CSF cerebellar hemisphere (%) | 3.2 ± 0.3 | 2.0 ± 1.3 | 8.5 ± 4.5** | 2.1 ± 1.3 |
| CSF pons (%) | 1.9 ± 0.1 | 2.5 ± 0.1 | 2.9 ± 0.1 | 2.4 ± 0.1 |
| Serum α-fetoprotein (μg/L) | n/a | n/a | 45 ± 53 (range: 9. 3 – 150, N = 6) | n/a |
| FARS score | 61 ± 26*** (range: 34 - 109) | 2 ± 2 | 59 ± 11*** (range: 44 – 75) | 3 ± 3 (N = 28) |
p=0.0033
p= 0.0015
p=0.0003
p<0.0001 (patient groups vs. their respective controls). Sample sizes are indicated if data are available from a subset of subjects in a group.
Atrophy
Figure 1 illustrates the sagittal and coronal T2-weighted images acquired in a control, a patient with FRDA and a patient with AOA2. In patients with FRDA, the spinal cord was visibly atrophied. The cerebellar cortical structures appeared intact on MRI, but mild atrophy was indicated by the CSF content in the vermis voxel (table 1). The pons was spared. In patients with AOA2, the entire cerebellum was visibly affected and characterized by severe atrophy confirmed by CSF content measurements (table 1). Pons did not show any detectable atrophy.
Figure 1.

T2-weighted images acquired in the mid-sagittal (left) and coronal planes (right) in a control subject (CTRL), a patient with FRDA (FRDA) and a patient with AOA2 (AOA2). Boxes indicate the voxel locations (1: pons; 2: vermis and 3: cerebellar hemisphere).
Metabolite analysis
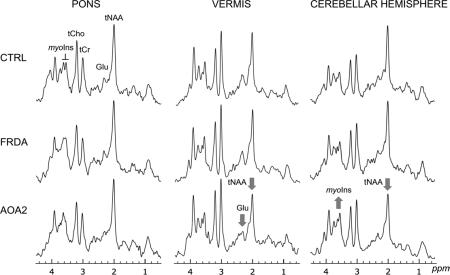
Typical spectra acquired in controls, patients with FRDA and patients with AOA2 are shown in Figure 2. Note that each brain region is characterized by a distinct spectral pattern. Some neurochemical differences were visible in individual spectra from patients with AOA2, in the vermis and the cerebellar hemispheres, notably the N-acetylaspartate (NAA, 2.01 ppm), myo-inositol (myo-Ins, 3.5 and 3.6 ppm) and glutamate (Glu, 2.1 ppm) peaks.
Figure 2. Short echo time 1H spectra acquired in the three regions (pons, vermis, cerebellar hemisphere) in a control, a patient with FRDA and a patient with AOA2.

tNAA, N-acetylaspartate + N-acetylaspartylglutamate; Glu, glutamate; tCr, creatine + phosphocreatine; tCho, choline-containing compounds; myo-Ins, myo-inositol. The arrows indicate visible differences in the patient spectrum compared to the corresponding control spectrum. Displayed spectra (STEAM, TE = 5 ms, TR = 4.5 s) are the sum of 128 transients after B0 and eddy-current correction. All spectra were processed identically, and weighted with the same Gaussian function prior to Fourier transformation. For the pons, the voxel size (VOI) was 4.1 ml; vermis, VOI = 6.3 ml; cerebellar hemisphere, VOI = 4.9 ml.
Figure 3 shows the metabolite concentrations that exhibited a significant difference or a trend for a difference in one or both patient groups relative to controls (for all metabolite concentrations, see table 2). In the vermis, the total concentration of N-acetylaspartate and N-acetylaspartylglutamate (tNAA) was lower in both the FRDA and AOA2 groups (by 11 and 17%, p=0.03 and 0.004, respectively) compared to their control groups. Myo-inositol concentration was significantly higher in both patient groups than controls (FRDA, +16%, p = 0.003 and AOA2, +19%, p = 0.004). The glutamate-to-glutamine ratio (Glu/Gln) was lower than controls in both pathologies (p = 0.03 for AOA2, p = 0.007 for FRDA), but due to higher glutamine concentration in FRDA (p = 0.01) and lower glutamate in AOA2 (p = 0.001).
Figure 3. Metabolite concentrations that differed (or showed a trend for a difference) between patient groups vs. controls in the three regions of interest.

All data are displayed as mean ± standard deviation. Gln, glutamine; Glu, glutamate; myo-Ins, myo-inositol; tNAA, N-acetylaspartate + N-acetylaspartylglutamate; tCr, creatine + phosphocreatine; GSH, glutathione. *p < 0.05; **p < 0.01; ***p < 0.005; ****p ≤ 0.001 (patient groups vs. their respective controls).
Table 2.
Metabolites concentrations measured in the three regions of interest in all groups.
| CTRL | FRDA | CTRL | AOA2 | ||
|---|---|---|---|---|---|
| VERMIS | Asc | 2.4 ± 0.6 | 2.4 ± 0.4 | 2.5 ± 0.8 | 2.9 ± 0.0 |
| Asp | 2.1 ± 0.5 | 1.9 ± 0.4 | 2.2 ± 0.4 | 2.9 ± 0.6 | |
| GABA | 1.9 ± 0.4 | 1.8 ± 0.5 | 1.9 ± 0.5 | 1.9 ± 0.8 | |
| Glca | 1.9 ± 0.6 | 1.8 ± 0.2 | 1.9 ± 0.9 | 2.0 ± 1.0 | |
| Gln | 3.4 ± 0.8 | 4.2 ± 0.5* | 3.6 ± 0.8 | 3.6 ± 1.0 | |
| Glu | 9.9 ± 1.5 | 9.5 ± 1.1 | 10.0 ± 1.5 | 7.8 ± 1.9**** | |
| GSH | 1.9 ± 0.5 | 2.0 ± 0.3 | 2.0 ± 0.5 | 2.3 ± 0.6 | |
| NAA | 10.9 ± 1.5 | 9.5 ± 1.0* | 10.9 ± 1.4 | 8.9 ± 1.7*** | |
| NAAG | 1.0 ± 0.2 | 1.1 ± 0.3 | 1.1 ±0.2 | 1.4 ± 0.3 | |
| tNAA | 11.8 ± 1.6 | 10.5 ± 1* | 11.8 ± 1.6 | 9.9 ± 1.7*** | |
| myo-Ins | 9.6 ± 1.3 | 11.1 ± 0.9*** | 9.7 ± 1.3 | 11.6 ± 2.0*** | |
| Lac | 1.6 ± 0.6 | 1.6 ± 0.8 | 1.6 ± 0.6 | - | |
| scyllo-Ins | 0.5 ± 0.2 | 0.3 ± 0.1 | 0.6 ± 0.3 | 0.9 ± 0.4 | |
| Tau | 3.6 ± 0.9 | 4.1 ± 0.5 | 3.8 ± 0.9 | 3.5 ± 1.3 | |
| tCho | 3.1 ± 0.5 | 3.2 ± 0.3 | 3.1 ± 0.5 | 3.2 ± 0.4 | |
| tCr | 12.8 ± 1.7 | 14.0 ± 1.3 | 13.2 ± 1.8 | 14.0 ± 1.8 | |
| | |||||
| CEREBELLAR HEMISPHERES | GABA | 1.7 ± 0.4 | 1.7 ± 1.0 | 1.7 ± 0.4 | - |
| Gln | 2.0 ± 0.6 | 1.7 ± 0.2 | 2.0 ± 0.6 | 2.4 ± 0.7 | |
| Glu | 6.8 ± 1.0 | 7.0 ± 0.8 | 6.8 ± 1.0 | 6.5 ± 1.7 | |
| GSH | 1.5 ± 0.4 | 1.5 ± 0.4 | 1.6 ± 0.4 | 1.7 ± 0.3 | |
| myo-Ins | 7.3 ± 1.0 | 8.1 ± 1.3 | 7.2 ± 1.0 | 8.7 ± 1.6 | |
| Lac | 1.4 ± 0.5 | 1.4 ± 0.3 | 1.4 ± 0.6 | 1.7 ± 0.6 | |
| tNAA | 12.2 ± 1.1 | 11.2 ± 1.0 | 12.3 ± 1.2 | 9.5 ± 1.7**** | |
| Glu+Gln | 8.0 ± 1.6 | 8.3 ± 1.1 | 8.0 ± 1.6 | 8.4 ± 2.0 | |
| tCho | 2.3 ± 0.3 | 2.5 ± 0.4 | 2.3 ± 0.3 | 2.6 ± 0.6 | |
| tCr | 8.0 ± 1.1 | 9.1 ± 0.9 | 8.1 ± 1.1 | 8.9 ± 1.3 | |
| Glc+Tau | 3.3 ± 1.6 | 3.4 ± 0.5 | 3.3 ± 1.6 | 3.9 ± 0.9 | |
| | |||||
| PONS | Asc | 3.1 ± 1.2 | - | 3.0 ± 1.1 | 3.3 ± 0.9 |
| Glu | 6.4 ± 1.1 | 6.6 ± 1.0 | 6.4 ± 1.4 | 6.1 ± 2.2 | |
| GSH | 1.6 ± 0.5 | 2.0 ± 0.5 | 1.6 ± 0.5 | 1.8 ± 0.8 | |
| myo-Ins | 9.9 ± 1.3 | 11.2 ± 2.2 | 10.0 ± 1.8 | 11.9 ± 2.0* | |
| Lac | 1.4 ± 0.3 | 1.5 ± 0.6 | 1.6 ± 0.6 | 1.8 ± 0.5 | |
| scyllo-Ins | 0.8 ± 0.3 | 0.7 ± 0.2 | 0.8 ± 0.4 | 0.7 ± 0.3 | |
| tNAA | 14.0 ± 1.3 | 13.7 ± 1.0 | 13.8 ± 1.3 | 12.5 ± 1.8 | |
| Glu+Gln | 7.3 ± 1.6 | 7.7 ± 1.4 | 7.5 ± 1.9 | 7.6 ± 2.3 | |
| tCho | 3.8 ± 0.6 | 3.9 ± 0.4 | 3.9 ± 0.5 | 3.7 ± 0.8 | |
| tCr | 7.5 ± 0.8 | 7.4 ± 1.0 | 7.6 ± 1.0 | 8.3 ± 1.5 | |
| Glc+Tau | 3.4 ± 1.0 | 4.0 ± 2.1 | 3.3 ± 1.0 | 3.9 ± 1.7 | |
Glucose and lactate concentrations in the brain were corrected for their concentration measured in CSF.
Asc, ascorbate; Asp, aspartate; GABA, γ-aminobutyric acid; Glc, glucose; Gln, glutamine; Glu, glutamte; GSH, glutathione; NAA, N-acetylaspartate; NAAG, N-acetylaspartylglutamate; myo-Ins, myo-inositol; Lac, lactate; scyllo-Ins, scyllo-inositol; tNAA, NAA + NAAG; Tau, taurine; tCho, choline compounds; tCr, total creatine (creatine+phosphocreatine).
p < 0.05
p < 0.005
p < 0.001 (See figure 3).
The patients with FRDA showed subtle alterations in the cerebellar hemispheres. A trend in lower tNAA concentration (-8%, p = 0.05) and a higher creatine + phosphocreatine concentration (tCr, +14%, p = 0.02) resulted in a lower tNAA / tCr ratio compared to controls (p = 0.002). Pons was spared in FRDA, although trends for higher myo-Ins (p = 0.05) and glutathione (GSH, p = 0.07) and slightly lower tNAA (p = 0.05) concentrations were observed, resulting in significantly higher GSH / tNAA ratio (p = 0.019). In the cerebellar hemisphere myo-Ins / tNAA ratio was higher (p = 0.01).
In patients with AOA2, the cerebellar hemisphere showed unequivocally lower tNAA (p < 0.0001) than controls. No other metabolites were affected. In the pons, the myo-Ins content was significantly higher than in controls (p = 0.02), along with a trend for lower tNAA (p = 0.05), resulting in a significantly lower tNAA / tCr (p = 0.016) and higher myo-Ins / tNAA (p = 0.0002) ratios.
Correlations of metabolite levels
In the FRDA group, no correlation was observed between any metabolites and the number of GAA repeats, the FARS score or the disease duration. However, a significant correlation between Glu and tNAA concentrations was observed in the vermis (r2 = 0.63, p = 0.018. Fig. 4a).
Figure 4. Scatter plots of correlations found in the FRDA and AOA2 groups.

Each marker corresponds to data obtained in one individual. In a) r2=0.634, p = 0.018. In b) r2 = 0.983, p < 0.0001. In c) vermis, r2 = 0.675, p = 0.012; cerebellar hemispheres (CH), r2 = 0.863, p < 0.0001.
In the AOA2 group, a correlation between myo-Ins and tCr was remarkably strong in the cerebellar hemisphere (r2=0.98, p < 0.0001), indicating that myo-Ins and tCr are likely involved in a common pathway (Fig. 4b). In both cerebellar regions, tNAA concentration correlated with the FARS score (Fig. 4c). In the cerebellar hemisphere, tNAA also correlated with the disease duration (not shown), although a multivariate analysis, on a greater number of patients, would be necessary to evaluate the separate contributions of each factor to the tNAA concentration.
3. Discussion
In this work, we report the neurochemical profiles of the cerebellum and pons in the two most frequent forms of recessive cerebellar ataxias, FRDA and AOA2. Patients with AOA2 showed more severe structural and neurochemical alterations in the cerebellum and the pons than patients with FRDA. However, despite striking anatomical differences, some of the metabolic alterations observed here were similar between the two populations: lower tNAA, trends or confirmed higher tCr and myo-inositol in the cerebellar regions, with these defects being overall more severe in AOA2 than in FRDA. Also in the vermis, both groups of patients exhibited a lower Glu/Gln compared to their respective controls, but interestingly this resulted from different effects: higher Gln in FRDA and lower Glu in AOA2. In both diseases, the pons was less affected or spared compared to the cerebellum.
CSF data
F2-isoprostane levels in CSF were comparable between patients and controls, for both FRDA and AOA2 patients. Similarly, Myers et al. showed that urinary F2-isoprostanes in patients with FRDA are not different from controls and concluded that these are not a useful biomarker for FRDA (Myers et al. 2008). The results we obtained in three patients with FRDA are consistent with this observation. Our results also indicate that CSF F2-isoprostanes are likely not a useful marker for AOA2 either although this remains to be confirmed in a higher number of patients.
In both groups of patients, CSF glucose concentration was higher than their respective control groups. Patients with FRDA are prone to diabetes and insulin resistance. Diabetes mellitus was an exclusion criterion in this study; a higher glucose level may then reflect insulin resistance in the three patients from whom CSF was collected. However, this preliminary result (N=3) needs to be confirmed with a higher number of patients. In AOA2, there has been no report on blood or CSF glucose in patients so far. Further investigations are needed to determine the origin of this elevated glucose level.
CSF lactate levels were comparable between the groups. Interestingly, we observed a correlation between lactate concentration and age in controls, confirming a previous observation (Yesavage et al. 1982).
Neurochemical Alterations in FRDA
In patients with FRDA, we report a lower tNAA in both the cerebellar hemispheres and vermis; a higher glutamine level in the vermis; a higher tCr concentration in the cerebellar hemisphere; and subtle trends for differences in the pons. These neurochemical differences were present despite the apparent structural integrity of the cerebellum in the voxel (Fig. 1) and demonstrate the advantage of assessing biochemical differences by MRS in the absence of differences detectable by conventional MRI. Our results are consistent with previous findings reporting a lower tNAA / tCr in the cerebellum and white matter (Franca et al. 2009, Guerrini et al. 2009, Mascalchi et al. 2002). Here, we show that the difference in the ratio is due to both lower tNAA and higher tCr levels. We were also able to detect differences in myo-Ins and Gln since these metabolites are detectable at high field. At variance with our study, Viau et al. reported in the vermis of 4 patients a lower tCho concentration and Guerrini et al. reported a higher tCr also in the vermis (Guerrini et al. 2009, Viau et al. 2005). A relatively small number of patients at various stages of the disease with different severity and technical differences (location of the voxel, quantification method) might account for such variations. This emphasizes the need for multi-center studies with technical consistency across institutions. Finally, in the pons, only trends were observed in higher myo-Ins and GSH and lower tNAA, resulting in significant differences in their ratios compared to controls. Such subtle changes may reflect the loss of gray matter volume in the dorsal pons (Franca et al. 2009) and the degeneration of the superior cerebellar peduncles (Oppenheimer 1979) contained in the voxel.
Neurochemical Alterations in AOA2
In patients with AOA2, we report 1H-MRS data in the brain for the first time. Changes observed in the cerebellum of these patients were similar to, albeit more severe than those in FRDA. The tNAA concentration is lower in both cerebellar regions and could be accounted for by loss of Purkinje cells (Criscuolo et al. 2006). We also observe higher myo-Ins than controls, consistent with the mild fibrous gliosis observed in the cerebellum (Criscuolo et al. 2006). The strong correlation between the tCr and myo-Ins concentrations in the cerebellar hemispheres in AOA2 (but not in control nor FRDA) supports an involvement of these metabolites in the same pathological pathway. Since tCr and myo-Ins may be markers of gliosis (Brand et al. 1993, Pouwels et al. 1998b, Vrenken et al. 2005), it is tempting to hypothesize that this common pathway is related to the gliotic process. However, this correlation was not observed in the vermis, where myo-Ins is significantly higher and gliosis is more severe (Criscuolo et al. 2006). Another mechanism may therefore account for this correlation. Interestingly, in the vermis, Glu/Gln was lower as in FRDA, but due to a lower glutamate level in this pathology. Finally, in the pons, we observed a higher myo-Ins level, along with trends for higher tCr and lower tNAA. To our knowledge, there is no histological report in the pons; however, the brainstem and spinal cord are slightly reduced in size (Asaka et al. 2006, Criscuolo et al. 2006), indicative of alterations in the tissue. Our results suggest a possible neuronal dysfunction (lower tNAA), disturbances in the homeostasis and metabolism of this structure and gliosis (higher myo-Ins and tCr). Further analyses with larger sample sizes are needed to confirm these results.
Glutamine and glutamate
In both pathologies, a lower Glu/Gln was observed in the vermis. However, this difference is due to higher glutamine in FRDA and lower glutamate in AOA2. Glutamate and glutamine are involved in the glutamatergic neurotransmission cycle. Glutamine is mainly found in astrocytes, while the glutamate pool is primarily localized in neurons (Hertz 2004). The higher glutamine could be due to gliosis in the vermis that has been reported in this region in FRDA (Oppenheimer 1979), consistent with the higher myo-Ins level observed in the same region. However, patients with AOA2 also exhibited a higher myo-Ins level in the vermis where gliosis has been reported (Criscuolo et al. 2006), and yet their vermian glutamine levels were comparable to controls. Attributing the higher glutamine, and/or the higher myo-Ins solely to a gliotic process might therefore be simplistic and these higher levels may also reflect impaired glutamatergic neurotransmission and/or altered mechanisms involving homeostasis. On the other hand, glutamate was low in the vermis of patients with AOA2. The major source of glutamate in the cerebellar cortex is granular neurons (Somogyi et al. 1986), which may degenerate with long-standing loss of Purkinje cells. That may be happening in the vermis first where atrophy is extensive, as lower tNAA is not accompanied by lower glutamate in the relatively less affected cerebellar hemisphere of the same patients. Supporting this hypothesis, a recent longitudinal study in mice with another form of ataxia reported a decrease in tNAA first, followed by a decrease in glutamate, suggesting that neuronal dysfunction must be more advanced before glutamate levels drop (Öz et al. 2010b). This sequence of events would be consistent with the drastic atrophy observed in the vermis of patients with AOA2. These results suggest that gliosis in FRDA and neuronal loss in AOA2 cannot account solely for the observations on glutamine and glutamate levels. It is interesting however that the vermian neurotransmission is likely affected in both pathologies but in different ways.
tNAA, a biomarker for neuronal dysfunction
In both pathologies, tNAA concentration was lower than controls in the cerebellum. Although its role is still under investigation, tNAA has been validated as a marker of neuronal viability and function (Clark 1998, Demougeot et al. 2001). Its concentration is variable in different regions of the brain but generally higher in white matter (WM) than in gray matter (GM) (Pouwels et al. 1998a). Therefore it is possible that a lower tNAA level may merely reflect a lower WM-to-GM ratio in the VOI. Because we studied VOI with the same dimensions in patients and controls, the cerebellar hemisphere VOI may have a lower WM-to-GM ratio in patients than controls due to WM loss (it is unlikely that the WM-to-GM ratio was different in patients in the other two VOI because atrophy results in a higher CSF content in vermis, rather than a different WM content, and we did not observe any indication for atrophy in the pons). However, our data in controls indicates similar tNAA levels in cerebellar GM (vermis) and WM (cerebellar hemispheres) (Fig. 3), therefore lower tNAA levels cannot be simply explained by a change in VOI composition. This argues in favor of tNAA as a functional marker of the tissue in these pathologies rather than a structural marker of gray and white matter volumes.
In addition, in AOA2, tNAA correlated with the FARS score in both cerebellar regions. Note that all concentrations were corrected for CSF content, and therefore do not merely reflect the degree of atrophy of the tissue. The tNAA-FARS correlation is particularly significant because atrophy appears not progressive on conventional MRI after disease onset in AOA2 (Anheim et al. 2009b). On the other hand, this correlation implies progressive cell loss/dysfunction after symptom onset. As such, the tNAA concentration in the cerebellum might provide a valuable biomarker for disease progression for which conventional MRI appears insufficient. We recently demonstrated that NAA, myo-Ins and Glu are markers of progressive cerebellar neurodegeneration in an ataxia mouse model (Öz et al. 2010b) and expect to observe similar progressive changes in MRS markers in future longitudinal studies with patients. While we did not observe a correlation of MRS markers with clinical measures in FRDA, future studies with larger sample sizes might reveal weak correlations that could not be detected in the current cross-sectional study.
Limitations
Future studies will require several improvements. First, a higher number of participants with FRDA and AOA2 will be necessary to define more precisely the changes characteristic of these diseases. Second, longitudinal studies are necessary to establish potential biomarkers of disease progression. Third, as high field magnets are spreading worldwide, studies at even higher fields (i.e. 7T) might unravel differences in a greater number of metabolites, hence potential biomarkers, with greater accuracy (Tkáč et al. 2009). While 4T scanners are not widely available, most of the results obtained in the current study are expected to be applicable to 3T scanners that are now available at almost all major universities, clinical centers and even some hospitals.
In conclusion, here we report neurochemical profiles of the cerebellum and brainstem of patients affected by the two most frequent forms of autosomal recessive ataxias. To our knowledge, we show for the first time 1H spectra acquired in patients with AOA2. Multiple neurochemical differences in each disease and a potential biomarker for disease progression in AOA2 were identified.
4. Experimental Procedure
Patients and Control Participants
Thirty-seven healthy volunteers (19 M / 18 F, mean age ± SD: 32 ± 12 years, range 19 - 61), 8 volunteers with FRDA (5 F / 3 M, 27 ± 9 years, range 15 - 41) and 9 volunteers with AOA2 (5 F / 4 M, 37 ± 9, range 26 - 53) participated in the study after giving written informed consent using procedures approved by the Institutional Review Board: Human Subjects Committee of University of Minnesota. FRDA and AOA2 diagnoses were confirmed by genetic testing, except for one AOA2 patient, where the genetic testing of a sibling confirmed the AOA2 diagnosis. The mutations identified in patients with AOA2 were the following: A1304G, A2975G and A6590G; 5264delC, A1945P and T4755G (Fogel et al. 2006); M2230T and AT insertion 2747-2748; 4 base pair (bp) deletion (GAGA) 5308-5311 and 2 bp deletion (CA) 6729-6730; C7103G (homozygous); and T5927G (Duquette et al. 2005). Ataxia severity was assessed using the FARS composite scores (Subramony et al. 2005). Briefly, FARS assesses bulbar, upper limb, lower limb, peripheral nerve, and upright stability/gait functions (maximum scores of 11, 36, 16, 26, and 28, respectively; maximum deficit composite score 117). Patients with Harrington rods were excluded as a magnet safety precaution. To avoid confounding factors that could influence metabolite levels, smoking and diabetes were exclusion criteria, and patients were asked to hold any antioxidant supplements for 3 weeks prior to the study because putative oxidative stress markers were assessed in the study. A subset of the volunteers underwent a lumbar puncture via the L3-4 intervertebral space under local anesthesia for CSF sample collection. Routine analyses for glucose and lactate were performed in these samples. For F2-isoprostanes measurement, CSF was centrifuged to remove cells and the supernatant was frozen in aliquots at -80°C. A negative ion chemical ionization GC/MS based method was then used to determine F2-isoprostane levels (Gross et al. 2005, Morrow et al. 1999). The study was performed according to the guidelines of the Declaration of Helsinki 1975.
MRS
All studies were performed with a 4 tesla magnet (Oxford Magnet Technology, Oxford, UK) with an INOVA console (Varian, Palo Alto, CA) and a TEM volume coil (Vaughan et al. 1994). Volume-of-interest (VOI) placement was based on sagittal and coronal multi-slice fast spin echo images. Spectra from vermis (1×2.5×2.5 cm3), cerebellar hemispheres (1.7×1.7×1.7 cm3) and pons (1.6×1.6×1.6 cm3) were acquired with a stimulated-echo acquisition mode (STEAM) sequence (echo time TE = 5 ms, mixing time TM = 42 ms, repetition time TR = 4.5 s, 128 averages) as detailed previously (Öz et al. 2010a, Öz et al. 2005). MR sessions lasted for 1 hour to 1 hour 15 minutes. Four-scan averages were corrected for B0, eddy current effects and summed. MRS data were collected in all three regions in patients with FRDA. For one patient with AOA2, data in the pons and the cerebellar hemisphere were discarded due to subject movement. In two other volunteers with AOA2, data in the vermis were discarded due to lipid contamination.
CSF content in the voxels was measured as an index of atrophy and for determination of metabolite concentrations in the tissue (Öz et al. 2010a). For this, unsuppressed water spectra were acquired at different TE values (5 to 5000 ms, with TR = 15 s for fully relaxed condition) and the integrals of the signals were fitted with a biexponential decay function (Ernst et al. 1993). To estimate tissue metabolite levels, CSF concentrations of all metabolites, except glucose and lactate, were assumed 0 (Kruse et al. 1985).
Spectral Quantitation
Metabolites were quantified using the LCModel (Linear Combination of Model spectra) method (Provencher 1993), as described before (Öz et al. 2010a, Öz et al. 2005). Briefly, the contribution of each metabolite to the spectra was estimated using frequency domain analysis. For this, model spectra of 19 metabolites (Öz et al. 2010a) were simulated based on previously reported chemical shifts and coupling constants (Govindaraju et al. 2000, Tkáč 2008). Macromolecule spectra were acquired using an inversion recovery sequence (TR = 2 s, inversion time = 0.675 s). The unsuppressed water signal was used as a quantitation reference assuming a water content of 72% for cerebellar hemispheres (primarily white matter) and 82% for vermis and pons (Siegel et al. 1999). Only results with Cramér-Rao lower bounds (CRLB) ≤ 50% were included in the analysis and only those metabolites with CRLB ≤ 50% in at least half of the spectra were included in the neurochemical profile of each region. If the correlation coefficient between two metabolites was consistently high (correlation coefficient < -0.5), their sum was reported, e.g. tNAA, tCr, tCho, Glc + Tau. Using these criteria, 10 metabolites were quantified in the pons, 11 in the cerebellar hemispheres and 15 in the vermis.
Statistical analysis
Patients with FRDA and those with AOA2 were different in age. Therefore, frequency matching on age was used and resulted in selecting controls ages 21-64 years for the AOA2 group and controls ages <45 years for the FRDA group. This resulted in two control groups, one for the FRDA and one for the AOA2 group. MRS data for each patient group vs. its control group were compared using ANCOVA, adjusting for age and gender, for each neurochemical concentration in each region separately. Secondary analyses using a linear mixed model to account for the potential correlation across regions within person were not different from results shown here. Because of a few outliers and skewness for some measures, analyses were repeated using a Wilcoxon test; results were again not different from those shown here. P-values shown have not been adjusted for multiple testing.
Acknowledgements
This work was supported by the Kory and Scott Tabor Ataxia Research Fund and NINDS grant R21NS056172 (to GÖ). The 4 T TEM coil was built with a grant (3761-9236-07) from the Minnesota Medical Foundation (to GÖ). The Center for MR Research is supported by National Center for Research Resources (NCRR) biotechnology research resource grant P41RR008079 and Neuroscience Center Core Blueprint Award P30NS057091. The General Clinical Research Center is supported by NCRR grant M01RR00400. The authors thank Drs. Susan Perlman (UCLA), Bernard Brais (CHUM), and David Lynch (UPenn) and their assistants for supporting this work by referring their patients to our institution. We also thank the patients and their families for participating in this study.
Abbreviations
- FRDA
Friedreich's ataxia
- AOA2
ataxia with oculomotor apraxia type 2
- ARCA
autosomal recessive cerebellar ataxia
- MRS
magnetic resonance spectroscopy
- FARS
Friedreich's Ataxia Rating Scale
- NAA
N-acetylaspartate
- NAAG
N-acetylaspartylglutamate
- tNAA
NAA+NAAG
- Glu
glutamate
- Gln
glutamine
- tCr
creatine + phosphocreatine
- tCho
choline-containing compounds
- myo-Ins
myo-inositol
- GSH
glutathione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Section. Disease-related Neuroscience
Research highlights
- Friedreich's ataxia (FRDA) and ataxia with oculomotor apraxia type 2 (AOA2) are the two most frequent forms of autosomal recessive cerebellar ataxias
- Biomarkers of the disease progression are lacking
- (1H) magnetic resonance spectroscopy (MRS) was used to assess the neurochemical profile of the pons, the cerebellar hemisphere and the vermis in patients with FRDA and AOA2 to identify potential biomarkers of these diseases
- Distinct neurochemical patterns were observed in the two patient populations
- In AOA2, total N-acetylaspartate levels in the cerebellum strongly correlated with the FARS score (p < 0.01)
- Studies with larger patient populations will determine if the alterations in metabolite levels observed here may be utilized to monitor disease progression and treatment

References
- Airoldi G, Guidarelli A, Cantoni O, Panzeri C, Vantaggiato C, Bonato S, Grazia D'Angelo M, Falcone S, De Palma C, Tonelli A, Crimella C, Bondioni S, Bresolin N, Clementi E, Bassi MT. Characterization of two novel SETX mutations in AOA2 patients reveals aspects of the pathophysiological role of senataxin. Neurogenetics. 2009;1:91–100. doi: 10.1007/s10048-009-0206-0. [DOI] [PubMed] [Google Scholar]
- Anheim M, Fleury M, Monga B, Laugel V, Chaigne D, Rodier G, Ginglinger E, Boulay C, Courtois S, Drouot N, Fritsch M, Delaunoy JP, Stoppa-Lyonnet D, Tranchant C, Koenig M. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2009a;1:1–12. doi: 10.1007/s10048-009-0196-y. [DOI] [PubMed] [Google Scholar]
- Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, Delaunoy JP, Fritsch M, Arning L, Synofzik M, Schols L, Sequeiros J, Goizet C, Marelli C, Le Ber I, Koht J, Gazulla J, De Bleecker J, Mukhtar M, Drouot N, Ali-Pacha L, Benhassine T, Chbicheb M, M'Zahem A, Hamri A, Chabrol B, Pouget J, Murphy R, Watanabe M, Coutinho P, Tazir M, Durr A, Brice A, Tranchant C, Koenig M. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009b;132:2688–98. doi: 10.1093/brain/awp211. [DOI] [PubMed] [Google Scholar]
- Asaka T, Yokoji H, Ito J, Yamaguchi K, Matsushima A. Autosomal recessive ataxia with peripheral neuropathy and elevated AFP: novel mutations in SETX. Neurology. 2006;66:1580–1. doi: 10.1212/01.wnl.0000216135.59699.9b. [DOI] [PubMed] [Google Scholar]
- Bernard V, Stricker S, Kreuz F, Minnerop M, Gillessen-Kaesbach G, Zuhlke C. Ataxia with oculomotor apraxia type 2: novel mutations in six patients with juvenile age of onset and elevated serum alpha-fetoprotein. Neuropediatrics. 2008;39:347–50. doi: 10.1055/s-0029-1214424. [DOI] [PubMed] [Google Scholar]
- Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH. Clinical, biochemical and molecular genetic correlations in Friedreich's ataxia. Hum Mol Genet. 2000;9:275–82. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- Brand A, Richter-Landsberg C, Leibfritz D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev Neurosci. 1993;15:289–98. doi: 10.1159/000111347. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Lodi R, Tonon C, D'Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, Stella AM, Butterfield DA. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia. J Neurol Sci. 2005;233:145–62. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–7. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Clark JB. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev Neurosci. 1998;20:271–6. doi: 10.1159/000017321. [DOI] [PubMed] [Google Scholar]
- Criscuolo C, Chessa L, Di Giandomenico S, Mancini P, Sacca F, Grieco GS, Piane M, Barbieri F, De Michele G, Banfi S, Pierelli F, Rizzuto N, Santorelli FM, Gallosti L, Filla A, Casali C. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology. 2006;66:1207–10. doi: 10.1212/01.wnl.0000208402.10512.4a. [DOI] [PubMed] [Google Scholar]
- Della Nave R, Foresti S, Tessa C, Moretti M, Ginestroni A, Gavazzi C, Guerrini L, Salvi F, Piacentini S, Mascalchi M. ADC mapping of neurodegeneration in the brainstem and cerebellum of patients with progressive ataxias. Neuroimage. 2004;22:698–705. doi: 10.1016/j.neuroimage.2004.01.035. [DOI] [PubMed] [Google Scholar]
- Della Nave R, Ginestroni A, Giannelli M, Tessa C, Salvatore E, Salvi F, Dotti MT, De Michele G, Piacentini S, Mascalchi M. Brain structural damage in Friedreich's ataxia. J Neurol Neurosurg Psychiatry. 2008;79:82–5. doi: 10.1136/jnnp.2007.124297. [DOI] [PubMed] [Google Scholar]
- Demougeot C, Garnier P, Mossiat C, Bertrand N, Giroud M, Beley A, Marie C. N-Acetylaspartate, a marker of both cellular dysfunction and neuronal loss: its relevance to studies of acute brain injury. J Neurochem. 2001;77:408–15. doi: 10.1046/j.1471-4159.2001.00285.x. [DOI] [PubMed] [Google Scholar]
- Duquette A, Roddier K, McNabb-Baltar J, Gosselin I, St-Denis A, Dicaire MJ, Loisel L, Labuda D, Marchand L, Mathieu J, Bouchard JP, Brais B. Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol. 2005;57:408–14. doi: 10.1002/ana.20408. [DOI] [PubMed] [Google Scholar]
- Ernst T, Kreis R, Ross BD. Absolute quantitation of water and metabolites in the human brain. I. Compartments and water. J Magn Reson. 1993;102:1–8. [Google Scholar]
- Fahey MC, Corben L, Collins V, Churchyard AJ, Delatycki MB. How is disease progress in Friedreich's ataxia best measured? A study of four rating scales. J Neurol Neurosurg Psychiatry. 2007;78:411–3. doi: 10.1136/jnnp.2006.096008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel BL, Perlman S. Novel mutations in the senataxin DNA/RNA helicase domain in ataxia with oculomotor apraxia 2. Neurology. 2006;67:2083–4. doi: 10.1212/01.wnl.0000247661.19601.28. [DOI] [PubMed] [Google Scholar]
- Franca MC, Jr., D'Abreu A, Yasuda CL, Bonadia LC, Santos da Silva M, Nucci A, Lopes-Cendes I, Cendes F. A combined voxel-based morphometry and 1H-MRS study in patients with Friedreich's ataxia. J Neurol. 2009;256:1114–20. doi: 10.1007/s00415-009-5079-5. [DOI] [PubMed] [Google Scholar]
- Gazulla J, Benavente I, Lopez-Fraile IP, Modrego P, Koenig M. Sensorimotor neuronopathy in ataxia with oculomotor apraxia type 2. Muscle Nerve. 2009;40:481–5. doi: 10.1002/mus.21328. [DOI] [PubMed] [Google Scholar]
- Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000;13:129–53. doi: 10.1002/1099-1492(200005)13:3<129::aid-nbm619>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Greco A, Minghetti L, Levi G. Isoprostanes, novel markers of oxidative injury, help understanding the pathogenesis of neurodegenerative diseases. Neurochem Res. 2000;25:1357–64. doi: 10.1023/a:1007608615682. [DOI] [PubMed] [Google Scholar]
- Gross M, Steffes M, Jacobs DR, Jr., Yu X, Lewis L, Lewis CE, Loria CM. Plasma F2-isoprostanes and coronary artery calcification: the CARDIA Study. Clin Chem. 2005;51:125–31. doi: 10.1373/clinchem.2004.037630. [DOI] [PubMed] [Google Scholar]
- Guerrini L, Belli G, Mazzoni L, Foresti S, Ginestroni A, Della Nave R, Diciotti S, Mascalchi M. Impact of cerebrospinal fluid contamination on brain metabolites evaluation with 1H-MR spectroscopy: a single voxel study of the cerebellar vermis in patients with degenerative ataxias. J Magn Reson Imaging. 2009;30:11–7. doi: 10.1002/jmri.21804. [DOI] [PubMed] [Google Scholar]
- Hertz L. Intercellular metabolic compartmentation in the brain: past, present and future. Neurochem Int. 2004;45:285–96. doi: 10.1016/j.neuint.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Michael SC, Knutson MD, Haile DJ, Qian J, Levi S, Santambrogio P, Garrick MD, Lamarche JB. The dentate nucleus in Friedreich's ataxia: the role of iron-responsive proteins. Acta Neuropathol. 2007;114:163–73. doi: 10.1007/s00401-007-0220-y. [DOI] [PubMed] [Google Scholar]
- Kruse T, Reiber H, Neuhoff V. Amino acid transport across the human blood-CSF barrier. An evaluation graph for amino acid concentrations in cerebrospinal fluid. J Neurol Sci. 1985;70:129–38. doi: 10.1016/0022-510x(85)90082-6. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Bouslam N, Rivaud-Pechoux S, Guimaraes J, Benomar A, Chamayou C, Goizet C, Moreira MC, Klur S, Yahyaoui M, Agid Y, Koenig M, Stevanin G, Brice A, Durr A. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127:759–67. doi: 10.1093/brain/awh080. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Brice A, Durr A. New autosomal recessive cerebellar ataxias with oculomotor apraxia. Curr Neurol Neurosci Rep. 2005;5:411–7. doi: 10.1007/s11910-005-0066-4. [DOI] [PubMed] [Google Scholar]
- Lodi R, Tonon C, Calabrese V, Schapira AH. Friedreich's ataxia: from disease mechanisms to therapeutic interventions. Antioxid Redox Signal. 2006;8:438–43. doi: 10.1089/ars.2006.8.438. [DOI] [PubMed] [Google Scholar]
- Mascalchi M, Cosottini M, Lolli F, Salvi F, Tessa C, Macucci M, Tosetti M, Plasmati R, Ferlini A, Tassinari CA, Villari N. Proton MR spectroscopy of the cerebellum and pons in patients with degenerative ataxia. Radiology. 2002;223:371–8. doi: 10.1148/radiol.2232010722. [DOI] [PubMed] [Google Scholar]
- Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schols L, Pandolfo M, Schulz JB, Pouget J, Calvas P, Shizuka-Ikeda M, Shoji M, Tanaka M, Izatt L, Shaw CE, M'Zahem A, Dunne E, Bomont P, Benhassine T, Bouslam N, Stevanin G, Brice A, Guimaraes J, Mendonca P, Barbot C, Coutinho P, Sequeiros J, Durr A, Warter JM, Koenig M. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225–7. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ., 2nd Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- Myers LM, Lynch DR, Farmer JM, Friedman LS, Lawson JA, Wilson RB. Urinary isoprostanes in Friedreich ataxia: lack of correlation with disease features. Mov Disord. 2008;23:1920–2. doi: 10.1002/mds.22038. [DOI] [PubMed] [Google Scholar]
- Nicolaou P, Georghiou A, Votsi C, Middleton LT, Zamba-Papanicolaou E, Christodoulou K. A novel c.5308_5311delGAGA mutation in Senataxin in a Cypriot family with an autosomal recessive cerebellar ataxia. BMC Med Genet. 2008;9:28. doi: 10.1186/1471-2350-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer DR. Brain lesions in Friedreich's ataxia. Can J Neurol Sci. 1979;6:173–6. doi: 10.1017/s0317167100119596. [DOI] [PubMed] [Google Scholar]
- Ormerod IE, Harding AE, Miller DH, Johnson G, MacManus D, du Boulay EP, Kendall BE, Moseley IF, McDonald WI. Magnetic resonance imaging in degenerative ataxic disorders. J Neurol Neurosurg Psychiatry. 1994;57:51–7. doi: 10.1136/jnnp.57.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öz G, Hutter D, Tkáč I, Clark HB, Gross MD, Jiang H, Eberly LE, Bushara KO, Gomez CM. Neurochemical alterations in spinocerebellar ataxia type 1 and their correlations with clinical status. Mov Disord. 2010a;25:1253–61. doi: 10.1002/mds.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öz G, Nelson CD, Koski DM, Henry PG, Marjanska M, Deelchand DK, Shanley R, Eberly LE, Orr HT, Clark HB. Noninvasive detection of presymptomatic and progressive neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2010b;30:3831–8. doi: 10.1523/JNEUROSCI.5612-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öz G, Tkáč I, Charnas LR, Choi IY, Bjoraker KJ, Shapiro EG, Gruetter R. Assessment of adrenoleukodystrophy lesions by high field MRS in nonsedated pediatric patients. Neurology. 2005;64:434–41. doi: 10.1212/01.WNL.0000150906.52208.E7. [DOI] [PubMed] [Google Scholar]
- Pagani E, Ginestroni A, Della Nave R, Agosta F, Salvi F, De Michele G, Piacentini S, Filippi M, Mascalchi M. Assessment of brain white matter fiber bundle atrophy in patients with Friedreich ataxia. Radiology. 2010;255:882–9. doi: 10.1148/radiol.10091742. [DOI] [PubMed] [Google Scholar]
- Palau F, Espinos C. Autosomal recessive cerebellar ataxias. Orphanet J Rare Dis. 2006;1:47. doi: 10.1186/1750-1172-1-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol. 2009;256(Suppl 1):9–17. doi: 10.1007/s00415-009-1003-2. [DOI] [PubMed] [Google Scholar]
- Pouwels PJ, Frahm J. Regional metabolite concentrations in human brain as determined by quantitative localized proton MRS. Magn Reson Med. 1998a;39:53–60. doi: 10.1002/mrm.1910390110. [DOI] [PubMed] [Google Scholar]
- Pouwels PJ, Kruse B, Korenke GC, Mao X, Hanefeld FA, Frahm J. Quantitative proton magnetic resonance spectroscopy of childhood adrenoleukodystrophy. Neuropediatrics. 1998b;29:254–64. doi: 10.1055/s-2007-973571. [DOI] [PubMed] [Google Scholar]
- Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30:672–9. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Schols L, Arning L, Schule R, Epplen JT, Timmann D. “Pseudodominant inheritance” of ataxia with ocular apraxia type 2 (AOA2). J Neurol. 2008;255:495–501. doi: 10.1007/s00415-008-0707-z. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Halasy K, Somogyi J, Storm-Mathisen J, Ottersen OP. Quantification of immunogold labelling reveals enrichment of glutamate in mossy and parallel fibre terminals in cat cerebellum. Neuroscience. 1986;19:1045–50. doi: 10.1016/0306-4522(86)90121-1. [DOI] [PubMed] [Google Scholar]
- Subramony SH, May W, Lynch D, Gomez C, Fischbeck K, Hallett M, Taylor P, Wilson R, Ashizawa T. Measuring Friedreich ataxia: Interrater reliability of a neurologic rating scale. Neurology. 2005;64:1261–2. doi: 10.1212/01.WNL.0000156802.15466.79. [DOI] [PubMed] [Google Scholar]
- Suraweera A, Becherel OJ, Chen P, Rundle N, Woods R, Nakamura J, Gatei M, Criscuolo C, Filla A, Chessa L, Fusser M, Epe B, Gueven N, Lavin MF. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol. 2007;177:969–79. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera A, Lim Y, Woods R, Birrell GW, Nasim T, Becherel OJ, Lavin MF. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet. 2009;18:3384–96. doi: 10.1093/hmg/ddp278. [DOI] [PubMed] [Google Scholar]
- Tazir M, Ali-Pacha L, M'Zahem A, Delaunoy JP, Fritsch M, Nouioua S, Benhassine T, Assami S, Grid D, Vallat JM, Hamri A, Koenig M. Ataxia with oculomotor apraxia type 2: a clinical and genetic study of 19 patients. J Neurol Sci. 2009;278:77–81. doi: 10.1016/j.jns.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Tkáč I. Refinement of the simulated basis set for LCModel analysis.. 16th Scientific Meeting of the International Society for Magnetic Resonance in Medicine; Toronto, Canada. 2008. p. 1624. [Google Scholar]
- Tkáč I, Öz G, Adriany G, Uğurbil K, Gruetter R. In vivo 1H NMR spectroscopy of the human brain at high magnetic fields: metabolite quantification at 4T vs. 7T. Magn Reson Med. 2009;62:868–79. doi: 10.1002/mrm.22086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan JT, Hetherington HP, Otu JO, Pan JW, Pohost GM. High frequency volume coils for clinical NMR imaging and spectroscopy. Magn Reson Med. 1994;32:206–18. doi: 10.1002/mrm.1910320209. [DOI] [PubMed] [Google Scholar]
- Viau M, Marchand L, Bard C, Boulanger Y. 1H magnetic resonance spectroscopy of autosomal ataxias. Brain Res. 2005;1049:191–202. doi: 10.1016/j.brainres.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Voncken M, Ioannou P, Delatycki MB. Friedreich ataxia-update on pathogenesis and possible therapies. Neurogenetics. 2004;5:1–8. doi: 10.1007/s10048-003-0170-z. [DOI] [PubMed] [Google Scholar]
- Vrenken H, Barkhof F, Uitdehaag BM, Castelijns JA, Polman CH, Pouwels PJ. MR spectroscopic evidence for glial increase but not for neuro-axonal damage in MS normal-appearing white matter. Magn Reson Med. 2005;53:256–66. doi: 10.1002/mrm.20366. [DOI] [PubMed] [Google Scholar]
- Yesavage JA, Holman CA, Sarnquist FH, Berger PA. Elevation of cerebrospinal fluid lactate with aging in subjects with normal blood oxygen saturations. J Gerontol. 1982;37:313–5. doi: 10.1093/geronj/37.3.313. [DOI] [PubMed] [Google Scholar]