

Abstract
AIMS
Although the standard treatment for primary central nervous system lymphoma (PCNSL) consists of three cycles of MBVP (methotrexate, BCNU, VP16, methylprednisolone) and radiotherapy, early failure of treatment may require modification of the treatment. However, our understanding of the outcome in such patients and of the factors involved in early failure of treatment is poor. In addition to known prognostic factors, we evaluated the influence of methotrexate (MTX) exposure on the response to MBVP chemotherapy in patients treated for PCNSL after the first two cycles.
METHODS
We retrospectively analyzed all patients with PCNSL treated with the MBVP regimen over the previous 10 years. Clinical, personal data and known prognostic factors were studied. The parameters of MTX exposure were estimated using a population pharmacokinetic approach with NONMEM. Objective response (OR), overall survival (OS) and failure-free survival (FFS) were evaluated in all patients.
RESULTS
Thirty-seven patients were studied. We observed lower FFS and OS (0.49 years) in patients who were not able to receive the planned treatment (group 1, n = 12) than in those who received three cycles (8.04 years) (group 2, n = 25). Known prognostic factors were comparable in both groups, but mean dose of MTX and mean AUC tended to be lower in patients who failed prematurely or showed no response after two cycles.
CONCLUSIONS
We found that patients who were early non-responders to MBVP chemotherapy had poor survival, without major influence of MTX exposure. It is thus probably unlikely that increasing the dose of MTX would improve outcome.
Keywords: dose–response relationship, methotrexate, pharmacokinetics, primary central nervous system lymphoma
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Although treated using the same high-dose methotrexate (HD-MTX)-based multiagent chemotherapy, patients with primary central nervous system lymphoma (PCNSL) have significant differences in outcome. However, little information has been published about factors influencing outcome in PCNSL. As it is known that the pharmacokinetics of MTX vary considerably between subjects leading to different exposure in patients receiving the same dose, it is important to evaluate its role in response to chemotherapy.
WHAT THIS STUDY ADDS
This study is the first to evaluate the exposure–response relationship in patients treated with MBVP chemotherapy. We found that patients who were early non-responders to MBVP chemotherapy had poor survival, whatever the salvage regimen. Tumour response at early evaluation was not associated with MTX pharmacokinetics and increasing the dose would probably not improve results.
Introduction
Primary central nervous system lymphoma (PCNSL) is a rare condition, although its incidence has increased during in the last 10 years [1–4]. It accounts for about 5% of all central nervous system (CNS) tumours. The standard treatment for newly diagnosed PCNSL in patients aged less than 60 years is high-dose methotrexate (HD-MTX)-based multi-agent chemotherapy followed by whole-brain radiation therapy (WBRT) [5]. In the elderly, chemotherapy alone is preferred since it has been described as effective as and less neurotoxic than radiotherapy or chemoradiotherapy [6]. The introduction of methotrexate, a drug which penetrates the blood brain barrier effectively has improved median survival from 10 to 16 months to more than 30 months [7]. Based on the experience of the French Groupe Ouest Est d'Etude des Leucémies et Autres Maladies du Sang (GOELAMS), MBVP (HD-methotrexate, BCNU, VP16, methylprednisolone) chemotherapy followed by RT at a dose of 40 Gy in responding patients represents the current standard treatment for PSNCL in our institution [8].
However, significant differences in outcome are observed between patients, whatever the therapy used. Age and performance status are universally accepted as prognostic factors [9, 10]. Ferreri et al. have provided new understanding in this area using multivariate analysis in a large cohort of patients with PCNSL [11]. They revealed an independent association between overall survival and age, performance status, LDH serum concentration, CSF protein concentration and involvement of deep structures of the brain. A prognostic score, obtained by adding each of these variables (assigned a score of 0 or 1, if absent or present), was significantly correlated with survival, thus making it possible to distinguish low-risk, low-intermediate and high-intermediate risk groups. The use of high dose methotrexate in 25% of the patients of this cohort was also independently associated with better survival, confirming the major role of this chemotherapeutic agent in PCNSL. As the efficacy of high dose methotrexate may be related to elevated serum concentrations favouring penetration of the CNS [12, 13], the authors also investigated the impact of MTX exposure on toxicity and outcome in their retrospective series of 45 patients with PSNCL treated according to various schedules [11]. They found that schedules leading to higher concentrations were associated with a better response with no higher incidence of toxicity. These results supported the use of a MTX dose >3 g m−2 administered as a 4 or 6 h infusion, every 3–4 weeks. The heterogeneity of the treatments analyzed did not allow the authors to study exposure–effect relationships within each treatment schedule. However, it is known that the pharmacokinetics of MTX vary considerably between subjects due to various factors, of which renal function and concurrent medications have the greatest impact [14–27]. Thus, although treated using the same schedule, different patients may have different exposure to the drug, which in turn may modify outcome.
In our institution, MBVP chemotherapy is the standard protocol for PSNCL. Although it is planned that all patients receive three cycles of MBVP, the attending physician may decide to cease or modify the treatment after two cycles, or earlier, for some of them, mainly because of insufficient efficacy. We have poor understanding of the outcome in such patients. Furthermore, factors involved in early failure of treatment have not been studied to date. In addition to already known prognostic factors, we evaluated the influence of methotrexate exposure on response to chemotherapy in patients treated with MBVP chemotherapy. We specifically focused on the exposure–response relationship after the first two cycles to study whether failure may be caused by lower levels of exposure.
Methods
Study population
We retrospectively analyzed all patients treated with MBVP chemotherapy for pathologically confirmed PCNSL between January 1995 and December 2005 in our institution. Patients were selected if it had been planned that they should receive three cycles of MBVP combined with radiotherapy.
Clinical and personal data were gathered by retrospective chart review, including age, sex, body weight, body surface area and creatinine clearance. Factors already known to be associated with outcome, i.e. performance status, CSF protein concentration, LDH serum concentration and involvement of deep structures or intraocular disease, were also included. Some of these prognostic factors were considered as nominal variables for our study as follows: age (≤60 and >60 years), performance status (PS 0–1 and 2–4), LDH serum concentration (elevated concentration ≥350 IU l−1, normal concentration <350 IU l−1).
Administration of treatment
All patients provided consent for chemotherapy and were admitted to the hospital for each cycle of treatment. Methotrexate was administered as part of MBVP polychemotherapy comprising high dose methotrexate (HD-MTX) at 3 g m−2, 60 mg m−2 methylprednisolone, 100 mg m−2 etoposide and 100 mg m−2 BCNU (Table 1). In patients aged >60 years with impaired creatinine clearance or poor performance status, the dose of MTX was reduced to 1 g m−2. In patients with impaired renal function, the dose was reduced in proportion to the decrease in creatinine clearance, in agreement with the recommendations of the product characteristics summary. All patients received adequate hydration and urine alkalinization during and following HD-MTX infusion. Toxicity was prevented by folinic acid rescue tailored according to methotrexate serum concentrations. Intrathecal methotrexate (15 mg), aracytine (40 mg) and methylprednisolone (40 mg) were administered to all patients throughout the treatment but no radiotherapy was performed at this stage. Clinical status was evaluated by the physician after each cycle. Normally, it is intended to evaluate tumour response after three cycles of chemotherapy. However, in practice, the physician may decide to evaluate the response after two cycles if he suspects an unfavourable outcome after clinical evaluation. Furthermore, some patients may die or be switched to another treatment without receiving the entire treatment. In patients still under treatment, tumour response was evaluated by iconography after they had completed two or three cycles of chemotherapy. Further treatment was decided according to tumour response. For partial or complete responders after three cycles, radiotherapy started 2 to 4 weeks after completing the last course of MBVP. The non-responders received two additional courses of methotrexate followed by radiotherapy or were switched to a salvage regimen.
Table 1.
Chemotherapy schedule, every 21 days
| Drug | Dose | Route and duration | Day |
|---|---|---|---|
| Methylprednisolone | 60 mg m−2 | p.o. | 1–5 |
| Methotrexate | 3 g m−2 | 6 h or 24 h infusion | 1, 15 |
| Leucovorin rescue | Tailored | i.v./p.o. | Tailored |
| Etoposide | 100 mg m−2 | i.v. | 1 |
| BCNU | 100 mg m−2 | i.v. | 1 |
Outcome measurement
Response was evaluated in all patients according to the World Health Organization criteria [28]. This was performed by clinical examination and by computed tomography or magnetic resonance imaging with contrast enhancement. Patients were classified as responders if they had complete or partial response. Complete response was defined by the disappearance of all disease-related symptoms if present before therapy, normalization of the biochemical abnormalities and disappearance of all detectable clinical and radiographic evidence of disease. Partial responders were characterized by a more than 50% decrease in the sum of the product of the greatest diameters (SPD) of the six largest dominant nodes or nodal masses [28].
Overall survival (OS) was defined as time from the start of treatment to death or to the date of the last follow-up control, whereas failure-free survival (FFS) was defined as time from the start of treatment to relapse or death or to the date of the last follow-up control.
Post-course toxicity was graded according to the National Cancer Institute criteria, common toxicity criteria (CTC). We also evaluated whether or not the theoretical interval between each MBVP course was respected.
Methotrexate monitoring
MTX concentrations were measured at specific times from the start of the infusion (48 h, 72 h and every 24 h if necessary, i.e. until the serum concentration was <0.2 µm) to determine adequate supportive care, including leucovorin rescue if the concentration exceeded a predefined threshold at each time-point.
Methotrexate serum concentrations were measured by a fluorescent polarization immunoassay (Abbott, FPIA-2) with a TDx-FLx® analyzer (Abbott Park, IL, USA).
Pharmacokinetic modelling
Methotrexate pharmacokinetic parameters were estimated using a population pharmacokinetic approach with NONMEM (version 5.1.1; Globomax LLC, Hanover, USA) and Wings for NONMEM software (WFN; http://sourceforge.net). A two-compartment pharmacokinetic model was fitted to the data, using the FOCE method and the NONMEM subroutine ADVAN3, parameterized in terms of clearance (CL), central compartment volume (V1), inter-compartment clearance (Q) and peripheral volume (V2) by the PREDPP subroutine library. Exponential and proportional model errors were used for inter-subject and residual variability, respectively. Standard errors were calculated with the COVARIANCE option of NONMEM. Graphic model diagnostics were performed with the following diagnostic plots: observed concentrations vs. population predicted concentrations, weighted residuals vs. time, individual predictions vs. DV and individual weighted residuals vs. time. A normalized prediction distribution error (NPDE) assessment of the final model was performed from 1000 Monte Carlo simulations. The distribution of the obtained NPDE was compared with the normal distribution [29]. Diagnostic plots were obtained with the R software (R version 2.5.1, R project, Auckland, USA).
Data from the present study were pooled with a previous database for NHL patients, treated according to the same schedules as ours and for whom more sampling times were available, particularly during the early period [30]. This enabled us to model accurately each point of the concentration profile for each subject despite using only their partial therapeutic drug monitoring data. The total number of courses analyzed was 263, from which 178 corresponded to the patients of the present study.
Individual pharmacokinetic parameters were obtained using the POSTHOC function of NONMEM. This allowed us to obtain an individual area under the curve for each course, from which we derived cumulative AUC (i.e. the sum of each individual AUC), mean AUC and AUC intensity for each patient. As AUC was directly obtained from estimated individual clearance, we analyzed the ηCL and εCL distribution to measure the extent of shrinkage [31]. AUC intensity was obtained as cumulative AUC divided by the number of weeks between the first administration and the time of the last methotrexate serum measurement for the last cycle. Exposure parameters were calculated after two cycles (four courses) in all patients, or from the exact number of courses in those who did not receive the two first cycles because of early failure.
Statistical analysis
Prognostic factors (age, performance status, LDH serum concentration, involvement of deep structures, intraocular disease, CSF protein concentration and presence of cells in CSF) were compared between patients who received three cycles and those who received two cycles or less, using the Chi Square test or Fisher's exact test.
Comparison of MTX dose (median dose, mean dose and cumulative dose) and of exposure parameters (mean AUC, cumulative AUC and AUC intensity) in patients who failed to respond at or before two cycles and those who were able to receive the planned treatment were based on the Wilcoxon test. Survival data were analyzed according to prognostic factors and methotrexate exposure parameters in patients of each group (≤ two or three cycles).
Cumulative survival curves were estimated by the Kaplan–Meier method. Comparison between patients of each group was made using the log-rank test.
In the subset of patients who received three cycles, prognostic factors (age, performance status, LDH serum concentration, involvement of deep structures, intraocular disease, CSF protein concentration and presence of cells in CSF) and methotrexate exposure parameters were compared between responders and non-responders using the Chi Square test or Fisher's exact test.
Analyses were performed using SAS software (SAS Institute Inc., Cary, NC, version 9.1).
Results
Patients and treatment
The total number of patients preselected for analysis on the basis of our criteria was 42. Five patients were not retained for analysis for the following reasons: secondary and not primary central nervous system lymphoma (n = 2), early blood stem cell graft before the second cycle of MBVP (n = 2), severe renal impairment before treatment precluding the administration of MTX (n = 1).
Thus, the study group consisted of 37 patients, including 51.0% females (Table 2). The median weight was 69 kg (47–92) and the body surface area 1.76 m2 (1.47–2.14). Twenty-six patients had at least one poor prognostic factor, and 67.6% of patients were aged over 60 years (median age: 66.6 years (38–86)). Twelve patients did not receive the three planned cycles; they were assigned to group 1. Some of them received two cycles and were evaluated just after, but others were switched to a salvage regimen or died before having received the two cycles. Twenty-five patients were able to receive three cycles of MBVP and they were assigned to group 2.
Table 2.
Characteristics of patients receiving onlytwo cycles of MBVP (Group 1), and those receiving threecycles of MBVP (Group 2)
| All patients (n = 37) | Group 1 (n = 12) | Group 2 (n = 25) | |||
|---|---|---|---|---|---|
| Variables | Subgroups | n (%) | n (%) | n (%) | P |
| Age | <60 years old | 12 (32) | 3 (25) | 9 (36) | 0.71 |
| >60 years old | 25 (68) | 9 (75) | 16 (64) | ||
| Performance status | 0–1 | 27 (73) | 8 (67) | 19 (76) | 0.69 |
| 2–4 | 10 (27) | 4 (33) | 6 (24) | ||
| Deep lesions | Yes | 14 (38) | 1 (8) | 13 (52) | 0.01 |
| No | 23 (62) | 11 (92) | 12 (48) | ||
| Intraocular disease | Yes | 9 (24) | 3 (25) | 6 (24) | 1 |
| No | 28 (76) | 9 (75) | 13 (52) | ||
| LDH serum concentration | Elevated | 14 (38) | 2 (17) | 12 (48) | 0.08 |
| Normal | 23 (62) | 10 (83) | 13 (52) | ||
| Cells in CSF | Yes | 7 (19) | 0 (0) | 7 (28) | 0.07 |
| No | 30 (81) | 12 (100) | 18 (72) | ||
| CSF protein concentration* | <0.4 g l−1 | 6 (17) | 2 (18) | 4 (17) | 1 |
| >0.4 g l−1 | 29 (83) | 9 (82) | 20 (83) |
Two CSF protein concentration values were not available, one in group 1 and one in group 2.
Overall, 178 methotrexate courses were analyzed and the patients received a median dose of 1990 mg m−2 (759–3035 mg m−2). Thirty-six courses were available in patients from group 1: six received the four courses, two received three courses, two received two courses and two received only one course. The median (range) dose of methotrexate was 1510 mg m−2 (966–3026). The theoretical dose of 3000 mg m−2 was administered in 13 courses (36.1%) and a reduced dosage was administered in the remaining 23. One hundred forty-two courses were analyzed in patients from group 2, the last course being administered after evaluation in eight of them. The median (range) dose was 2010 mg m−2 (759–3035). The theoretical dose of 3000 mg m−2 was administered in 70 courses (49%) and a reduced dosage was administered in the remaining 72.
Response to treatment
Twenty-nine (78%) patients were classified as responders, 44.8% of whom had a complete response.
Seven of the patients in group 1 (58%) had progressive disease and five (42%) had a partial response. Two patients received CYVE salvage chemotherapy (cytarabine 2 g m−2 day−1 on days 2 to 5 as a 3 h infusion plus 50 mg m−2 day−1 on days 1 to 5 as a 12 h infusion, and VP16 200 mg m−2 day−1 on days 2 to 5 as a 2 h infusion) and four received salvage radiotherapy. The disease progressed in the six remaining patients and they died before being switched to another treatment.
Evaluation at the end of treatment for patients who received the entire MBVP protocol (group 2, n = 25) showed that only one patient (4%) had progressive disease, 11 (44%) had a partial response and 13 (52%) had a complete response. The responding patients (with complete or partial response) then received radiotherapy. The patient with progressive disease died 1 month after ending treatment.
Survival
Eighteen patients died (48.7%), with 13 deaths (72%) occurring during the first 2 years after diagnosis, 10 belonging to group 1 (83%) and three (12%) to group 2. Of the patients who received the entire treatment, three patients relapsed after MBVP treatment (12%) and five patients were lost to follow-up (20%).
The median follow-up was 6 months (2.4–58.8) for patients of group 1 vs. 26.4 months (15.6–32.4) for patients receiving the entire MBVP protocol. The median survival was evaluated at 0.49 years (0.22–4.92) in group 1 vs. 8.04 years (2.16–NE) in group 2 (P < 0.001). The upper limit of median survival was non estimable (NE) because of insufficient data (Figure 1).
Figure 1.
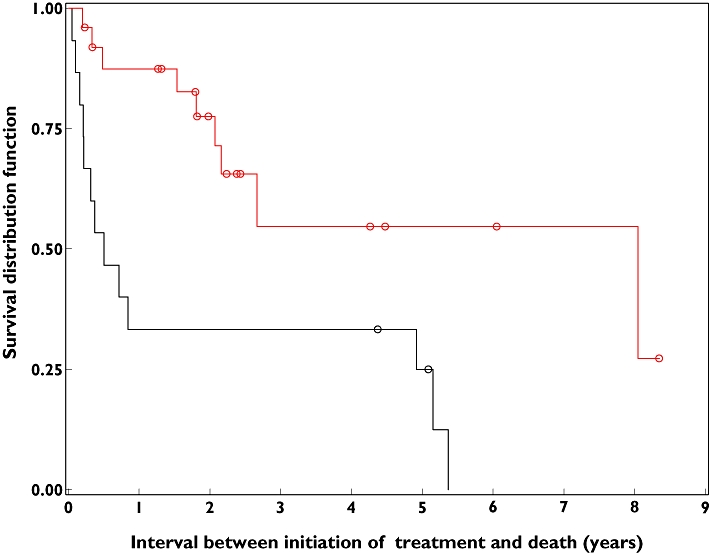
Kaplan–Meier curves representing overall survival of all patients. Black curves correspond to group 1, red curves correspond to group 2 and empty circles represent censored data
Methotrexate exposure
Population modelling was performed on a total of 704 serum samples, 475 being from our patients. A two-compartment model best fitted the data. Intra-patient variability was often observed, and the inclusion of creatinine clearance (CLcr) at each course and of inter-occasion variability produced a decrease in the objective function from −782.1 to −1033.6 and a decrease in the inter-individual variability of CL from 54.7 to 35.3% as compared with the base model (Table 3). Importantly, we obtained identical population pharmacokinetics parameters as those previously published using a subset of our data [30]. Individual weighted residues were between −1 and +1 for all observed concentrations. The shape of the distribution of the NPDE is presented on Figure 2 (Figure 2). The test of normality was significant but this is often observed when analyzing a great number of observations. Apart from a few outliers, the scatter plot of the NPDE vs. time did not pinpoint a particular deficiency in the model (m = 0.109, P = 0.062 and v = 1.074, P = 0.168) (Figure 2).
Table 3.
Final parameters estimates of the final model
| Parameter | Population estimate of θ | Standard error of estimate | Relative standard error (CV%) | BSVa (%) |
|---|---|---|---|---|
| CL = θi × CLcr | ||||
| θ1 | 0.0716 | 9.0 | 22.5 | 28.7 |
| θ2 | 0.0788 | 10.3 | 32.3 | 31.0 |
| θ3 | 0.0740 | 11.0 | 32.4 | 28.4 |
| θ4 | 0.0700 | 12.5 | 72.2 | 34.1 |
| θ5 | 0.0744 | 7.6 | 40.7 | 25.2 |
| θ6 | 0.0684 | 11.8 | 31.2 | 36.5 |
| V1 (l) | 22.7 | 19.5 | 32.3 | 41.2 |
| V2 (l) | 3.46 | 11.6 | 23.5 | 53.9 |
| Residual error (CV %) | 35.2 | 9.0 | 10.9 | – |
θi: between occasion value of θ, as evaluated on the six first courses. CLcr in ml min−1 and CL in l h−1.
between subject variability.
Figure 2.
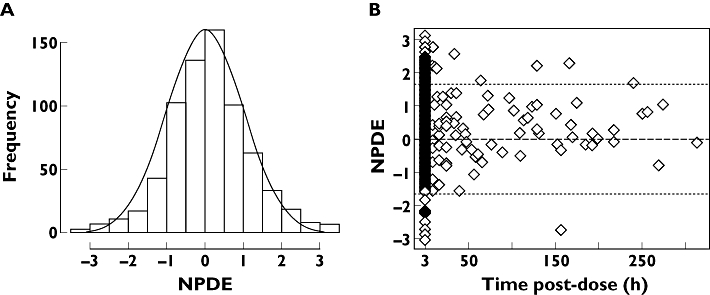
Distribution plot (A) of the Normalized Prediction Distribution Error (NPDE) and scatter plot of the NPDE vs. time (B) after 1000 Monte Carlo simulations
Shrinkage analysis for CL showed mean ηCL shrinkage of 23% (13–38) and ε-shrinkage of 37%.
Factors influencing response to treatment
The patients in group 2 were those for whom the physician judged that they were able to receive the entire course of MBVP chemotherapy. In contrast, patients in group 1 had clinical signs of evolution or poor response. Unexpectedly, a statistically higher incidence of deep lesions was found in patients in group 2 than in group 1 (P < 0.01). The other prognostic factors were not different between the two groups.
Although patients from group 1 received a lower median (range) dose of methotrexate [1510 mg m−2 (966–3026)] than those of group 2 [1983 mg m−2 (999–3000)] after two cycles, the difference was not significant (P = 0.18). The cumulative dose was higher in patients in group 2 compared with those in group 1 (19 200 mg vs. 12 600 mg, P = 0.02) due to the greater number of courses administered per patient in group 2. Accordingly a higher cumulative AUC was found in group 2 compared with group 1 (P = 0.01). Interestingly, patients in group 1 had a trend to have lower mean AUC. We found no significant difference in AUC intensity (Table 4).
Table 4.
Exposure parameters based on the first two cycles for patients of group 2 and on all available courses for patients of group 1
| Total population (n = 37) | Group 1 (n = 12) | Group 2 (n = 25) | |
|---|---|---|---|
| Exposure parameter | Median (1st–3rd quartile) | Median (1st–3rd quartile) | Median (1st–3rd quartile) |
| AUCc (mg l−1 h) | 2459 (1515–3123) | 1954 (823–3135) | 2468 (1915–3088) |
| AUCm (mg l−1 h) | 617 (475–778) | 571 (486–802) | 622 (436–735) |
| AUCi (mg l−1 h weeks−1) | 422 (276–534) | 484 (347–587) | 416 (257–512) |
AUCc, cumulative AUC; AUCi, AUC intensity; AUCm, mean AUC.
Within group 2, methotrexate dose, systemic exposure and prognostic factors were not different between responders and non-responders and had no influence on the FFS.
Severe toxicity
Forty-one percent of courses were associated with toxicity, including a high rate of haematotoxicity (39.6% of courses) and also renal toxicity (2.0% of courses) and mucositis (0.5%). The toxicity rate during the first two cycles of MBVP was not statistically different between the two groups (46% in group 1 vs. 37% in group 2). Grade 3-4 toxicity also occurred, including 25.8% of haematotoxicity, 1.5% renal toxicity and 0.5% mucositis. Half of the patients who had grade 3-4 renal toxicity returned to normal renal function within a few weeks of ending treatment. Six courses were delayed, one because of grade 4 haematotoxicity and the others for reasons that were not medical.
MTX overexposure, defined by a MTX serum concentration >2 µm 48 h after administration, occurred in six (3.1%) courses, three in group 1 and three in group 2.
Discussion
This study reports outcomes of patients with PSCNL treated by standard MBVP chemotherapy. The analysis showed therapeutic outcomes similar to those found in the literature [11]. However, two distinct groups of patients were identified, based on their ability or inability to receive the three planned cycles. Some factors had previously been shown to have a prognostic value in PSNCL. However, they were identified in large cohorts of patients treated according to various modalities, including radiotherapy (RT) alone, RT followed by chemotherapy, chemotherapy alone and chemotherapy followed by RT. To our knowledge, no study has evaluated prognostic factors in patients receiving homogenous treatment. We thus focused on evaluating factors that may have contributed to the divergent outcomes in our group of patients.
In the patients in our study who received two or less MBVP cycles on the basis of the physician's judgment, then secondarily evaluated by iconography, failure was confirmed in 58% of cases, the others being only partial responders.
These patients (group 1) had a very poor outcome compared with patients who were able to receive the three planned cycles. Thus, the patients showing an early response to the MBVP protocol after two cycles had a higher survival rate, whatever the salvage chemotherapy used in patients from group 1. Identifying factors involved in early failure would therefore have important consequences.
When considering the whole population, we observed that most of the patients were administered a decreased dose of methotrexate compared with the theoretical dose. This was mainly explained by the fact that the patients were elderly (67% of patients were more than 60 years old) or had altered renal function. Moreover, mean dose effectively received did not influence the early response to treatment, as we found no significant difference between patients who were able to continue and those for whom treatment was prematurely withdrawn due to unfavourable evolution.
As similar doses may produce variable systemic exposure, we also analyzed whether the pharmacokinetic parameters of MTX differ between the two groups. We used a population pharmacokinetic approach to describe MTX pharmacokinetics and analyzed the covariates possibly associated with variable exposure. Using this approach, it is possible to estimate exposure to a drug much more precisely than by using only isolated point-concentrations. The concentrations measured in our patients as part of therapeutic drug monitoring, and pooled with those from an independent database, were best described by a two-compartment model. The major interest of pooling our data to an independent database was to estimate better AUC by adding early concentrations not available for our patients. We also included inter-occasion variability in the model, to compensate for the marked differences observed in some patients from one course to another despite the unchanged MTX dose.
This pharmacokinetic model was used to estimate individual AUC at each course, from which we derived individual cumulative AUC, individual mean AUC and individual AUC intensity for the first two cycles. As individual AUC is directly obtained from individual clearance estimated from the model (POSTHOC), the extent of shrinkage of ηCL gives useful information to assess the relevance of the subsequent exposure–response analysis. Savic et al.[31] indicated that, when shrinkage is higher than 20–30%, individual parameter estimation may be affected. Our result for ηCL shrinkage (23%) was not different from theoretically expected, considering the few observations per subject in our data set. However, the extent of shrinkage that we obtained, although not very high, could have limited precise evaluation of parameter–covariate relationships as well as brought a degree of uncertainty on the estimation of individual CL. As expected, MTX exposure varied markedly between patients. Intraindividual variability was evident in some patients while pharmacokinetic parameters were stable in others. Creatinine clearance was identified as a covariate influencing MTX clearance. As methotrexate exposure (mean AUC) was not statistically different between patients who were able to receive the entire treatment and those whose treatment was stopped just after or before two cycles, our results indicate that pharmacokinetic variability does not play a significant role in the early response to treatment and that increasing the dose of MTX to target a higher AUC would thus probably not improve results. PCNSL is a rare condition and we were not able to include more patients in this monocentric study. Despite being not significant, our results indicate that, when evaluated at the same time point, patients in group 1 had a trend to have lower mean exposure than those who were able to pursue treatment. Hopefully, our results will open the way for others to evaluate this relationship in a larger cohort as it would have important consequences in terms of dosage adjustment. Our results also confirmed that dosage adjustment in patients >60 years old or with impaired renal function yielded similar exposure to patients treated with the standard dose, i.e it compensated for the decreased clearance.
A quantitative marker of response, i.e. change in tumour size from baseline and the end of cycle 2, would have been very useful to establish exposure–response relationships. Karrison et al. used both a dichotomous index (whether or not a 50% decrease in tumour area had occurred) and a quantitative one (the ratio of the tumour area taken at a fixed time point compared with the tumour area at the start of protocol treatment) to evaluate response in 46 patients with advanced gastric cancer [32]. Such a quantitative evaluation of response was not possible in our study since, on these retrospective data, the accurate information of the change in tumour size was not available. Response was thus evaluated from both clinical examination and computed tomography or magnetic resonance imaging with contrast enhancement, from which a percentage of response was determined (total/partial/non-responders). The results of our study should thus be interpreted from a pragmatic point of view, as it aimed at answering the question of whether or not MTX exposure has an impact on response, the latter being evaluated by standard methods that are used in routine patient care.
Using already known prognostic factors, the involvement of deep lesions was unexpectedly higher in patients who were able to receive three cycles than in those who failed after receiving ≤ two cycles. However, our population was very homogeneous, with a high rate of responders, and this could have limited the possibility of confirming the role of prognostic factors.
Creatinine concentrations and blood cell count were available for each course. The toxicity rate observed in our study was in the same range as previously reported in the literature for MBVP chemotherapy combined with radiotherapy [33]. However, we did not study the relationship between parameters of exposure to MTX and toxicity because the low rate of grade 3-4 toxicity hindered proper statistical evaluation.
In conclusion, we found that patients who were early non-responders to MBVP chemotherapy had poor survival, whatever the salvage regimen. Patients who were able to receive the entire MBVP chemotherapy had a good survival rate. We were not able to identify factors influencing response after two cycles, even if early MTX exposure tended to be lower in non-responders. The exact role of pharmacokinetic variability in PCNSL seems to be more difficult to establish than in other cancers.
Competing interests
There are no competing interests to declare.
REFERENCES
- 1.Bastard C, Tilly H, Lenormand B, Bigorgne C, Boulet D, Kunlin A, Monconduit M, Piguet H. Translocations involving band 3q27 and Ig gene regions in non-Hodgkin's lymphoma. Blood. 1992;79:2527–31. [PubMed] [Google Scholar]
- 2.Lister A, Abrey LE, Sandlund JT. Central nervous system lymphoma. Hematology Am Soc Hematol Educ Program. 2002:283–96. doi: 10.1182/asheducation-2002.1.283. [DOI] [PubMed] [Google Scholar]
- 3.Surawicz TS, McCarthy BJ, Kupelian V, Jukich PJ, Bruner JM, Davis FG. Descriptive epidemiology of primary brain and CNS tumors: results from the Central Brain Tumor Registry of the United States, 1990–1994. Neuro Oncol. 1999;1:14–25. doi: 10.1093/neuonc/1.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy S, Josephson SA, Fridlyand J, Karch J, Kadoch C, Karrim J, Damon L, Treseler P, Kunwar S, Shuman MA, Jones T, Becker CH, Schulman H, Rubenstein JL. Protein biomarker identification in the CSF of patients with CNS lymphoma. J Clin Oncol. 2008;26:96–105. doi: 10.1200/JCO.2007.12.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeAngelis LM, Seiferheld W, Schold SC, Fisher B, Schultz CJ. Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: Radiation Therapy Oncology Group Study 93–10. J Clin Oncol. 2002;20:4643–8. doi: 10.1200/JCO.2002.11.013. [DOI] [PubMed] [Google Scholar]
- 6.Hoang-Xuan K, Taillandier L, Chinot O, Soubeyran P, Bogdhan U, Hildebrand J, Frenay M, De Beule N, Delattre JY, Baron B. Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: a multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol. 2003;21:2726–31. doi: 10.1200/JCO.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 7.Plotkin SR, Batchelor TT. Primary nervous-system lymphoma. Lancet Oncol. 2001;2:354–65. doi: 10.1016/S1470-2045(00)00390-9. [DOI] [PubMed] [Google Scholar]
- 8.Desablens B, Gardembas M, Delwail V, Le Mevel A, Escoffre-Barbe M, Leprise PY, Briere J, Dutel JL, Harousseau JL, Raphael M, Colombat P. Primary CNS lymphomas: long term results of the GOELAMS LCP 88 trial with a focus on neurological complications among 152 patients. Ann Oncol. 1999;10:40. abstract. [Google Scholar]
- 9.Abrey LE, Ben-Porat L, Panageas KS, Yahalom J, Berkey B, Curran W, Schultz C, Leibel S, Nelson D, Mehta M, DeAngelis LM. Primary central nervous system lymphoma: the Memorial Sloan-Kettering Cancer Center prognostic model. J Clin Oncol. 2006;24:5711–15. doi: 10.1200/JCO.2006.08.2941. [DOI] [PubMed] [Google Scholar]
- 10.Corry J, Smith JG, Wirth A, Quong G, Liew KH. Primary central nervous system lymphoma: age and performance status are more important than treatment modality. Int J Radiat Oncol Biol Phys. 1998;41:615–20. doi: 10.1016/s0360-3016(97)00571-3. [DOI] [PubMed] [Google Scholar]
- 11.Ferreri AJ, Guerra E, Regazzi M, Pasini F, Ambrosetti A, Pivnik A, Gubkin A, Calderoni A, Spina M, Brandes M, Ferrarese F, Rognone A, Govi S, Dell'Oro S, Locatelli M, Villa E, Reni M. Area under the curve of methotrexate and creatinine clearance are outcome-determining factors in primary CNS lymphomas. Br J Cancer. 2004;90:353–8. doi: 10.1038/sj.bjc.6601472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balis FM, Savitch JL, Bleyer WA, Reaman GH, Poplack DG. Remission induction of meningeal leukemia with high-dose intravenous methotrexate. J Clin Oncol. 1985;3:485–9. doi: 10.1200/JCO.1985.3.4.485. [DOI] [PubMed] [Google Scholar]
- 13.Pitman SW, Frei E., III Weekly methotrexate-calcium leucovorin rescue: effect of alkalinization on nephrotoxicity; pharmacokinetics in the CNS; and use in CNS non-Hodgkin's lymphoma. Cancer Treat Rep. 1977;61:695–701. [PubMed] [Google Scholar]
- 14.Adams JD, Hunter GA. Drug interaction in psoriasis. Australas J Dermatol. 1976;17:39–40. doi: 10.1111/j.1440-0960.1976.tb00587.x. [DOI] [PubMed] [Google Scholar]
- 15.Beorlegui B, Aldaz A, Ortega A, Aquerreta I, Sierrasesumega L, Giraldez J. Potential interaction between methotrexate and omeprazole. Ann Pharmacother. 2000;34:1024–7. doi: 10.1345/aph.19094. [DOI] [PubMed] [Google Scholar]
- 16.Breedveld P, Zelcer N, Pluim D, Sonmezer O, Tibben MM, Beijnen JH, Schinkel AH, van Tellingen O, Borst P, Schellens JH. Mechanism of the pharmacokinetic interaction between methotrexate and benzimidazoles: potential role for breast cancer resistance protein in clinical drug-drug interactions. Cancer Res. 2004;64:5804–11. doi: 10.1158/0008-5472.CAN-03-4062. [DOI] [PubMed] [Google Scholar]
- 17.Dean R, Nachman J, Lorenzana AN. Possible methotrexate-mezlocillin interaction. Am J Pediatr Hematol Oncol. 1992;14:88–9. [PubMed] [Google Scholar]
- 18.Frenia ML, Long KS. Methotrexate and nonsteroidal antiinflammatory drug interactions. Ann Pharmacother. 1992;26:234–7. doi: 10.1177/106002809202600219. [DOI] [PubMed] [Google Scholar]
- 19.Joerger M, Huitema AD, van den Bongard HJ, Baas P, Schornagel JH, Schellens JH, Beijnen JH. Determinants of the elimination of methotrexate and 7-hydroxy-methotrexate following high-dose infusional therapy to cancer patients. Br J Clin Pharmacol. 2006;62:71–80. doi: 10.1111/j.1365-2125.2005.02513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karpf DM, Kirkegaard AL, Evans AM, Nation RL, Hayball PJ, Milne RW. Effect of ketoprofen and its enantiomers on the renal disposition of methotrexate in the isolated perfused rat kidney. J Pharm Pharmacol. 2003;55:1641–6. doi: 10.1211/0022357022287. [DOI] [PubMed] [Google Scholar]
- 21.Najjar TA, Abou-Auda HS, Ghilzai NM. Influence of piperacillin on the pharmacokinetics of methotrexate and 7-hydroxymethotrexate. Cancer Chemother Pharmacol. 1998;42:423–8. doi: 10.1007/s002800050840. [DOI] [PubMed] [Google Scholar]
- 22.Relling MV, Fairclough D, Ayers D, Crom WR, Rodman JH, Pui CH, Evans WE. Patient characteristics associated with high-risk methotrexate concentrations and toxicity. J Clin Oncol. 1994;12:1667–72. doi: 10.1200/JCO.1994.12.8.1667. [DOI] [PubMed] [Google Scholar]
- 23.Ronchera CL, Hernandez T, Peris JE, Torres F, Granero L, Jimenez NV, Pla JM. Pharmacokinetic interaction between high-dose methotrexate and amoxycillin. Ther Drug Monit. 1993;15:375–9. doi: 10.1097/00007691-199310000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Thyss A, Milano G, Kubar J, Namer M, Schneider M. Clinical and pharmacokinetic evidence of a life-threatening interaction between methotrexate and ketoprofen. Lancet. 1986;1:256–8. doi: 10.1016/s0140-6736(86)90786-5. [DOI] [PubMed] [Google Scholar]
- 25.Titier K, Lagrange F, Pehourcq F, Moore N, Molimard M. Pharmacokinetic interaction between high-dose methotrexate and oxacillin. Ther Drug Monit. 2002;24:570–2. doi: 10.1097/00007691-200208000-00018. [DOI] [PubMed] [Google Scholar]
- 26.Tracy TS, Krohn K, Jones DR, Bradley JD, Hall SD, Brater DC. The effects of a salicylate, ibuprofen, and naproxen on the disposition of methotrexate in patients with rheumatoid arthritis. Eur J Clin Pharmacol. 1992;42:121–5. doi: 10.1007/BF00278469. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto K, Sawada Y, Matsushita Y, Moriwaki K, Bessho F, Iga T. Delayed elimination of methotrexate associated with piperacillin administration. Ann Pharmacother. 1997;31:1261–2. doi: 10.1177/106002809703101022. [DOI] [PubMed] [Google Scholar]
- 28.Cheson BD, Horning SJ, Coiffier B, Shipp MA, Fisher RI, Connors JM, Lister TA, Vose J, Grillo-López A, Hagenbeek A, Cabanillas F, Klippensten D, Hiddemann W, Castellino R, Harris NL, Armitage JO, Carter W, Hoppe R, Canellos GP. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 29.Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: the npde add-on package for R. Comput Methods Programs Biomed. 2008;90:154–66. doi: 10.1016/j.cmpb.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Faltaos DW, Hulot JS, Urien S, Morel V, Kaloshi G, Fernandez C, Xuan H, Leblond V, Lechat P. Population pharmacokinetic study of methotrexate in patients with lymphoid malignancy. Cancer Chemother Pharmacol. 2006;58:626–33. doi: 10.1007/s00280-006-0202-0. [DOI] [PubMed] [Google Scholar]
- 31.Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558–69. doi: 10.1208/s12248-009-9133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karrison TG, Maitland ML, Stadler WM, Ratain MJ. Design of phase II cancer trials using a continuous endpoint of change in tumor size: application to a study of sorafenib and erlotinib in non small-cell lung cancer. J Natl Cancer Inst. 2007;99:1455–61. doi: 10.1093/jnci/djm158. [DOI] [PubMed] [Google Scholar]
- 33.Poortmans PM, Kluin-Nelemans HC, Haaxma-Reiche H, Van't Veer M, Hansen M, Soubeyran P, Taphoorn M, Thomas J, Van den Bent M, Fickers M, Van Imhoff G, Rozewicz C, Teodorovic I, van Glabbeke M. High-dose methotrexate-based chemotherapy followed by consolidating radiotherapy in non-AIDS-related primary central nervous system lymphoma: European Organization for Research and Treatment of Cancer Lymphoma Group Phase II Trial 20962. J Clin Oncol. 2003;21:4483–8. doi: 10.1200/JCO.2003.03.108. [DOI] [PubMed] [Google Scholar]