

Abstract
The chemotaxis of differentiated HL60 cells stably expressing CXCR2 was examined in a microfluidic gradient device where the steepness of the CXCL8 chemokine gradient was varied from 2 pg/ml/μm (0–1 ng/ml over a width of 500 μm) to 50 pg/ml/μm (0–25 ng/ml over 500 μm). The differentiated HL60 cells stably expressing CXCR2 exhibited little chemotaxis in response to a 0–1 ng/ml gradient, but displayed an increasing chemotactic response as the gradient steepness increased from 0 to 5, 0 to 10, and 0 to 25 ng/ml, demonstrating that steepness of gradient is a major determinant of the relative ability of cells to persistently migrate up a chemotactic gradient. When HL60 cells expressed CXCR2 mutated in the C terminus LLKIL motif (IL to AA), ligand-induced internalization of receptors was reduced 50%, whereas cell migration along the gradient of CXCL8 was completely lost. Although both mutant and wild-type receptors could mediate Akt and Erk activation in response to CXCL8, the level of activation of these two kinases was much lower in the cell line expressing the mutant receptors. These data imply that the IL amino acid residues in the LLKIL motif are very important for activation of the signal transduction cascade, which is necessary for cells to sense the chemokine gradient and respond with chemotaxis. Moreover, because mutation of the IL residues in the LLKIL motif resulted in only 50% reduction in receptor internalization, and a 50% reduction in Akt and Erk phosphorylation, but a complete loss of chemotactic response, the data imply that IL amino acid residues in the LLKIL motif are key either for amplification or oscillation of crucial signaling events or for establishment of a threshold for signals required for chemotaxis.
Chemotaxis is a process by which cells move directionally up a gradient of chemokine or chemoattractant. Chemotaxis plays an important role in the immune response through recruitment of leukocytes to the inflammatory sites (1, 2), in embryonic development where stem cells move directionally to the site of organogenesis (3), and in cancer metastasis (4–7), whereby tumor cells preferentially chemotax to specific target organs.
The phosphorylation and internalization of chemokine receptors are very important for modulation of receptor function, although the role of chemokine receptor internalization for chemotaxis remains controversial. Some studies revealed that receptor internalization was not required for certain chemoattractant receptors to mediate chemotaxis (8–12), whereas others reported that inhibition of receptor internalization also inhibited chemotaxis (13–17). We previously reported that the CXCR2 chemokine receptor mutated in the LLKIL motif, which binds adaptor protein-2 (AP-2)3 and heat shock 70 interacting protein (HIP), displayed compromised ligand-triggered receptor internalization and mediated impaired chemotaxis in HEK293 cells (18, 19). In contrast, Richardson et al. (12) demonstrated that, in rat basophilic leukemia (RBL-2H3) cells, a C-terminal-truncated CXCR2 did not undergo ligand-triggered internalization but still mediated robust chemotaxis in response to one of its ligands, CXCL8. However, this same C-terminal-truncated receptor underwent internalization equivalent to the wild-type CXCR2 in HEK293 cells (18). The discrepancy of the same receptor displaying different behavior in different cell lines could be due to different cellular levels of the components used for receptor internalization. For example, it has been reported that the levels of the β-arrestin adaptor proteins are high in leukocytes but very low in HEK293 cells (20). AP-2 and β-arrestin are two adaptor proteins that play very important roles in G-protein-coupled receptor internalization. AP-2 is a complex that can bind to the leucine-rich motif of the receptor through its μ-subunit, and its β-subunit binds to clathrin to form clathrin-coated vesicles. Additionally, AP-2 can be recruited to the receptor as a result of recruitment of AP-2 to β-arrestin, which binds the phosphorylated C-terminal domain of CXCR2.
Because of the cell type differences in expression of receptor regulatory proteins, it is helpful to study CXCR2 internalization in relation to chemotaxis in cells such as neutrophils that naturally express the receptor. However, due to the difficulties of maintaining native neutrophils and the technical challenge of transfecting or transducing expression vectors for mutant receptors into neutrophils, we chose to study HL60 cells. HL60 cells are human promyelocytic leukemia cells (21) that have been extensively studied because they can be manipulated to differentiate into monocytes or neutrophils (22–25). In this report two HL60 cell lines stably expressing either wild-type CXCR2 or the LLKIL motif mutant form of CXCR2 (IL/AA) were constructed from the parental HL60 cell line that naturally expresses very low levels of CXCR2, even after Me2SO-induced differentiation along the granulocytic linage. Chemotaxis assays were performed using a microfabricated device, the microfluidic gradient device. The microfluidic gradient device has several advantages over traditional methods of generating a chemokine gradient (26). With this device, stable chemokine gradients can be delivered and manipulated in a variety of ways, thereby providing a very powerful tool to study different aspects of chemotaxis, such as the effects of the chemokine concentration on chemotaxis (27). Moreover, cells can be visualized microscopically during the migration process. In this report, the response of these stable HL60 cell lines to a CXCL8 gradient delivered in the microfluidic gradient device was tested to determine the role of the IL residues of the LLKIL motif and ligand-triggered receptor internalization in mediating intracellular signaling and chemotaxis.
MATERIALS AND METHODS
Cell Lines and Reagents
The HL60 cell line was kindly provided by Henry Bourne, University of California at San Francisco. The retroviral vector, pMSCV, was purchased from Clontech (Mountain View, CA). RPMI1640 was purchased from Invitrogen. Antibodies used in FACS and Western blot were purchased from following companies: phycoerythrin (PE)-conjugated anti-CXCR2 antibody (PE-CXCR2, CDw128b), BD Pharmingen (San Diego, CA); rabbit polyclonal antibody to CXCR2 previously developed and characterized in our laboratory (28), rabbit polyclonal antibodies to Erk (Lys-23), Akt1/2 (His-136), and mouse monoclonal antibody to CXCR2 (E2) and phosphorylated Erk (E4), Santa Cruz Biotechnology (Santa Cruz, CA); polyclonal rabbit antibody to phosphorylated Akt (Thr-308), Cell Signaling Technology (Beverly, MA); horseradish peroxidase-conjugated secondary antibodies to mouse or rabbit, Chemicon International (Temecula, CA); IR-conjugated secondary antibodies, IRdye 800, Rockland Immunochemicals, Inc. (Gilbertsville, PA); and Alexa 680, Molecular Probes, Inc. (Eugene, OR). ECL reagents were purchased from Amersham Biosciences, and x-ray films were from Eastman Kodak (Rochester, NY). CXCL8 was purchased from PeproTech (Rocky Hill, NJ). Radiolabeled CXCL8 (125I, 2200 Ci/mmol) was purchased from PerkinElmer Life Science. Human fibronectin was purchased from BD Biosciences, and proteinase inhibitor cocktails were from Sigma-Aldrich.
HL60 Cell Culture and Differentiation
HL60 cells were induced to differentiate along the neutrophilic lineage as previously described (29). Briefly, cells were cultured in RPMI 1640 medium supplemented with 20 mM HEPES (pH 7.4), 10% fetal bovine serum (Atlanta Biologicals, Atlanta, GA), L-glutamine, 100 units/ml PennStrep (Mediatech, Inc., Herndon, VA). Cells were subcultured every 3–4 days to a cell density of 106 cells/ml. To differentiate HL60 cells along the granulocytic lineage, cells were inoculated at a density of 2 × 105 cells/ml in antibiotic-free medium containing 1.3% Me2SO (endotoxin-free, Sigma) and cultured for 6–7 days.
Transduction of HL60 Cells with a Retroviral Construct
The human CXCR2 DNA sequence was amplified by PCR with the following pair of primers: 5′ primer, 5′-GCGAATTCATGGAAGATTTTAACATGG-3′, and 3′ primer, 5′-TACTCGAGTTAGAGAGTAGTGGAAGTG-3′. IL/AA mutant CXCR2 was amplified from a mutated CXCR2 template that was previously generated by point mutation of IL to AA in the LLKIL motif (18). The amplified DNA fragments were cloned into the retroviral vector pMSCV. The retroviral construct containing CXCR2 was cotransfected with the DNA construct expressing the viral envelop protein into a packaging cell line, GP2–293 (BD Biosciences-Clontech). The retroviral particles were collected from the cultured medium of packaging cells and used to infect HL60 cells. The cell lines stably expressing CXCR2 were selected for resistance to neomycin by inclusion of 1 mg/ml G418 (Mediatech, Inc.) in the culture medium.
Flow Cytometry Analysis
Differentiated HL60 cells (5 × 105cells) were washed with 0.1% BSA in PBS and resuspended in 100 μl of BSA/PBS. PE-conjugated anti-CXCR2 antibody (1:50 dilution, 0.2 mg/ml) was added, and cells were allowed to sit on ice for 1 h. Cells were subsequently washed three times with ice-cold 0.1% BSA/PBS and resuspended in 500 μl of 0.1% BSA/PBS for flow cytometry (FACScan, BD Biosciences). For the receptor internalization assay, cells were pre-treated with ligand, CXCL8 (100 ng/ml), for different time periods before staining with PE-conjugated anti-CXCR2 antibody. For cell sorting, cells were treated the same as for FACS, except that 1% fetal bovine serum in PBS was used instead of 0.1% BSA/PBS to enhance cell survival.
Radioligand Binding Assay
Differentiated HL60 cells (5 × 105 cells) were washed with binding buffer (1% BSA in serum-free RPMI 1640 medium) and resuspended in 200 μl of binding buffer containing 0.1 nM 125I-CXCL8 and different concentrations of unlabeled CXCL8 (from 0 to 100 nM). After sitting on ice for 4 h, cells were washed with binding buffer three times and lysed in 200 μl of lysis buffer (1% SDS and 0.1 N NaOH). Radioactivity in the cell lysate was determined by a γ-counter (Cobra II, Global Medical Instrumentation, Inc., Ramsey, MN). For the receptor internalization assay, 5 × 105 cells were washed with binding buffer and resuspended in 100 μl of binding buffer containing total 25 ng/ml CXCL8 (1.2 ng/ml 125ICXCL8 and 23.8 ng/ml unlabeled CXCL8). After a 3-h incubation on ice with ligand, 1 ml of ice-cold binding buffer was added to the incubation tubes to terminate ligand binding, and the cells were washed twice with fresh binding buffer. The receptor internalization was initiated by incubating the cells at 37 °C for different periods of time. The non-internalized ligand bound to cells was removed from cell membrane by 1 ml of acid wash (0.2 M acidic acid and 0.5 M NaCl) and followed by a wash with binding buffer. The cells were lysed in 200 μl of lysis buffer, and the radioactivity was determined on a γ-counter. The data were analyzed by the computer software Prism (GraphPad Software, Inc., San Diego, CA).
Immunostaining
Differentiated HL60 cells were allowed to adhere to fibronectin (100 μg/ml) pre-coated coverslips for 10 min at 37° C. After being stimulated with CXCL8 (25 ng/ml) for different periods of time at 37° C, cells were fixed in 4% paraformaldehyde for 10 min at room temperature. Fixed cells were washed three times with PBS, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked with 1% BSA in PBS for 30 min. Polyclonal rabbit antibody against human CXCR2 was applied to the cells for 30 min, washed with PBS, and Cy3-conjugated secondary antibody anti-rabbit IgG was applied. Confocal microscopy was carried out with an LSM510 Zeiss inverted microscope (Carl Zeiss Microimage, Germany).
Chemotaxis Assay in Microfluidic Gradient Device
Microfluidic gradient devices were made at the Vanderbilt Institute for Integrative Biosystems Research and Education as described previously (30). Devices were pre-coated with fibronectin (100 μg/ml) in modified Hanks' buffer (mHBSS) (20 mM HEPES, pH7.2, 150 mM NaCl, 4 mM KCl, 1.2 mM MgCl2 and 10 mg/ml glucose) (29) for 1 h at room temperature and washed briefly with mHBSS before use. Differentiated HL60 cells were washed and resuspended in mHBSS at 3 × 106 cells/ml, and injected into the pre-coated device. Cells were seeded in the device for 5 min at 37° C and 5% CO2 and placed in a pre-warmed temperature-controlled chamber of the inverted microscope (Axiovert 200M, Carl Zeiss Microimage). The two input tubes of the device were connected to syringes filled with CXCL8 in mHBSS containing 1% human serum albumin and buffer alone without CXCL8, respectively. The injection of the solutions from syringes into the device was driven by a syringe pump (Harvard PHD2000, Harvard Apparatus, Holliston, MA), first at 50 μl/min to quickly fill the tubing and at 1 μl/min to maintain a CXCL8 gradient in the main channel of the device. The live cell microscopic images were taken every 20 s for a period of 30 min by a charge-coupled device camera (Hamamatsu, Japan) controlled by the computer software Openlab (Improvision, Lexington, MA). The data were analyzed by using the Openlab and Metamorph (Molecular Devices, Downingtown, PA) to trace the cell movements.
Chemotaxis Assay in a Modified Boyden Chamber
A Boyden chamber (Neuroprobe Inc., Gaithersburg, MD) was assembled as the manufacturer suggested, and a 5-μm pore filter was used for differentiated HL60 cells. 100,000 cells were loaded into each well in the top chamber, and cells were allowed to migrate through the filter into the bottom chamber containing varying concentrations of CXCL8 for 3 h at 37°C and 5% CO2. Cells that migrated into the bottom chamber were collected by centrifugation and counted with hemocytometer.
Cell Adhesion Assay
A 96-well plate was precoated with mHBSS buffer, 1% BSA in mHBSS buffer or 100 μg/ml fibronectin for 2 h at room temperature, and washed with mHBSS buffer before loading the cells. Differentiated HL60 cells were washed and resuspended in mHBSS buffer at a density of 2 × 105 cells/ml. A 100-μl aliquot of these cells was loaded for each well pre-coated as above. Cells were allowed to adhere for 20 min at 37° C and 5% CO2, and 200 μl of paraformaldehyde (3.8%) was added directly to the cells in each well to fix adhered cells followed by mHBSS wash to remove non-adhered cells. The numbers of cells in each well were counted under the microscope (10× objective, Axiovert 200M, Carl Zeiss Micro-image) for ten randomly selected fields.
Western Blot
Differentiated HL60 cells were washed, resuspended in mHBSS, and treated with 100 ng/ml CXCL8 or buffer alone. After CXCL8 stimulation for different periods of time, cells were lysed with an equal volume of 2× RIPA buffer (100 mM Tris-Cl, pH7.5, 300 mM NaCl, 2% Nonidet P-40, 1% sodium deoxycholate, and 0.2% SDS supplemented with proteinase inhibitor mixture). The lysate was sonicated for 10 s at output 20 (Sonifier 250, Branson Ultrasonics Corp., Danbury, CT) and centrifuged at 16,000 × g for 5 min. The supernatant was mixed with 5× SDS sample buffer and heated at 95 °C for 10 min. 20-μg aliquots of protein were loaded to each lane of a 10% SDS-PAGE, and samples were electrophoresed at 100 V for 2 h. Resolved proteins were electrotransferred onto nitrocellulose membrane at 100 V for 1 h. Membranes were blocked in 5% nonfat Carnation skim milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, and 0.05% Tween 20) for 1 h at room temperature and then immunoblotted with primary and secondary antibodies. For horseradish peroxidase-conjugated secondary antibody, substrates from ECL or ECL advance were applied on the membrane for 1 or 5 min, respectively, and exposed to x-ray film. In some cases, infrared-conjugated secondary antibodies were used, and the membrane was scanned in Odyssey scanner (LICOR Biosciences, Lincoln, NE). Quantitation of signal intensities was performed in an Odyssey scanner. For each set of samples in a time course, the signal intensity from the untreated control (time 0) were set to be 1, and all the other samples in the time course were normalized to the value of each control.
GTPase Activity Assay
Crude cell membrane preparations were obtained from differentiated HL60 cells by swelling in hypotonic buffer (20 mM HEPES, pH 7.4, 2 mM MgCl2,1mM EDTA, and 10 mM NaF supplemented with proteinase inhibitor mixture) for 10 min on ice, followed by homogenization in a Dounce homogenizer. Nuclei were removed by low speed centrifugation at 400 × g for 10 min, and the plasma membranes were precipitated by ultracentrifugation at 40,000 rpm (SW50.1 rotor, 150,000 × g) for 30 min. The membrane pellet was resuspended in hypotonic buffer at a protein concentration of 1 μg/μl. GTPase activity assays were carried out according to the protocol described previously by Richardson et al. (31). Briefly, 20 μg of membrane proteins (20 μl) was mixed with 25 μl of GTPase activity assay buffer (25 mM HEPES, pH 7.4, 125 mM NaCl, 20 mM MgCl2, 0.5 mM EGTA, 1 mM dithiothreitol, 0.1 μM GTP, 0.5 mM ATP, 0.1% BSA, 0.4 mg/ml creatine kinase, 5 mM creatine phosphate, and 50 nM [γ-32P]GTP) and 5 μl of CXCL8 (20 ng/μl). Reaction tubes were incubated at 30 °C for different periods of time, and the reaction was terminated by adding 750 μl of ice-cold 5% charcoal solution in PBS. After 15 min on ice, the charcoal was precipitated by centrifugation at 16,000 × g at 4 °C for 3 min. 400 μl of supernatant was removed, mixed with 2 ml of scintillation mixture, and the radioactivity incorporation was determined by scintillation counting (LS6500, Beckman Coulter, Inc., Fullerton, CA).
RESULTS
Characterization of CXCR2-expressing HL60 Cells
Differentiated HL60 cells induced to differentiate with Me2SO normally gain neutrophil-like characteristics after 6–7 days of treatment. The cells change their morphology from round to somewhat stellate, become easily adherent to the fibronectin or collagen matrix, and develop the capability to chemotax toward the chemoattractant fMLP, indicating the expression of the receptor for this chemoattractant. The endogenous expression of CXCR2 in differentiated HL60 cells studied here was tested by flow cytometry and by ligand binding assay with radiolabeled CXCL8 (125I-CXCL8), and the results revealed little, if any, expression of CXCR1 or CXCR2 on these cells (Fig. 1A). There was no chemotactic response to CXCL8 as tested in a microfluidic chamber, although the cells did exhibit chemotaxis in response to fMLP (supplemental videos S1 and S2). Because the expression of CXCR2 after Me2SO-induced differentiation of HL60 cells revealed virtually no CXCR1 or CXCR2 expression, we transduced these cells with a retroviral expression vector for the wild-type (WT) or the IL/AA mutant CXCR2 to study the function of this receptor in neutrophil-like cells. Stable HL60 cell lines expressing either WT or the IL/AA mutant CXCR2 were constructed by retroviral delivery and selection under G418 (1 mg/ml). The expression of CXCR2 was confirmed by FACS with PE-conjugated anti-CXCR2 antibody, and CXCR2-expressing cells were sorted to obtain a population of cells with a similar expression level of CXCR2 in the two cell lines (Fig. 1A). The ligand binding affinities (Kd) for WT and IL/AA mutant receptor in stable HL60 cell lines were calculated to be 1.61 ± 0.29 nM and 1.51 ± 0.63 nM, respectively, indicating that the mutation of the receptor did not change the ligand affinity (Fig. 1B). The Kd value for CXCR2 in differentiated HL60 cells was very close to that in human neutrophils (~1 nM).
FIGURE 1. Construction of stable HL60 cell lines expressing WT CXCR2 or internalization-defective CXCR2 (IL/AA).

FACS analysis (A, left panel) used to test the membrane expression of receptors with PE-conjugated antibody to CXCR2 shows the equivalent expression of receptors in differentiated HL60 cells from two polyclonal cell lines. Ligand binding assay with 125I-CXCL8 (specific activity, 2200 Ci/mmol) shows very little CXCR1 or CXCR2 expression in differentiated HL60 parental cells (A, right panel). The binding affinity was measured by competition assay (B) with radiolabeled ligand (0.1 nM 125I-CXCL8) (specific activity, 2200 Ci/mmol) and analyzed by the Prism program. The Kd value for each cell line was calculated from the results of three individual experiments.
Differentiated HL60 Cells Expressing IL/AA Mutant CXCR2 Exhibit Compromised Receptor Internalization but Normal Adhesivity to Fibronectin
We previously reported that the LLKIL motif of CXCR2 is required for optimal ligand-triggered receptor internalization in HEK293 cells (18). This motif binds to AP-2 as well as heat shock-70 interacting protein (HIP). The confocal images of immunostaining on the differentiated HL60 cells expressing either WT CXCR2 or IL/AA mutant CXCR2 revealed that, in WT CXCR2-expressing cells, the internalization of receptor could be detected as early as 1–2 min upon 25 ng/ml CXCL8 stimulation and reached a maximum at 30 min after ligand stimulation when tested over a 90-min incubation period. However, in cells expressing the IL/AA mutant of CXCR2, the internalization of receptor was much delayed and never reached the same level of internalization as in cells expressing WT CXCR2 (Fig. 2A).
FIGURE 2. Internalization of CXCR2 in cells expressing wild-type or mutant CXCR2.

A, differentiated HL60 cells expressing either WT CXCR2 (upper panel) or IL to AA mutant CXCR2 (lower panel) were allowed to adhere fibronectin-coated (100 mg/ml) coverslips, and the CXCR2 receptors were induced to internalize by stimulation with CXCL8 (25 ng/ml) for different periods of time at 37 °C. Immunostaining was performed to detect CXCR2 expression in the cells after fixation. Representative confocal images are shown from two experiments with similar results. The numbers shown in each image indicated the time in minutes for which cells were stimulated with ligand before being fixed, and the scale bar is 10 μm. B, radiolabeled ligand (0.1 nM 125I-CXCL8, specific activity, 2200 Ci/mmol) was allowed to bind to the CXCR2 receptors on differentiated HL-60 cells stably expressing either wild-type CXCR2 (open bars) or IL to AA mutant CXCR2 (solid bars) on ice as described under “Materials and Methods.” After non-bound 125I-CXCL8 was removed by washing, ligand-stimulated internalization of receptor was allowed to proceed by incubating the cells at 37 °C for the times indicated. Internalization was terminated by transferring cells back to ice, and non-internalized ligand was removed by acid wash. The radioactivity was counted on a Gamma counter, and the percent internalization of receptor was calculated as a ratio of internalized radiolabeled ligand divided by the total binding without acid wash. The graph was plotted from the means of three individual experiments performed in duplicate.
Receptor internalization was also tested by a ligand binding assay containing radiolabeled ligand (125I-CXCL8) to allow more accurate quantitation. In cells expressing WT CXCR2, the ligand-triggered receptor internalization occurred quickly, reaching 70% in 10 min and almost 90% in 30 min at 37° C. In contrast, for the HL60 cells expressing the IL/AA mutant receptor, receptor internalization was greatly inhibited. Only ~35% of the receptor was internalized after 10-min incubation, and 45% internalized after 30-min incubation at 37° C (Fig. 2B). The analysis of variance indicated that the time course for receptor internalization was significantly different (p = 0.005) between the two cell lines.
Chemotaxis of Differentiated HL60 Cells Stably Expressing CXCR2 under Different Gradient Steepness
As little as 1% difference in the chemokine concentration across a cell is believed to be sufficient to induce neutrophil chemotaxis (32). We used a microfluidic gradient device to test the hypothesis that chemotactic response is directly proportional to the steepness of the chemokine gradient. With this device, well controlled chemokine gradients with varying steepness can be delivered into the chamber where cells are located with a constant flow driven by a syringe pump. Here, chemotaxis assays were performed in the microfluidic gradient devices using differentiated HL60 cells stably expressing WT CXCR2. The movements of the cells in the microfluidic gradient device are driven by two forces, shear force from flow and chemotactic force from the chemokine gradient. The trajectories of cell movements are dependent on the balance of these two forces on the cells. We have previously tested the affect of shear force on cell movement with different flow rates and discussed the possibility of estimating the chemotactic force generated by external chemokine gradients by observing the trajectory of cell movements when detoured by shear force (30). We now show that cell movement was also influenced by varying the steepness of the chemokine gradient. When the steepness of the CXCL8 gradient is low (slope = 2 pg/ml/μm), only a small fraction of the cells (4 out of 70) exhibited chemotaxis. When the cells were presented with a CXCL8 gradient of sharp steepness (slope = 50 pg/ml/μm), over 90% of the cells plated (58 out of 64) demonstrated robust chemotaxis. Moreover, in the range of gradient steepness tested (from 2 to 50 pg/ml/μm in the gradient slope) with a flow rate of 2 μl/min (866 μm/s), the steeper the gradient, the more the angle of the cell's trajectory of movement turned toward the direction of chemokine gradient, indicating increased ability of migrating cells to persistently stay on course (Table 1, Fig. 3, and supplemental videos S3–S6).
TABLE 1. Chemotaxis of HL-60 cells stably expressing CXCR2 in CXCL8 gradients of varying steepness.
Differentiated HL-60 cells were seeded in a fibronectin precoated microfluidic device. Chemotaxis was assayed under conditions where the steepness of CXCL8 gradient was varied, and time-lapse videos were taken every 30 s for 30 min. The total number of cells in the field and the number of cells showing chemotaxis were counted manually. Cells consistently migrating up the CXCL8 gradient were considered as chemotaxing cells. (See supplemental videos S1–S4 for details).
| Gradient | Steepness of gradient (slope) | Total cells observed | Number of cells in field that chemotaxed | Percentage |
|---|---|---|---|---|
| ng/ml | pg/ml/μm | % | ||
| 0–1 | 2 | 70 | 4 | 5.7 |
| 0–5 | 10 | 64 | 10 | 15.6 |
| 0–10 | 20 | 39 | 18 | 46.2 |
| 0–25 | 50 | 64 | 58 | 90.6 |
FIGURE 3. Chemotaxis of differentiated HL-60 cells stably expressing CXCR2 under different gradient steepness.

Varying amounts of CXCL8 in modified Hanks' buffer or Hanks' buffer alone were infused through separate tubing portals into the microfluidic gradient device at a flow rate of 1 μl/min. Depending upon the concentration of the input CXCL8, CXCL8 gradients of varied steepness were generated in the main channel of the microfluidic gradient device. The real-time video of cell movement was recorded, and the data were analyzed by the Metamorph program to trace the movement of 8–12 cells for each experiment. A, trajectories of the cells under each steepness of gradient; and B, mean trajectory of cell movement for each experiment. The solid line represents the best linear fit for each mean trajectory. The cell movement was driven by two forces, flow and chemotactic force. The trajectory of cell movement represents the net result of these two forces. With the increase of the steepness of chemokine gradient, the angle of the trajectories was more in line with the direction of the gradient. (See supplemental videos S3–S6.)
Loss of Chemotaxis in HL60 Cells Stably Expressing IL/AA Mutant CXCR2
To test the hypothesis that reduction in CXCR2 internalization would produce an equivalent diminution in CXCR2-mediated chemotaxis, differentiated HL60 cells stably expressing either WT CXCR2 or the IL/AA mutant CXCR2 were tested for chemotaxis in response to a gradient of CXCL8 from 0 to 50 ng/ml across a 500-μm wide channel. ~80–90% of the cells expressing WT CXCR2 were found to have consistent directional movement toward the chemokine gradient, whereas the cells expressing the IL/AA mutant receptor did not undergo chemotaxis (Fig. 4A and supplemental videos S7 and S8). Differentiated HL60 cells stably expressing WT or IL/AA mutant CXCR2 were also observed with a traditional Boyden chamber, and similar results were obtained (Fig. 4B). Cell adhesion plays an important role in cell migration. We performed cell adhesion assays for differentiated HL60 cells on either fibronectin (100 μg/ml)-coated surface, 1% BSA-coated surface, or surface with no coating. We observed no difference in adhesivity between the cells expressing WT CXCR2 and IL/AA mutant CXCR2 (Fig. 4C). Therefore, loss of adhesivity does not account for the defectiveness of chemotaxis in cells expressing IL/AA mutant CXCR2. In addition, the adhesivity of differentiated HL60 cells to fibronectin (100 μg/ml) was much lower than the adhesivity to a non-coated surface (possibly due to electrostatic adhesion of cells to plastic surface), and the differentiated HL60 cells were only able to move on the fibronectin-coated surface. The cells also adhered to collagen IV (20 –100 μg/ml), but the cells did not migrate on collagen IV.
FIGURE 4. Chemotaxis of HL-60 cells stably expressing WT or mutant CXCR2 (IL/AA).

A, differentiation of HL60 cells stably expressing either wild-type (upper panel) or mutant CXCR2 (lower panel) was induced by 1.3% Me2SO for 7 days. Cells were allowed to adhere to a fibronectin-coated (100 mg/ml) microfluidic gradient device. Chemotaxis was assayed in a 0–50 ng/ml CXCL8 gradient, and time-lapse videos were taken every 20 s for 30 min. Partial images (five cells for each cell line) are shown in the figure, and the numbers in each image indicate the time lapse in minutes at which the image was taken. (See supplemental videos S7 and S8 for detail). B, chemotaxis was performed with a modified Boyden chamber with differentiated HL60 cells stably expressing either WT CXCR2 or IL to AA mutant CXCR2. C, adhesion assays were performed with differentiated HL60 cells on either non-coating, 1% BSA, or fibronectin (100 μg/ml). The number of cells in a microscope field (200× magnification) were counted for 10 randomly selected fields for each treatment. The error bars are ± S.E.
Effects of CXCL8 on Cell Motility in Cells Expressing WT or Mutant CXCR2
Ligand-induced effects on cell motility in HL60 cells expressing WT or the IL/AA mutant CXCR2 were investigated to determine whether the intact LLKIL motif is needed for CXCR2-mediated chemokinesis. Chemokinesis is distinguished from chemotaxis by the following parameters: 1) chemokinesis can occur in response to an evenly distributed chemical, whereas in contrast, chemotaxis occurs only in response to a gradient of chemotactic substance; 2) chemokinesis involves random movement changes, whereas chemotaxis is a process in which cells demonstrate consistent directional movement. The checkerboard assay is often used to test the chemokinesis of the cells. In this report, we used the microfluidic gradient device to measure the chemokinesis. The assay was configured to examine cell motility in the absence of chemokine for the first 10 min. With well controlled constant flow regulation by the syringe pump of the microfluidic device, CXCL8 (25 ng/ml) applied through both inputs was delivered into the main channel, and cell motility changes were recorded in response to this evenly distributed CXCL8. We measured motility changes in response to CXCL8 by tracking the cell movement with the computer software Metamorph. The advantage of using the microfluidic gradient device over checkerboard assay in assessing chemokinesis is that the exact rate changes of cell movement can be observed with the same group of cells immediately before and after the uniform ligand stimulation (25 ng/ml). A major benefit of this procedure is that we can measure how quickly the cells respond to the chemokine stimulation. When cells expressing WT CXCR2 or IL/AA mutant CXCR2 were tested for chemokinesis under identical conditions, we found that cell motility was significantly increased (p = 0.002, Student's t test) in response to uniform delivery of CXCL8 to cells expressing WT receptor. For cells expressing the IL/AA mutant CXCR2 receptor, the motility was slightly, but not significantly increased (p = 0.19, Student's t test) after uniform ligand delivery. Failure to see a significant response was probably due to the higher basal cell motility (cell movement without CXCL8) in HL60 cells expressing the IL/AA CXCR2. There was also no significant difference in CXCL8-induced chemokinesis between two cell lines (p = 0.45, Student's t test) (Fig. 5 and supplemental videos S9 and S10). These data suggest that the defective chemotaxis in response to the CXCL8 gradient in cells expressing mutant CXCR2 was not due to defective cell motility but is probably due to impaired directional response to the gradient.
FIGURE 5. Cell motility changes after stimulation with CXCL8 (25 ng/ml) (chemokinesis).
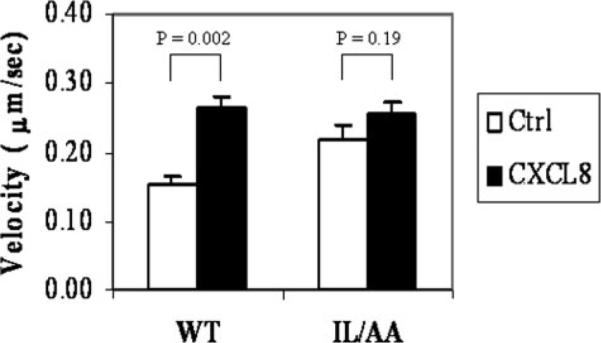
Chemokinesis was tested for these two cell lines with the microfluidic gradient device controlled to run with Hanks' buffer only for ~10 min, and then 25 ng/ml CXCL8 was allowed to flow into the channel. Time-lapse videos were recorded every 20 s to monitor the cell movement. Trajectories of 10 individual cells were traced for each cell line, and the mean velocities were calculated from these traces. In cells expressing wild-type (WT) CXCR2, the basal velocity was lower than the mutant receptor-expressing cells (open bars). However, upon ligand treatment, the cell motility was accelerated significantly (chemokinesis) in wild-type receptor-expressing cells, reaching a velocity of 0.25 μm/s, corresponding to the same velocity of the mutant receptor-expressing cells (solid bars). (See supplemental videos S9 and S10 for details.)
Effects of LLKIL Mutation on CXCL8-induced MAPK and PI3K Activation
The MAPK and PI3K pathways are two major signal transduction pathways activated by CXCR2 in response to its ligand. PI3K plays an important role in cell polarization and in sensation of the direction of chemoattractant gradient during chemotaxis (33–36). Although it is still controversial whether the MAPK pathway is required for chemotaxis (37–39), Erk activation is a very important landmark for the activation of a G-protein-coupled receptor (40, 41). We chose these two signal transduction pathways to test the activation of downstream signals mediated by WT or IL/AA mutant CXCR2 by testing the ligand-induced phosphorylation of Erk and Akt for MAPK and PI3K activities, respectively. Ligand activation of Erk and Akt in differentiated HL60 cells expressing WT or mutant CXCR2 receptors were compared over a time course of 5 min. Although both cell lines showed activation of Erk and Akt in response to 100 ng/ml CXCL8 stimulation, the amplitude of the activation in cells expressing IL/AA mutant CXCR2 was much lower than that in cells expressing WT CXCR2 (Fig. 6). In Fig. 6A, the raw data from these Western blots showed slightly more basal Erk and Akt phosphorylation, but, when normalized for slight differences in receptor expression level, there was no significant difference in basal Akt or Erk phosphorylation between mutant and WT CXCR2-expressing cells. Statistical tests showed a significant difference between two cell lines in activation of Akt (p = 0.0007) and Erk (p = 0.004, analysis of variance). Two possible reasons for low activation of Erk and Akt in cells expressing IL/AA mutant CXCR2 are reduced ligand binding or weaker G-protein coupling to the mutant receptors. We show here in Fig. 1B that there is no difference in ligand binding affinity between WT and IL/AA mutant CXCR2. The GTPase activity assay also showed little difference between WT and IL/AA mutant CXCR2, indicating that there is no defect in G-protein coupling/uncoupling in the IL/AA mutant receptor-expressing cells (Fig. 7). These data suggest that the IL residues of the LLKIL motif in the C-terminal domain of CXCR2 are required for appropriate amplification of intracellular signals in response to ligand.
FIGURE 6. Impaired activation of intracellular signal molecules in response to ligand by cells expressing IL/AA mutant CXCR2.

Differentiated HL-60 cells expressing either wild-type or IL/AA mutant CXCR2 were stimulated with 100 ng/ml CXCL8 at room temperature for the times indicated and mixed with 2× RIPA buffer to lyse the cells. After electrophoresis as described under “Materials and Methods,” Western blot was performed with antibodies for p-Akt (Thr-308) and pERK, and the membrane was reblotted with total Akt and ERK antibodies after stripping. A graph illustrating results from densitometric scans of Western blots from three independent experiments is shown in panel B with Akt (left) and Erk (right).
FIGURE 7. GTPase activity assay for WT and IL/AA mutant CXCR2.
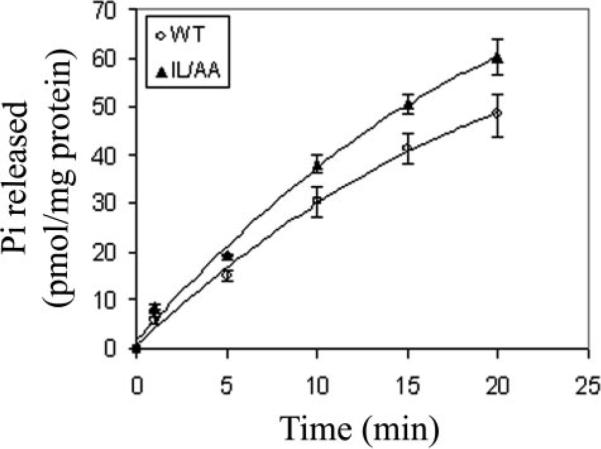
Crude cell membrane preparations were isolated from differentiated HL-60 cells expressing either wild-type or IL/AA mutant CXCR2. The GTPase activities were tested after stimulation with CXCL8 (2 μg/ml) by measuring 32Pi release in the reactions at 30 °C as described under “Materials and Methods” for the times indicated. The error bar is mean ± S.E. from three independent experiments.
DISCUSSION
Use of model cell lines like HEK293 to study the function of CXCR2 has contributed greatly to the understanding of some aspects of CXCR2 functions, such as signal transduction and receptor internalization. The ease with which this model cell line can be transfected makes it very useful. However, because the expression of signaling and trafficking components vary considerably between different cell lines, the results and conclusions obtained from one cell line may not hold for other cell types. Moreover, the characteristic slow movement of HEK293 cells makes them less suitable for modeling chemotaxis of highly motile cells such as neutrophils. It is known that CXCR2 plays an important role in mediating neutrophil chemotaxis during inflammation. Therefore, for our studies described here the promyelocytic cell line HL60 was selected to study the functional role of CXCR2 and activation of intracellular signal molecules in chemotaxis, because HL60 cells can be induced to differentiate into neutrophil-like cells. Differentiated HL60 cells acquire typical characteristics of native neutrophils in the expression of membrane surface markers and the ability to perform chemotaxis and degranulation. The parental HL60 cell line studied in this report did not express significant levels of CXCR2 or CXCR1, and these parental cells did not respond to CXCL8 stimulation. Therefore, transduction of these cells with retroviral expression vectors for either WT CXCR2 or the IL/AA mutant form of CXCR2 led to the establishment of an appropriate in vitro model to investigate the functional requirement of the IL residues of the LLKIL motif in CXCR2.
Delivery of a stable and consistent chemokine gradient has been a real challenge for researchers studying chemotaxis. The microfluidic gradient device used in our studies provides a new approach to solve this problem. With this device, the differentiated HL60 cells stably expressing WT CXCR2 were tested for chemotaxis under conditions where the steepness of the CXCL8 gradient was varied. Although a few cells (5.7%) were observed to chemotax in a gradient where the steepness was 2 pg/ml/μm(~0.2 pM/μm), a majority of the cells exhibited chemotaxis when the gradient steepness was 50 pg/ml/μm(~5 pM/μm). We also observed that the efficiency of chemotaxis in differentiated HL60 cells stably expressing WT CXCR2 was lower when the gradient ranged from 25 to 50 ng/ml CXCL8 (data not shown) than when the gradient ranged from 0 to 25 ng/ml, although both these gradients have the same slope (~5 pM/μm). We observed the optimal chemotaxis when the mean CXCL8 concentration across the gradient was 1.25 nM. In another study where human neutrophils were assayed, the most efficient chemotaxis was induced in the microfluidic gradient device with the mean CXCL8 concentration of 3 nM across the gradient (27). Our studies with the microfluidic device show that both steepness of gradient and mean ligand concentration affect the ability of cells to persist in their migration up a gradient.
These results are consistent with those from the early studies using diffusion-based chemotaxis assay, such as Zigmond or Boyden chambers. Based on the reports from Zigmond (32) and others (42, 43), polymorphonuclear leukocytes can detect as little as a 1% difference in the concentration of the chemoattractant peptide N-formylmethionylmethionylmethione over the cell length (10 μm) to orient along the gradient (10–20 nM over a 1-mm bridge width in a Zigmond chamber). However, polymorphonuclear leukocytes oriented much more efficiently (>90%) when the difference in the concentration of the gradient of chemotactic peptide was 10% (gradient 1–10 nM over a 1-mm bridge width) (31).
In contrast to cells expressing WT CXCR2, which exhibited strong chemotaxis in response to the CXCL8 gradient (0–25 ng/ml), cells stably expressing IL/AA mutant CXCR2 only showed random movements in response to the same CXCL8 gradient. We previously reported that IL/AA and other LLKIL mutant forms of CXCR2 showed compromised ligand-triggered receptor internalization and chemotactic response to CXCL8 gradient in HEK293 cells (18). We now show that also in HL60 cells the IL/AA mutant receptor exhibits compromised ligand-triggered receptor internalization and cannot efficiently mediate chemotactic response to the CXCL8 gradient. Ligand binding assays and GTPase activity assays showed no difference in the ligand binding affinity and G-protein-coupling capability between HL60 expressing the WT and IL/AA mutant CXCR2. However, HL60 cells expressing IL/AA mutant receptor exhibited much lower activation in MAPK and PI3K pathways in response to ligand, compared with those from the cells expressing WT CXCR2. This blunted response is likely, because the IL/AA CXCR2-expressing cells were missing an amplification component of signaling due to loss of AP-2 adaptor association with receptor at the LLKIL sequence. These results are different from our previous findings in HEK293 cells, where the same level of Erk activation was observed in cells expressing IL/AA mutant receptor and cells expressing WT CXCR2 (19). This result may be due to differences in levels of adaptor protein between cell types.
It has been speculated previously that if the receptor does not undergo ligand-stimulated internalization, the receptor stays on membrane longer and is potentially activated to a greater extent or for a longer period of time by the ligand (11, 44). This increased length of ligand binding should generate stronger downstream signals. Studies with other desensitization-compromised receptors (non-phosphorylated receptor or intracellular C-tail truncated receptor) in RBL cells confirmed that the cells expressing these internalization-compromised receptors exhibited increased activation of downstream signals, such as calcium mobilization, GTPase activity, phosphatidylinositol hydrolysis and hexosaminidase release (44). Studies with the CC chemokine receptor CCR5 in RBL cells also showed that the internalization-compromised receptor mutants exhibited higher calcium mobilization and slightly higher Erk activation in response to ligand than WT CCR5-expressing cells (11). Moreover, although β-arrestins play a very important role in the receptor desensitization and internalization, they are not required for CCR5 receptor-meditated MAPK activation (11). Here we show that in contrast to studies with CCR5 (11) and studies with CXCR2 in RBL cells (44), mutation of the LLKIL motif of both CXCR2 only partially compromises CXCR2 internalization and the Akt/Erk activation in response to ligand but totally ablates CXCL8-mediated chemotaxis monitored by the microfluidic gradient device. These data suggest that, although there may be a direct relationship between ligand-induced receptor internalization and signal transduction, the magnitude of two events is not directly related to chemotactic responses.
It has been reported that when excess ligand induces full desensitization and internalization of membrane chemokine receptors, this generates a stop signal for cell migration as postulated by Rose et al. (45). However, we show here that ligand concentrations as low as 25 ng/ml induce receptor internalization, and, when this event is compromised only 50%, chemotaxis is totally ablated. Furthermore, the intracellular trafficking of internalized receptors engages several protein-protein interactions, such as the binding of β-arrestins, AP-2, HIP, and PP2A to CXCR2, and these binding events modify many signaling activities. For example, the binding of β-arrestins to the β2-adrenergic receptor increases Src and MAPK activation (46–48). More recently, arrestins were also found to play a role in suppressing CXCR2-mediated wound healing and in vivo neutrophils chemotaxis (49).
The question arises then, how does the LLKIL motif modulate chemotaxis? Is its effect on chemotaxis directly linked to the impaired receptor internalization and strength of signal generated? Our data suggest that the IL amino acid residues in the LLKIL motif of CXCR2 are crucial for mediating downstream signaling events in HL60 cells. Although the chemokinesis in response to exogenous CXCL8 in the cells expressing IL/AA mutant CXCR2 was not different from that in cells expressing WT CXCR2, the directional movement up the gradient was greatly reduced in cells expressing IL/AA mutant receptor as compared with cells expressing WT CXCR2. The compromised ligand-induced activation of downstream signaling in cells expressing IL/AA mutant CXCR2 may be partially responsible for the reduced chemotaxis of cells expressing this receptor in response to the CXCL8 gradient. These data suggest that the IL amino acid residues of the LLKIL motif are absolutely required for CXCL8-mediated chemotaxis, even though its absence only partially impairs receptor internalization and Erk/Akt activation. There are two possible explanations for why loss of the IL residues from the LLKIL motif in CXCR2 results in loss of chemotaxis in response to CXCL8: 1) AP-2/HIP adaptor binding to the intact LLKIL motif of CXCR2 is required for amplifying and oscillating the cellular responses to CXCL8, and loss of the IL residues of this motif blocks the receptor association with AP2/HIP and thus blocks the threshold amplification and oscillation of signals needed for chemotactic responses; 2) CXCR2 internalization is required for amplifying and oscillating the cellular responses to CXCL8 needed to maintain a chemotactic response to chemokine gradient. Because the IL residues of the LLKIL motif are required for optimal CXCR2 internalization, loss of this motif results in a substantial reduction in receptor internalization and thus chemotaxis. One could argue against the later possibility, because, although CXCR1 internalizes slowly and at a reduced percentage (60% for CXCR1 versus 90% for CXCR2 in 30 min after ligand stimulation in RBL cells), it is as efficient in mediating chemotaxis as the rapidly internalizing CXCR2. However, the total percentage of ligand-mediated internalization for the IL/AA mutant CXCR2 was only ~45% after 30 min of ligand stimulation, considerably less than observed for CXCR1 (60%). These data suggest that a threshold level of receptor internalization may be required, but the rate of internalization is not a regulator of the chemotactic response.
Clearly, much more work in the field is needed to further explain how varying the threshold of chemokine receptor internalization or receptor binding to adaptor molecules affects chemotaxis. Our data suggest a multifactorial model will be required to explain how these regulatory events work in real time. Use of microfluidic gradient devices to monitor these events will provide a valuable tool for the mathematical modeling that will be needed to fully explain these interactions.
Supplementary Material
Acknowledgments
We thank Henry Bourne for kindly providing us HL60 cells in this study. We thank Cathy Alford and Melanie Smith from Veterans Affairs Medical Center (Nashville, TN) for cell sorting and flow cytometry. We thank Linda Horton and Yingchun Yu for the help in tissue culture. We thank Ricardo Richardson for his advice on GTPase activity assays and Jingshong Xu from Henry Bourne's laboratory for information on the chemotaxis assay in HL60 cells.
This work was supported by National Institutes of Health Grants CA34590 (to A. R.) and P50 CA113007, a Senior Research Career Scientist Award from the Dept. of Veterans Affairs (to A. R.), the Vanderbilt Institute for Integrative Biosystems Research and Education and the Vanderbilt Academic Venture Capital Fund (to J. W.), and the Vanderbilt Ingram-Cancer Center Grant CA68485 (to Harold Moses). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The on-line version of this article (available at http://www.jbc.org) contains supplemental videos S1–S10.
The abbreviations used are: AP-2, adaptor protein 2; CXCR2, CXC chemokine receptor 2; CXCL8, CXC ligand 8 or interleukin 8; HL60, human leukemia; Erk, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; Akt, protein kinase B; LLKIL, binding domain on CXCR2 for AP-2 and HIP; fMLP, formylmethionylleucylphenylalanine; HIP, heat shock 70 interacting protein; FACS, fluorescence-activated cell sorting; PE, phycoerythrin; BSA, bovine serum albumin; PBS, phosphate-buffered saline; mHBSS, modified Hanks' balanced salt solution; WT, wild type; PI3K, phosphatidylinositol 3-kinase.
REFERENCES
- 1.Harris H. Physiol. Rev. 1954;34:529–562. doi: 10.1152/physrev.1954.34.3.529. [DOI] [PubMed] [Google Scholar]
- 2.Hersh EM, Bodey GP. Annu. Rev. Med. 1970;21:105–132. doi: 10.1146/annurev.me.21.020170.000541. [DOI] [PubMed] [Google Scholar]
- 3.Behar TN, Schaffner AE, Colton CA, Somogyi R, Olah Z, Lehel C, Barker JL. J. Neurosci. 1994;14:29–38. doi: 10.1523/JNEUROSCI.14-01-00029.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 5.Payne AS, Cornelius LA. J. Invest. Dermatol. 2002;118:915–922. doi: 10.1046/j.1523-1747.2002.01725.x. [DOI] [PubMed] [Google Scholar]
- 6.Chay CH, Cooper CR, Gendernalik JD, Dhanasekaran SM, Chinnaiyan AM, Rubin MA, Schmaier AH, Pienta KJ. Urology. 2002;60:760–765. doi: 10.1016/s0090-4295(02)01969-6. [DOI] [PubMed] [Google Scholar]
- 7.Cooper CR, Chay CH, Gendernalik JD, Lee HL, Bhatia J, Taichman RS, McCauley LK, Keller ET, Pienta KJ. Cancer. 2003;97(Suppl. 3):739–747. doi: 10.1002/cncr.11181. [DOI] [PubMed] [Google Scholar]
- 8.Arai H, Monteclaro FS, Tsou CL, Franci C, Charo IF. J. Biol. Chem. 1997;272:25037–25042. doi: 10.1074/jbc.272.40.25037. [DOI] [PubMed] [Google Scholar]
- 9.Kim JY, Soede RD, Schaap P, Valkema R, Borleis JA, Van Haastert PJ, Devreotes PN, Hereld D. J. Biol. Chem. 1997;272:27313–27318. doi: 10.1074/jbc.272.43.27313. [DOI] [PubMed] [Google Scholar]
- 10.Hsu MH, Chiang SC, Ye RD, Prossnitz ER. J. Biol. Chem. 1997;272:29426–29429. doi: 10.1074/jbc.272.47.29426. [DOI] [PubMed] [Google Scholar]
- 11.Kraft K, Olbrich H, Majoul I, Mack M, Proudfoot A, Oppermann M. J. Biol. Chem. 2001;276:34408–34418. doi: 10.1074/jbc.M102782200. [DOI] [PubMed] [Google Scholar]
- 12.Richardson RM, Marjoram RJ, Barak LS, Snyderman R. J. Immunol. 2003;170:2904–2911. doi: 10.4049/jimmunol.170.6.2904. [DOI] [PubMed] [Google Scholar]
- 13.Perez HD, Elfman F, Marder S, Lobo E, Ives HE. J. Clin. Invest. 1989;83:1963–1970. doi: 10.1172/JCI114105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samanta AK, Oppenheim JJ, Matsushima K. J. Biol. Chem. 1990;265:183–189. [PubMed] [Google Scholar]
- 15.Sambrano GR, Coughlin SR. J. Biol. Chem. 1999;274:20178–20184. doi: 10.1074/jbc.274.29.20178. [DOI] [PubMed] [Google Scholar]
- 16.Fernandis AZ, Cherla RP, Chernock RD, Ganju RK. J. Biol. Chem. 2002;277:18111–18117. doi: 10.1074/jbc.M200750200. [DOI] [PubMed] [Google Scholar]
- 17.Roland J, Murphy BJ, Ahr B, Robert-Hebmann V, Delauzun V, Nye KE, Devaux C, Biard-Piechaczyk M. Blood. 2003;101:399–406. doi: 10.1182/blood-2002-03-0978. [DOI] [PubMed] [Google Scholar]
- 18.Fan GH, Yang W, Wang XJ, Qian Q, Richmond A. Biochemistry. 2001;40:791–800. doi: 10.1021/bi001661b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sai J, Fan GH, Wang D, Richmond A. J. Cell Sci. 2004;117:5489–5496. doi: 10.1242/jcs.01398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng ZJ, Zhao J, Sun Y, Hu W, Wu YL, Cen B, Wu GX, Pei G. J. Biol. Chem. 2000;275:2479–2485. doi: 10.1074/jbc.275.4.2479. [DOI] [PubMed] [Google Scholar]
- 21.Collins SJ, Gallo RC, Gallagher RE. Nature. 1977;270:347–349. doi: 10.1038/270347a0. [DOI] [PubMed] [Google Scholar]
- 22.Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. Proc. Natl. Acad. Sci. U. S. A. 1978;75:2458–2462. doi: 10.1073/pnas.75.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallagher RE, Bilello PA, Ferrari AC, Chang CS, Yen RW, Nickols WA, Muly EC., 3rd. Leuk. Res. 1985;9:967–986. doi: 10.1016/0145-2126(85)90067-0. [DOI] [PubMed] [Google Scholar]
- 24.Bhalla K, Hindenburg A, Taub RN, Grant S. Cancer Res. 1985;45:3657–3662. [PubMed] [Google Scholar]
- 25.Cross AK, Richardson V, Ali SA, Palmer I, Taub DD, Rees RC. Cytokine. 1997;9:521–528. doi: 10.1006/cyto.1996.0196. [DOI] [PubMed] [Google Scholar]
- 26.Li Jeon N, Baskaran H, Dertinger SK, Whitesides GM, Van de Water L, Toner M. Nat. Biotechnol. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- 27.Lin F, Nguyen CM, Wang SJ, Saadi W, Gross SP, Jeon NL. Biochem. Biophys. Res. Commun. 2004;319:576–581. doi: 10.1016/j.bbrc.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 28.Mueller SG, Schraw WP, Richmond A. J. Biol. Chem. 1994;269:1973–1980. [PubMed] [Google Scholar]
- 29.Servant G, Weiner OD, Neptune ER, Sedat JW, Bourne HR. Mol. Biol. Cell. 1999;10:1163–1178. doi: 10.1091/mbc.10.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker GM, Sai J, Richmond A, Stremler M, Chung CY, Wikswo JP. Lab. Chip. 2005;5:611–618. doi: 10.1039/b417245k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richardson RM, DuBose RA, Ali H, Tomhave ED, Haribabu B, Snyderman R. Biochemistry. 1995;34:14193–14201. doi: 10.1021/bi00043a025. [DOI] [PubMed] [Google Scholar]
- 32.Zigmond SH. J. Cell Biol. 1977;75:606–616. doi: 10.1083/jcb.75.2.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parent CA, Devreotes PN. Science. 1999;284:765–770. doi: 10.1126/science.284.5415.765. [DOI] [PubMed] [Google Scholar]
- 34.Firtel RA, Chung CY. BioEssays. 2000;22:603–615. doi: 10.1002/1521-1878(200007)22:7<603::AID-BIES3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 35.Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, Takuwa Y, Sugimoto N, Mitchison T, Bourne HR. Cell. 2003;114:201–214. doi: 10.1016/s0092-8674(03)00555-5. [DOI] [PubMed] [Google Scholar]
- 37.Knall C, Worthen GS, Johnson GL. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3052–3057. doi: 10.1073/pnas.94.7.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuhler GM, Knol GJ, Drayer AL, Vellenga E. J. Leukoc. Biol. 2005;77:257–266. doi: 10.1189/jlb.0504306. [DOI] [PubMed] [Google Scholar]
- 39.Fujita T, Zawawi KH, Kurihara H, Van Dyke TE. Cell Signal. 2005;17:167–175. doi: 10.1016/j.cellsig.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Jones SA, Moser B, Thelen M. FEBS Lett. 1995;364:211–214. doi: 10.1016/0014-5793(95)00397-r. [DOI] [PubMed] [Google Scholar]
- 41.Van Lint J, Van Damme J, Billiau A, Merlevede W, Vandenheede JR. Mol. Cell Biochem. 1993;128:171–177. doi: 10.1007/BF01076768. [DOI] [PubMed] [Google Scholar]
- 42.Grimes GJ, Barnes FS. Exp. Cell Res. 1973;79:375–385. doi: 10.1016/0014-4827(73)90457-6. [DOI] [PubMed] [Google Scholar]
- 43.Ramsey WS. Exp. Cell Res. 1974;86:184–187. doi: 10.1016/0014-4827(74)90669-7. [DOI] [PubMed] [Google Scholar]
- 44.Richardson RM, Pridgen BC, Haribabu B, Ali H, Snyderman R. J. Biol. Chem. 1998;273:23830–23836. doi: 10.1074/jbc.273.37.23830. [DOI] [PubMed] [Google Scholar]
- 45.Rose JJ, Foley JF, Murphy PM, Venkatesan S. J. Biol. Chem. 2004;279:24372–24386. doi: 10.1074/jbc.M401364200. [DOI] [PubMed] [Google Scholar]
- 46.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 47.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 48.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Proc. Natl. Acad. Sci. U. S. A. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su Y, Raghuwanshi SK, Yu Y, Nanney LB, Richardson RM, Richmond A. J. Immunol. 2005;175:5396–5402. doi: 10.4049/jimmunol.175.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.