

Abstract
Escherichia coli has three DNA damage-inducible DNA polymerases: DNA polymerase II (Pol II), DNA polymerase IV (Pol IV), and DNA polymerase V (Pol V). While the in vivo function of Pol V is well understood, the precise roles of Pol IV and Pol II in DNA replication and repair are not as clear. Study of these polymerases has largely focused on their participation in the recovery of failed replication forks, translesion DNA synthesis, and origin-independent DNA replication. However, their roles in other repair and recombination pathways in E. coli have not been extensively examined. This study investigated how E. coli’s inducible DNA polymerases and various DNA repair and recombination function together to convey resistance to 4-nitroquinoline-1-oxide (NQO), a DNA damaging agent that produces replication blocking DNA base adducts. The data suggest that full resistance to this compound depends upon an intricate interplay among the activities of the inducible DNA polymerases and recombination. The data also suggest new relationships between the different pathways that process recombination intermediates.
1. Introduction
Cells are constantly exposed to DNA damaging agents from both endogenous and exogenous sources. Various chemicals that mimic naturally occurring genotoxic agents are used to study cellular responses to DNA damage under controlled conditions. One example is 4-nitroquinoline-1-oxide (NQO), a mutagen and carcinogen that, in both mammals and bacteria, is only reactive after metabolic activation [1, 2]. Activated NQO reacts with DNA in vivo to form several different lesions, including adducts at the C8 and N2 positions of guanine and the N6 position of adenine [3, 4]. 50 to 80% of the lesions produced by NQO are quinoline adducts to the N2 position of guanine (N2-dG adducts) [5]. N2-dG adducts similar to those caused by NQO block DNA replication by E. coli DNA polymerase I (Pol I) in vitro [6], and NQO treatment stalls replication forks in avian cells in vivo [7]. In addition to persistent N2-dG adducts, NQO also produces unstable 8-hydroxyguanine lesions [8] that may lead to DNA double-strand breaks (DSBs) [4]. NQO exposure may also cause DSBs when replication forks encounter single strand nicks or gaps formed during repair of NQO-induced lesions.
In E. coli and other organisms the major pathway for repair of NQO-induced DNA lesions is nucleotide excision repair (NER) [9–12]. During NER, the single-strand of DNA immediately surrounding the lesion is excised and the resulting gap is filled in, usually by Pol I [13, 14]. If Pol I is absent, DNA repair synthesis still occurs after UV irradiation and is attributed to the activities of DNA polymerases II and III (Pol II, Pol III) [15]; however, the contributions made by E. coli’s other DNA polymerases in repair synthesis have not been thoroughly examined. Because of the UV mimetic properties of NQO, other genes required for UV resistance, especially those involved in homologous recombination, are also likely required for NQO resistance [16–19].
In addition to DNA repair activities, cells also possess a mechanism to tolerate DNA damage, called translesion DNA synthesis (TLS), which allows damaged DNA to continue to be replicated until it is repaired. E. coli has three DNA polymerases capable of TLS, DNA polymerase II (Pol II), DNA polymerase IV (Pol IV), and DNA polymerase V (Pol V) [20]. Each of these polymerases has a specific repertoire of lesions that its active site can accommodate during TLS. Because NQO is a popular tool to study the roles of specialized DNA polymerases, (for examples, see [6, 21]), it is important to fully understand the relationships among the other pathways involved in NQO resistance and the activities of these polymerases. In the study presented here, we used semi-quantitative assays to analyze the requirements for E. coli’s damage-induced TLS DNA polymerases, Pol II, Pol IV, and Pol V, for NQO resistance. We also examined the functional relationships between homologous recombination, NER and these specialized DNA polymerases in conferring NQO resistance. Our data support the hypothesis that Pol IV acts prior to NER, probably in TLS; however, our results also suggest a recombination-dependent function for Pol IV. In addition, our data reveal some new relationships between different pathways for recombination.
2. Materials and Methods
2.1 Bacterial strains and plasmids
The bacterial strains used in this study are E. coli K-12 derivatives and are described in Table 1. Genetic manipulations used standard techniques as described [22]. Antibiotics were used at the following concentrations: chloramphenicol (Cm), 10μg/ml; tetracycline (Tc), 20μg/ml; kanamycin (Kn), 30μg/ml. Standard P1vir transduction was used for strain construction; the donor and recipient strains are provided in Table 1. All episomes are derivatives of F’128, carrying the proAB+ genes; episomes were mated into Pro− recipient strains by selecting for Pro+ transconjugants. The order of genetic manipulations was transduction and then mating. Additional details of some constructions are provided with Table 1.
Table 1.
E. coli strains used in this study.
| Strain | Relevant phenotype or genotype | Recipient | Source of allele | Reference |
|---|---|---|---|---|
| P90C | F− ara Δ(gpt-lac)5 thi | [54, 55] | ||
| AM887 | Δ(ruvA-ruvC)65 eda57::Cm | [31] | ||
| N3055 | uvrA277::Tn10 | Lloyd, R.G. | ||
| JC10289 | Δ(recA-srl)306 srlR301::Tn10 | [56] | ||
| A354 | recB21 argA::Tn10 sbcA | F.W. Stahl | ||
| WA576 | recF400::Tn5 | [57] | ||
| JJC2135 | recO1504::Tn5 | [58] | ||
| N2731 | recG258::Tn10Kn | [17] | ||
| N4452 | ΔrecG265::Cm | [59] | ||
| OH1000 | recQ::Cm | [60] | ||
| JW1172 | ΔumuD::Kn | [61] | ||
| YG7207 | ΔdinB::Kn | [62] | ||
| FC36 | F− ara Δ (gpt-lac)5 thi RifR (Pro−) | [55] | ||
| FC40 | FC36/ F’ Pro+ Φ(lacI33-lacZ) | [55] | ||
| FC348 | FC40 Δ(recA-srl)306 srlR301::Tn10 | [63] | ||
| FC404 | FC40 recB21 | [63] | ||
| FC438 | FC40 recG162 zib363::Tn10 | [63] | ||
| FC465 | FC40 recG258::Tn10Kn | [63] | ||
| FC485 | FC40 ruvA60::Tn10 | [63] | ||
| FC521 | FC40 recG258::Tn10Kn ruvA60::Tn10 | [63] | ||
| FC573 | FC40 Δ(ruvA-ruvC)65 | [63] | ||
| FC581 | FC40 Δ(ruvC)64::Kn | [63] | ||
| FC1354 | FC40 Δd i n B::Kn on chromosome and episome | [64] | ||
| FC1408 | FC40 ΔrecG265::Cm | [65] | ||
| FC1418 | FC40 lexA1 (Ind−) | [64] | ||
| PFB59 | F− parent of PFB60 | [66] | ||
| PFB60 | FC40 Δp o l B 1 | [66] | ||
| PFB208 | FC40 ΔpolB1 ΔdinB::Kn on chromosome and episome | PFB59 | YG7207 | This study |
| PFB236 | F− ara Δ (gpt-lac)5 thi RifR ΔdinB::Zeo | [65] | ||
| PFB241 | Met− donor of F’ Φ(lacI33-lacZ) ΔdinB::Zeo | This study | ||
| PFB243 | FC40 ΔdinB::Zeo on chromosome and episome | [65] | ||
| PFB286 | FC40 Δ(umuDC)::Erm | [67] | ||
| PFB287 | FC40 ΔpolB1 Δ(umuDC)::Erm | PFB286 | PFB60 | This study |
| PFB876 | FC40 ΔdinB::Kn Δ(umuDC)::Erm | PFB286 | YG7207 | This study |
| PFB878 | FC40 ΔdinB::Kn ΔpolB1 Δ(umuDC)::Erm | PFB876 | PFB60 | This study |
| PFB284 | FC36 Δ(umuDC)::Erm | This study | ||
| PFB873 | FC36 ΔdinB::Zeo Δ(umuDC)::Erm | This study | ||
| FC1536 | FC36 ΔumuD::Kn | FC36 | JW1172 | This study |
| PFB646 | FC40 ΔrecG265::Cm ΔdinB::Zeo on chromosome and episome | PFB243 | FC1408 | This study |
| PFB755 | FC40 uvrA277::Tn10 | FC40 | N3055 | This study |
| PFB756 | PFB243 uvrA277::Tn10 | PFB243 | N3055 | This study |
| PFB820 | FC40 lexA1 ΔdinB::Zeo on chromosome and episome | PF2015 | PFB243 | This study |
| PFB822 | FC40 ΔumuD::Kn | FC40 | JW1172 | This study |
| PFB881 | FC40 Δ(ruvA-ruvC)65ΔdinB::Zeo on chromosome and episome | PFB236 | AM887a | This study |
| PFB879 | FC40 recB21 ΔdinB::Zeo on chromosome and episome | PFB236 | A354b | This study |
| PFB827 | FC40 recF400 ΔdinB::Zeo on chromosome and episome | B243 | WA576 | This study |
| PFG404 | FC40 recF400::Tn5 | FC40 | WA576 | This study |
| PFG406 | FC40 recQ::Cm | FC40 | OH1000 | This study |
| PFG484 | FC40 recO1504::Tn5 | FC40 | JJC2135 | This study |
To introduce the Δ(ruvA-ruvC)65 allele, PFB236 was transduced to Cm resistance using a P1vir lysate grown on AM887 and transductants were screened for UV sensitivity. The episome from PFB241 was introduced by mating as described in the Materials and Methods (Section 2.1).
To introduce the recB21 allele, PFB236 was transduced to Tc resistance using a P1vir lysate grown on A354 and transductants were screened for UV sensitivity. The episome from PFB241 was introduced by mating as described in the Materials and Methods (Section 2.1).
2.2 NQO sensitivity assays
The stock solution of 4-nitroquinoline-1-oxide (NQO) (Sigma N8141) was 10mM in N,N-dimethylformamide (DMF) (Sigma-Aldrich 227056). NQO in DMF is stable at −20º for at least one month. Because NQO is light and water sensitive, plates were made immediately as needed and kept in the dark. Sensitivity to NQO was measured as follows: cultures of each strain to be tested were grown overnight in LB broth [22] at 37º with aeration. These cultures were then diluted to 10−6 by serial 10-fold dilutions in 0.85% NaCl. 10μL aliquots of each dilution were spotted on an LB agar plate and on an identical plate containing the indicated concentration of NQO. Plates were incubated at 37º in the dark and photographed after 18 to 36 hours, depending on the growth rate of the cells. We typically tried several NQO concentrations and chose the one that gave the clearest results (although in no case were the results different). To confirm the reproducibility of the results, each experiment was repeated at least two additional times with independently-grown cultures of each strain. Plates shown are representative examples of the results.
The assay we used is similar to those reported previously for NQO and nitrofurazone [21, 23, 24] in which various dilutions of stationary-phase cells are plated on LB plates containing the agent and the number of colony-forming units that subsequently appear are evaluated. In preliminary experiments we found that cells were insensitive to killing by NQO unless they were grown in its presence. We also found that the plate-spotting procedure yielded more reproducible results than growing the cells in liquid medium containing NQO. Some of the strains used grew poorly on minimal medium, so rich medium was used throughout to avoid spurious viability problems. When the NQO sensitivities of some representative strains were testing on minimal medium the results did not differ.
2.3 Analysis of cellular DNA content by flow cytometry
Cultures were grown overnight in LB broth at 37º, diluted 1000-fold in fresh LB broth, and grown to an optical density (OD600) of 0.3 for the wild-type strain and 0.1 for the recA mutant strain. Since both cultures were in exponential growth, this difference in OD600 should not impact the results. The cultures were divided in half; one half served as a control and the other half was exposed either to 10μM NQO or to 40J/m2 of UV light. To stop transcription and thus replication initiation, and to block cell division, 250μg/mL of rifampicin (Sigma, R-3501) and 10μg/mL of cephalexin (Sigma, C4895) were added to the cells. The cells were incubated for three hours to allow the completion of any ongoing DNA replication, which is unaffected by rifampicin and cephalexin [25, 26]. Cells were fixed by adding 0.5mL of the cultures to 4.5mL of 78% ice-cold ethanol to yield a final concentration of 70% ethanol. Each sample was centrifuged, the pellet rinsed twice with 10mL of 1X phosphate buffered saline (PBS), and stored at 4º C until analysis. Cells were stained with propidium iodide in a solution of 0.2mg/mL RNase (Sigma, R4875), 0.01% Triton X-100 (Thermo Fisher Scientific, Inc., Waltham, MA), and 3μM propidium iodide (Sigma, P4864) in 1X PBS [27, 28] and Christiane Hassel (personal communication).
DNA content was analyzed with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Approximately 50,000 cells were analyzed at between 300 and 900 cells per second. The FL2-Width (FL2-W) was used to measure cell size and FL2-Area (FL2-A) was used to measure total propidium iodide fluorescence (DNA content). Data analysis was performed using CellQuant Software (BD Biosciences, San Jose, CA).
3. Results
3.1 Moderate NQO exposure does not cause extensive genomic DNA degradation
Exposure of a recA mutant strain of E. coli to UV irradiation results in rapid chromosome degradation and death, a phenotype known as “rec-less death” [29, 30]. We used flow cytometric analysis of propidium iodide stained cells (see Section 2.3) to determine if the concentrations of NQO used in our assays would cause chromosome degradation in recombination-proficient cells. As shown Figure 1, exposure of a recombination proficient strain, P90C (the parent strain of FC36 and FC40) to 40J/m2 of UV irradiation resulted in a marked increase in the number of cells with fluorescence intensities at values below the normal peaks at 200 and 400 (arbitrary units), indicating that, as expected, chromosomal DNA was degraded. Treatment of the same strain with 10μM NQO had a similar but much milder effect. In contrast, treatment of recA mutant cells with the same doses of UV and NQO caused more significant alterations in the fluorescence profiles, indicating that in the absence of RecA, both treatments induced extensive DNA degradation (Figure 1B). Since the rifampicin treatment used to inhibit replication initiation in these flow-cytometric experiments prevents the induction of the SOS response, the level of DNA degradation sustained by DNA repair-competent cells during the experiments described below would be even less than that indicated in Figure 1. Thus, the degree of damage-dependent DNA degradation occurring upon exposure to the standard NQO concentration used in our assays was mild.
Figure 1. Effects of UV irradiation and NQO exposure on DNA content in wild-type and recombination-defective cells.

The DNA content of cells treated with either NQO (10μM) or irradiated with UV light (40J/m2) was analyzed using flow cytometry of propidium iodide stained cells (see Section 2.3). Approximately 50,000 cells were analyzed and cell count is plotted against relative fluorescent intensity(in arbitrary units). E. coli strain P90C is the parent strain of FC40 and its derivatives used in this study (Table 1). (A) UV irradiation and NQO treatment have different effects on the DNA content in recombination competent cells. (B) Both UV irradiation and NQO treatment reduce the DNA content and broaden the fluorescent peaks in cells defective for recombination.
3.2 SOS-regulated genes contribute to NQO resistance
As shown in Figure 2A, at 10 μM NQO a recA mutant strain is at least five orders of magnitude more sensitive to NQO than a wild-type strain. This result suggests that SOS induction and/or recombination are required for NQO resistance, probably to prevent DNA degradation (Figure 1) and cell death (the requirements for recombination are tested below). The SOS-regulated genes dinB and umuC are known to contribute to NQO resistance [6]; however, other SOS-regulated genes may also be involved. To test this hypothesis, we measured the NQO sensitivity of a strain carrying the lexA1 allele that prevents SOS induction. As shown in Figure 2A, at 10 μM NQO a lexA1 mutant strain was two orders of magnitude more sensitive to10 μM NQO NQO than a strain deleted for dinB, indicating that some SOS-regulated gene or genes in addition to dinB+ are involved in NQO resistance. Interestingly, a lexA1 ΔdinB double mutant strain was about an order of magnitude more sensitive to 10 μM NQO NQO than the lexA1 mutant strain, indicating that even when the dinB+ gene is repressed, the cellular level of Pol IV is sufficient to convey some NQO resistance. These results also suggest, but do not prove, that Pol IV may participate in a pathway for NQO resistance that does not involve other SOS-regulated genes, which will be further explored below.
Figure 2. RecA/LexA and nucleotide excision repair each contribute to NQO resistance.

Spot tests were performed as described in Section 2.2; only the 10−6 dilution from the untreated LB plate is shown. Strains in (A) and in (B) were tested on different plates and the corresponding untreated LB plate is shown. The same overnight cultures were used for all the plates shown. FC40 (wild-type); ΔdinB = PFB243; ΔrecA = FC348; lexA1 = FC1418; ΔdinB lexA1 = PFB820; uvrA = PFB755; ΔdinB uvrA = PFB756.
3.3 Pol IV acts in NER-dependent and NER-independent pathways
If Pol IV and NER act in the same pathway to confer NQO resistance, then loss of Pol IV and loss of NER should be epistatic. As shown in Figure 2A, a strain deleted for dinB was about two orders of magnitude more sensitive to 10 μM NQO NQO than a wild-type strain; a strain with a null mutant allele of uvrA, which encodes an essential component of the NER pathway, was more than five orders of magnitude more sensitive to 10 μM NQO NQO than a wild-type strain; and, a uvrA ΔdinB double mutant strain was less than one order of magnitude more sensitive to 10 μM NQO NQO than the uvrA single mutant strain. This last result was confirmed by exposing the same strains to half the concentration of NQO (5μM) (Figure 2B). Because loss of Pol IV reduces NQO resistance even in the absence of UvrA, Pol IV clearly can act in a NER-independent pathway, such as repair of the DSBs that can result from NQO damage [4]. However, because the effect of loss of Pol IV is reduced in the absence of UvrA, Pol IV and NER may also work in a common pathway to confer NQO resistance.
3.4 Damage-inducible DNA polymerases contribute to NQO resistance
Previous results indicated that Pol V contributes to NQO resistance [6, 31]; however, as shown in Figure 3A, we found that a Δ(umuDC) mutant strain was not more sensitive to 12.5μM NQO than a wild-type strain. To determine if Pol IV and Pol V act together, we tested a Δ(umuDC) ΔdinB double mutant strain and found that it was not more sensitive to 12.5μM NQO than the ΔdinB mutant strain. Because FC40 carries two copies of the dinB+ gene (one on the chromosome and one on the episome), it was possible that the increased amount of Pol IV in these strains could obscure Pol V’s contribution to NQO resistance. However, as shown in Figure 3B, similar results were obtained at a slightly lower NQO concentration (10 μM) with F− strains that have only one copy of the dinB+ gene. Thus, our results suggest that Pol V contributes little to NQO resistance in an otherwise wild-type strain.
Figure 3. Damage-induced DNA polymerases contribute to NQO resistance.

Spot tests were performed as described in Section 2.2; only the 10−6 dilution from the untreated LB plate is shown. A higher NQO concentration (12.5μM) was used in (A) as the dinB mutant strain used in this experiment, which has a larger deletion in the region of dinB, causes a milder NQO sensitivity phenotype than the dinB allele used elsewhere (ΔdinB::Zeo), which carries a smaller, non-polar, deletion. (A) FC40 (wild-type); ΔdinB = FC1354; ΔpolB = PFB60; Δ (umuDC) = PFB286; ΔdinB ΔpolB = PFB208; ΔdinB Δ (umuDC) = PFB876; ΔpolB ΔumuDC) = PFB287; ΔdinB ΔpolB ΔumuDC) = PFB876. (B) FC36 (F− parent for FC40); ΔdinB = PFB236; ΔumuDC) = PFB284; ΔdinB Δ(umuDC) = PFB873; ΔumuD = FC1536.
DNA polymerase II (polB) is E. coli’s third damage-inducible DNA polymerase. As shown in Figure 3A, a strain lacking Pol II (ΔpolB) was more sensitive to 12.5μM NQO than either the wild-type strain or the ΔdinB mutant strain. The ΔpolB ΔdinB double mutant strain was more sensitive than either of the single mutant strains, suggesting that Pol II and Pol IV confer resistance to NQO independently. Interestingly, the ΔpolB Δ (umuDC) double mutant was about as sensitive to NQO as the ΔpolB mutant strain (Figure 3A), suggesting little contribution of Pol V to NQO resistance in Pol II deficient cells. Taken together, these results suggest that Pol II is the major damage-inducible polymerase conferring NQO resistance, but that Pol IV can also act in a different pathway from Pol II to confer NQO resistance.
Finally, as shown in Figure 3B, a strain with a non-polar deletion of the umuD gene was not more sensitive to 10 μM NQO than a wild-type strain; thus neither UmuD nor UmuD’ appear to act alone to confer NQO resistance.
3.5 Recombination contributes to NQO resistance
Recombination-dependent DNA repair pathways often result in the formation of Holliday junctions; thus, if the requirement for RecA shown above is for recombination and not just for induction of the SOS response, enzymes that process Holliday junctions should contribute to NQO resistance. The RuvABC complex migrates and resolves Holliday junctions during homologous recombination [32]. As shown in Figure 4, strains carrying either the ruvA60 mutant allele or the Δ(ruvA-ruvC) mutant allele (both of which are polar on ruvB [33]) were three orders of magnitude more sensitive to10 μM NQO than was the wild-type strain. In the Δ(ruvA-ruvC) mutant background, loss of Pol IV had little, if any, effect on NQO sensitivity. This result suggests that Pol IV acts in a pathway with RuvABC to confer NQO resistance. Interestingly, the mutant strain carrying the Δ(ruvA-ruvC) allele was somewhat less sensitive to 10 μM NQO than the ΔruvC64 single mutant strain. One explanation for this result is that in the absence of RuvC, RuvAB binds and sequesters Holliday junctions non-productively, blocking processing by another protein, such as RecG.
Figure 4. Holliday junction processing is required for full NQO resistance.
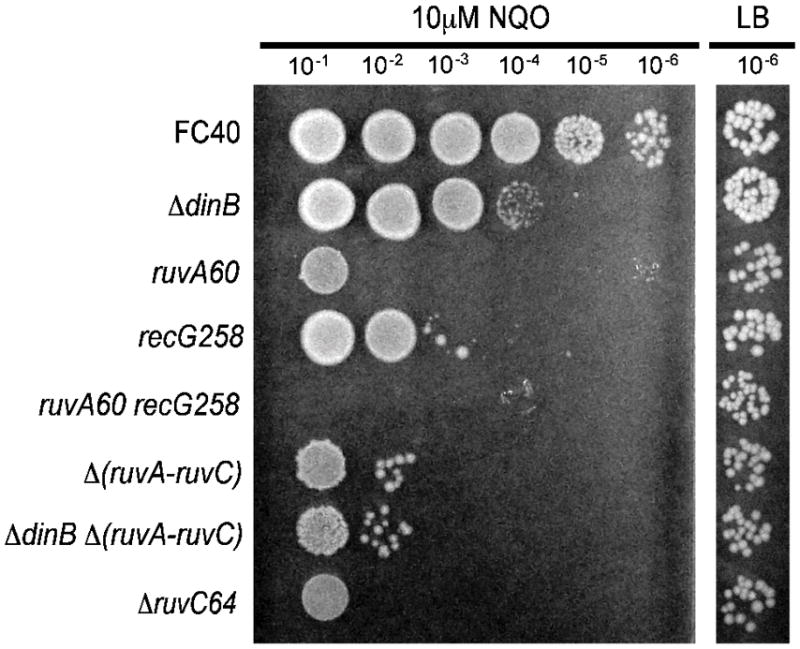
Spot tests were performed as described in Section 2.2; only the 10−6 dilution from the untreated LB plate is shown. All assays shown were performed on the same plates. FC40 (wild-type); ΔdinB = PFB243; ruvA = FC485; recG258 = FC465; ruvA60 ΔrecG265 = FC521; Δ (ruvA-ruvC) = FC573; ΔdinB Δ(ruvA-ruvC) = PFB881; Δ(ruvC) = FC581.
E. coli RecG enzyme can also perform Holliday junction migration, but, unlike the RuvABC complex, RecG does not have an associated resolvase activity [34, 35]. As shown in Figure 4, the recG258 mutant strain was two orders of magnitude more sensitive to 10 μM NQO than the wild-type strain. To confirm that this effect was not allele specific, strains with two other recG mutant alleles (recG162 and ΔrecG265) were tested, and all three strains had similar NQO sensitivities (Figure 5). As shown in Figure 4, a ruvA60 recG258 double mutant strain was unable to grow on 10μM NQO, suggesting that Holliday junction processing by one of the two pathways is a critical step in the repair of NQO-induced lesions. Loss of Pol IV in the ΔrecG265 mutant background caused little, or no, increase in NQO sensitivity (Figure 5A), suggesting that Pol IV functions in a RecG-dependent pathway.
Figure 5. The RecBCD pathway contributes to NQO resistance.

Spot tests were performed as described in Section 2.2. Because of the reduced viability of the recB21 mutant, both the 10−5 and the 10−6 dilution from the untreated LB plate is shown. The same overnight cultures were used for all the plates shown. FC40 (wild-type); ΔdinB = PFB243; recG258 = FC465; recG162 = FC438; ΔrecG265 = FC1408; ΔdinB ΔrecG265 = PFB646; recB21 = FC404; ΔdinB recB21 = PFB879.
3.6 Both RecBCD and RecFOR contribute to NQO resistance
As discussed in the Introduction, NQO exposure can result in DSBs. As shown in Figure 5A, a strain with a mutation in recB, which encodes an essential component of the RecBCD pathway for DSB repair, was about three orders of magnitude more sensitive to 10 μM NQO than the wild-type strain. To test if Pol IV and RecBCD function together, we assayed the sensitivity of a ΔdinB recB double mutant strain. As shown in Figures 5A & B, even at the low concentration of 5 μM NQO, loss of Pol IV had little or no effect on the NQO sensitivity of the recB mutant strain. Thus, Pol IV appears to have, at most, only a small role in conferring NQO resistance in the absence of recB-dependent double-strand break repair.
When UV-induced lesions block replication forks, re-initiation of DNA replication downstream of the lesion can result in single-strand gaps [36, 37]. Because NQO similarly blocks replication forks, NQO treatment should also cause single-strand gaps. The classical role of the RecFOR proteins are to initiate the recombinational repair of single-strand gaps by displacing single-strand binding protein (SSB) and facilitating RecA loading [38–41]. A competing model is that RecFOR, in conjunction with RecJ and RecQ, reactivate the replication forks that are stalled at DNA lesions [42]. The RecFOR proteins also are required for recA-stimulation of Pol V TLS past UV-induced lesions [43]. As shown in Figure 6, recF or recO mutant strains were about three orders of magnitude more sensitive to 10 μM NQO than the wild-type strain. Since loss of the RecQ helicase caused only a minor increase in NQO sensitivity (Figure 6), RecFOR probably confers NQO resistance via its role in single-strand gap repair. A ΔdinB recF double mutant strain was only slightly more sensitive to 10 μM NQO than the recF single mutant strain and no more sensitive than the recO single mutant strain. These results suggest that Pol IV may function in a RecFOR-dependent recombinational pathway that confers NQO resistance.
Figure 6. The RecFOR pathway also contributes to NQO resistance.

Spot tests were performed as described in Section 2.2; only the 10−6 dilution from the untreated LB plate is shown. FC40 (wild-type); ΔdinB = PFB243; recF = PFG404; recO = PFG484; recQ = PFG406; ΔdinB recF = PFB827.
4. Discussion
The full extent of the functions performed by the damage-induced DNA polymerases in vivo remains undefined. In some cases, single polymerases appear to have several functions. For example, the eukaryotic DNA polymerases η and κ (homologs of E. coli’s Pol V and Pol IV, respectively) carry out TLS past DNA base adducts [44, 45], and also can extend invading 3’-termini during homologous recombination [46–48]. Likewise Pol IV has the capacity for TLS and to aid in strand invasion [48]. That these enzymes are multifunctional complicates the design of experimental methods to detect their activities. In order to effectively use resistance to genotoxic agents to assess DNA polymerase activity, it is crucial to understand the functional relationships between the polymerase and other factors that contribute to resistance and to understand which specific activity of the polymerase confers resistance. To this end, we have investigated the genetic requirements for resistance to the genotoxic agent NQO, which is frequently used to analyze DNA polymerase activity.
The major determinants of NQO resistance are NER and recombination. Since loss of each of these pathways has a greater effect on NQO resistance than loss of any one of the damage-inducible DNA polymerases, the pathways must function with alternative polymerases (including Pol I and III). Nonetheless, in vitro Pol IV can bypass DNA lesions of the type induced by NQO [6], and Pol IV contributes to survival to this agent [6] (Figure 2). The results of our analysis of the relationship between loss of Pol IV and loss of NER suggest that Pol IV and NER act together to confer NQO resistance. The simplest interpretation for this result is that Pol IV carries out TLS at sites of NQO-induced damage, allowing uninterrupted chromosome replication, and that the damage is subsequently repaired by NER. However, the lack of epistasis between loss of NER and loss of Pol IV (Figure 2) indicates that Pol IV participates in one or more other pathways for NQO resistance. Our data indicate that homologous recombination by the RecBCD pathway (Figure 5) and the RecFOR pathway (Figure 6) also contributes to NQO resistance, and that Pol IV appears to play a role in both of these pathways.
RecFOR is postulated to be involved in at least three DNA repair pathways: “long-patch excision repair” (reviewed in [19]) the recombinational repair of single-strand gaps that are formed when DNA replication resumes downstream of lesions (daughter-strand gap repair) [49, 50], and replication restart [42, 51]. Each of these pathways requires DNA synthesis, and thus could provide a role for Pol IV, but models differ in the requirements for the resolution of Holliday junctions. Since long patch excision repair requires both homologous recombination and NER proteins, a role for Pol IV in this pathway could explain the overlapping roles of NER and recombination in NQO resistance. Such a function for Pol IV may correspond to the requirement for Pol κ, the Pol IV homolog, in NER of UV-light induced damage in mice [52].
Our results partially confirm the UV mimetic properties of NQO since both NER and recombination functions are major contributors to resistance to NQO- and UV- induced DNA damage. However, there are differences in the specifics of the pathways involved. No UV-sensitive phenotype for Pol IV has been reported, and thus it plays no essential role in conveying UV resistance. Of the three damage inducible polymerases, only Pol V conveys resistance to UV damage (see for example reference [24]) whereas Pol II and Pol IV, but not Pol V, convey resistance to NQO (Figure 3). However, the possible role of Pol IV in recombinational repair of UV-induced DNA damage has not been adequately addressed.
Nitrofurazone (NFZ) is a DNA damaging agent that causes lesions at the N2 position of guanine residues that are chemically similar to those caused by NQO. Pol IV, and to a much lesser extent Pol V, are important factors in the resistance of E. coli to NFZ [6]. A recent study by Ona and colleagues [24] confirmed that Pol IV (but not Pol II or Pol V) contributed to NFZ resistance, but found that NFZ resistance was much more dependent on functional NER and recombination pathways (specifically the uvrA+, recA+, recBC+, and recF+ genes) than on Pol IV. Ona and colleagues further showed that loss of all three damage-inducible polymerases, Pol II, Pol IV, and Pol V, resulted in about a 10-fold increase in sensitivity to NFZ in a uvrA+ strain but had only a slight effect in a uvrA mutant strain. This result suggests that Pol II, Pol IV, and Pol V can substitute for each other in a NER-dependent pathway for NFZ resistance. Because loss of Pol IV, but not Pol II or Pol V, alone, increased NFZ sensitivity, Pol IV may also function in one or more NER-independent (presumably recombinational, although this was not tested) pathway for NFZ resistance, consistent with our results with NQO. However, our results differ in indicating a significant role for Pol II as well as Pol IV. Indeed, we found that overproduction of Pol II from a multicopy plasmid can partially complement the NQO sensitivity of a ΔdinB mutant strain (data not shown), indicating that Pol II and Pol IV can substitute for each other in conveying resistance to NQO.
In our assays, loss of Pol V did not increase sensitivity to NQO (Figure 3). Thus, in our wild-type strain, Pol II and Pol IV are sufficient to deal with the DNA damage caused by NQO. However, a previous study found that both Pol IV and Pol V contributed to NQO resistance [6]. This discrepancy is not due to a difference in copy number of the dinB+ gene in our F’ strains, as isogenic F− strains also showed no effect of loss of Pol V (Figure 3). Further research will be required to determine the cause of this difference.
The data presented in this study start to clarify the genetic requirements and their epistatic relationships for resistance to NQO, a genotoxic agent commonly used to assess DNA repair in E. coli. An understanding of these relationships is critical to address the functions of specific proteins, including DNA polymerases, in DNA repair pathways.
Acknowledgments
We thank R.G. Lloyd, S.T. Lovett, B. Michel, J.H. Miller, H. Shinagawa, F.W. Stahl, W. Wackernagel, M. Goodman, and R. Woodgate for generously providing strains used in this study. We also acknowledge the National BioResource Project (NIG, Japan) for supplying the KEIO collection of E. coli mutant strains. We thank Shera Lesly and Rachel Baker for constructing some of the strains used in this study. Christiane Hassel at Indiana University Flow Cytometry Core Facility provided assistance for the flow cytometry experiments. We are also grateful for the comments and suggestions of Kimberly A.M. Storvik and the other members of our laboratory during the course of this work. This research was supported by the following grants from the US National Institutes of Health: GM065175 (to PLF) and T32 GM007757 (to ABW and KMH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nakahara W, Fukuoka F, Sakai S. Carcinogenic quinoline derivatives. Gan. 1957;48:568–9. [PubMed] [Google Scholar]
- 2.Nakahara W, Fukuoka F, Sugimura T. Carcinogenic action of 4-nitroquinoline-N-oxide. Gan. 1957;48:129–37. [PubMed] [Google Scholar]
- 3.Galiègue-Zouitina S, Bailleul B, Ginot YM, Perly B, Vigny P, Loucheux-Lefebvre MH. N2-guanyl and N6-adenyl arylation of chicken erythrocyte DNA by the ultimate carcinogen of 4-nitroquinoline-1-oxide. Cancer Res. 1986;46:1858–63. [PubMed] [Google Scholar]
- 4.Galiègue-Zouitina S, Bailleul B, Loucheux-Lefebvre MH. Adducts from in vivo action of the carcinogen 4-hydroxyaminoquinoline-1-oxide in rats and from in vitro reaction of 4-acetoxyaminoquinoline-1-oxide with DNA and polynucleotides. Cancer Res. 1985;45:520–5. [PubMed] [Google Scholar]
- 5.Menichini P, Fronza G, Tornaletti S, Galiègue-Zouitina S, Bailleul B, Loucheux-Lefebvre MH, Abbondandolo A, Pedrini AM. In vitro DNA modification by the ultimate carcinogen of 4-nitroquinoline-1-oxide: influence of superhelicity. Carcinogenesis. 1989;10:1589–93. doi: 10.1093/carcin/10.9.1589. [DOI] [PubMed] [Google Scholar]
- 6.Jarosz DF, V, Godoy G, Delaney JC, Essigmann JM, Walker GC. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–8. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 7.Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell. 2008;30:519–29. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 8.Kohda K, Tada M, Kasai H, Nishimura S, Kawazoe Y. Formation of 8-hydroxyguanine residues in cellular DNA exposed to the carcinogen 4-nitroquinoline-1-oxide. Biochem Biophys Res Commun. 1986;139:626–32. doi: 10.1016/s0006-291x(86)80036-5. [DOI] [PubMed] [Google Scholar]
- 9.Ishii Y, Kondo S. Comparative analysis of deletion and base-change mutabilities of Escherichia coli B strains differing in DNA repair capacity (wild-type, uvrA-, polA-, recA-) by various mutagens. Mutat Res. 1975;27:27–44. doi: 10.1016/0027-5107(75)90271-7. [DOI] [PubMed] [Google Scholar]
- 10.Ikenaga M, Ichikawa-Ryo H, Kondo S. The major cause of inactivation and mutation by 4-nitroquinoline-1-oxide in Escherichia coli: excisable 4-NQO-purine adducts. J Mol Biol. 1975;92:341–56. doi: 10.1016/0022-2836(75)90233-8. [DOI] [PubMed] [Google Scholar]
- 11.Ikenaga M, Ishii Y, Tada M, Kakunaga T, Takebe H. Excision-repair of 4-nitroquinoline-1-oxide damage responsible for killing, mutation, and cancer. Basic Life Sci. 1975;5B:763–71. doi: 10.1007/978-1-4684-2898-8_54. [DOI] [PubMed] [Google Scholar]
- 12.Ikenaga M, Takebe H, Ishii Y. Excision repair of DNA base damage in human cells treated with the chemical carcinogen 4-nitroquinoline-1-oxide. Mutat Res. 1977;43:415–27. doi: 10.1016/0027-5107(77)90062-8. [DOI] [PubMed] [Google Scholar]
- 13.Gross J, Gross M. Genetic analysis of an E. coli strain with a mutation affecting DNA polymerase. Nature. 1969;224:1166–8. doi: 10.1038/2241166a0. [DOI] [PubMed] [Google Scholar]
- 14.Campbell JL, Soll L, Richardson CC. Isolation and partial characterization of a mutant of Escherichia coli deficient in DNA polymerase II. Proc Natl Acad Sci USA. 1972;69:2090–4. doi: 10.1073/pnas.69.8.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. American Society for Microbiology; Washington, D.C: [Google Scholar]
- 16.Wang TV, Smith KC. Effect of recB21, uvrD3, lexA101 and recF143 mutations on ultraviolet radiation sensitivity and genetic recombination in delta uvrB strains of Escherichia coli K-12. Mol Gen Genet. 1981;183:37–44. doi: 10.1007/BF00270135. [DOI] [PubMed] [Google Scholar]
- 17.Lloyd RG, Buckman C. Genetic analysis of the recG locus of Escherichia coli K-12 and of its role in recombination and DNA repair. J Bacteriol. 1991;173:1004–11. doi: 10.1128/jb.173.3.1004-1011.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Courcelle J, Hanawalt PC. Participation of recombination proteins in rescue of arrested replication forks in UV-irradiated Escherichia coli need not involve recombination. Proc Natl Acad Sci USA. 2001;98:8196–202. doi: 10.1073/pnas.121008898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith KC. Recombinational DNA repair: the ignored repair systems. Bio Essays. 2004;26:1322–6. doi: 10.1002/bies.20109. [DOI] [PubMed] [Google Scholar]
- 20.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu Rev Microbiol. 2006;60:231–53. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 21.Beuning PJ, Simon SM, Godoy VG, Jarosz DF, Walker GC. Characterization of Escherichia coli translesion synthesis polymerases and their accessory factors. Meth Enzymol. 2006;408:318–40. doi: 10.1016/S0076-6879(06)08020-7. [DOI] [PubMed] [Google Scholar]
- 22.Miller JH. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: [Google Scholar]
- 23.Heltzel JM, Maul RW, Scouten Ponticelli SK, Sutton MD. A model for DNA polymerase switching involving a single cleft and the rim of the sliding clamp. Proc Natl Acad Sci USA. 2009;106:12664–9. doi: 10.1073/pnas.0903460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ona KR, Courcelle CT, Courcelle J. Nucleotide excision repair is a predominant mechanism for processing nitrofurazone-induced DNA damage in Escherichia coli. J Bacteriol. 2009;191:4959–65. doi: 10.1128/JB.00495-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boye E, Lobner-Olesen A. Bacterial growth control studied by flow cytometry. Res Microbiol. 1991;142:131–5. doi: 10.1016/0923-2508(91)90020-b. [DOI] [PubMed] [Google Scholar]
- 26.Bahloul A, Boubrik F, Rouvière-Yaniv J. Roles of Escherichia coli histone-like protein HU in DNA replication: HU-beta suppresses the thermosensitivity of dnaA46ts. Biochimie. 2001;83:219–29. doi: 10.1016/s0300-9084(01)01246-9. [DOI] [PubMed] [Google Scholar]
- 27.Skarstad K, Steen HB, Boye E. Escherichia coli DNA distributions measured by flow cytometry and compared with theoretical computer simulations. J Bacteriol. 1985;163:661–8. doi: 10.1128/jb.163.2.661-668.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steen HB. Staining and measurement of DNA in bacteria. Methods Cell Biol. 2001;64:539–51. doi: 10.1016/s0091-679x(01)64028-7. [DOI] [PubMed] [Google Scholar]
- 29.Horii Z, Suzuki K. Degradation of the DNA of Escherichia coli K12 rec- (JC1569b) after irradiation with ultraviolet light. Photochem Photobiol. 1968;8:93–105. doi: 10.1111/j.1751-1097.1970.tb05976.x. [DOI] [PubMed] [Google Scholar]
- 30.Horii ZI, Suzuki K. Degradation of the DNA of recA mutants of Escherichia coli K-12 after irradiation with ultraviolet light. II. Further studies including a recA uvrA double mutant. Photochem Photobiol. 1970;11:99–107. doi: 10.1111/j.1751-1097.1970.tb05976.x. [DOI] [PubMed] [Google Scholar]
- 31.Galiègue-Zouitina S, Daubersies P, Loucheux-Lefebvre MH, Bailleul B. Mutagenicity of N2 guanylarylation is SOS functions dependent and reminiscent of the high mutagenic property of 4-NQO. Carcinogenesis. 1989;10:1961–6. doi: 10.1093/carcin/10.10.1961. [DOI] [PubMed] [Google Scholar]
- 32.Dickman MJ, Ingleston SM, Sedelnikova SE, Rafferty JB, Lloyd RG, Grasby JA, Hornby DP. The RuvABC resolvasome. Eur J Biochem. 2002;269:5492–501. doi: 10.1046/j.1432-1033.2002.03250.x. [DOI] [PubMed] [Google Scholar]
- 33.Mandal TN, Mahdi AA, Sharples GJ, Lloyd RG. Resolution of Holliday intermediates in recombination and DNA repair: Indirect suppression of ruvA, ruvB, and ruvC mutations. J Bacteriol. 1993;175:4325–4332. doi: 10.1128/jb.175.14.4325-4334.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGlynn P, Lloyd RG. Genome stability and the processing of damaged replication forks by RecG. Trends Genet. 2002;18:413–419. doi: 10.1016/s0168-9525(02)02720-8. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Mahdi AA, Briggs GS, Lloyd RG. Promoting and avoiding recombination: contrasting activities of the Escherichia coli RuvABC Holliday junction resolvase and RecG DNA translocase. Genetics. 2010 doi: 10.1534/genetics.110.114413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol. 1968;31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]
- 37.Heller RC, Marians KJ. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006;439:557–62. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 38.Umezu K, Chi NW, Kolodner RD. Biochemical interaction of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc Natl Acad Sci USA. 1993;90:3875–9. doi: 10.1073/pnas.90.9.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Umezu K, Kolodner RD. Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J Biol Chem. 1994;269:30005–13. [PubMed] [Google Scholar]
- 40.Cox MM. Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol. 2007;42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- 41.Sakai A, Cox MM. RecFOR and RecOR as distinct RecA loading pathways. J Biol Chem. 2009;284:3264–72. doi: 10.1074/jbc.M807220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Courcelle J, Hanawalt PC. RecA-dependent recovery of arrested DNA replication forks. Annu Rev Genet. 2003;37:611–46. doi: 10.1146/annurev.genet.37.110801.142616. [DOI] [PubMed] [Google Scholar]
- 43.Fujii S, Isogawa A, Fuchs RP. RecFOR proteins are essential for Pol V-mediated translesion synthesis and mutagenesis. EMBO J. 2006;25:5754–63. doi: 10.1038/sj.emboj.7601474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogi T, Shinkai Y, Tanaka K, Ohmori H. Pol kappa protects mammalian cells against the lethal and mutagenic effects of benzo[a]pyrene. Proc Natl Acad Sci USA. 2002;99:15548–53. doi: 10.1073/pnas.222377899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kusumoto R, Masutani C, Iwai S, Hanaoka F. Translesion synthesis by human DNA polymerase eta across thymine glycol lesions. Biochemistry. 2002;41:6090–9. doi: 10.1021/bi025549k. [DOI] [PubMed] [Google Scholar]
- 46.McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC. Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell. 2005;20:783–92. doi: 10.1016/j.molcel.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Rajao MA, Passos-Silva DG, DaRocha WD, Franco GR, Macedo AM, Pena SD, Teixeira SM, Machado CR. DNA polymerase kappa from Trypanosoma cruzi localizes to the mitochondria, bypasses 8-oxoguanine lesions and performs DNA synthesis in a recombination intermediate. Mol Microbiol. 2009;71:185–97. doi: 10.1111/j.1365-2958.2008.06521.x. [DOI] [PubMed] [Google Scholar]
- 48.Lovett ST. Replication arrest-stimulated recombination: Dependence on the RecA paralog, RadA/Sms and translesion polymerase, DinB. DNA Repair (Amst) 2006;5:1421–7. doi: 10.1016/j.dnarep.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 49.Wang TV, Smith KC. recF-dependent and recF recB-independent DNA gap-filling repair processes transfer dimer-containing parental strands to daughter strands in Escherichia coli K-12 uvrB. J Bacteriol. 1984;158:727–9. doi: 10.1128/jb.158.2.727-729.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galitski T, Roth JR. Pathways for homologous recombination between chromosomal direct repeats in Salmonella typhimurium. Genetics. 1997;146:751–67. doi: 10.1093/genetics/146.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 52.Ogi T, Lehmann AR. The Y-family DNA polymerase kappa (pol kappa) functions in mammalian nucleotide-excision repair. Nat Cell Biol. 2006;8:640–2. doi: 10.1038/ncb1417. [DOI] [PubMed] [Google Scholar]