

Abstract
Idiopathic inflammatory myopathies (IIMs), comprising of polymyositis, dermatomyositis, and inclusion-body myositis, are characterized by muscle weakness and various types of inflammatory changes in muscle cells. They also show non-inflammatory changes, including perifascicular atrophy, mitochondrial changes, and amyloid protein accumulation. It is possible that some molecules/mechanisms bridge the extracellular inflammatory stimulation and intracellular non-inflammatory changes. One such mechanism, Ca2+ influx leading to calpain activation has been proposed. In this study, we demonstrated that post-treatment with calpeptin (calpain inhibitor) attenuated intracellular changes to prevent apoptosis (Wright staining) through both mitochondrial pathway (increase in Bax:Bcl-2 ratio) and endoplasmic reticulum stress pathway (activation of caspase-12), which were induced by interferon-gamma (IFN-γ) stimulation in rat L6 myoblast cells. Our results also showed that calpeptin treatment inhibited the expression of calpain, aspartyl protease cathepsin D, and amyloid precursor protein. Thus, our results indicate that calpain inhibition plays a pivotal role in attenuating muscle cell damage from inflammatory stimulation due to IFN-γ, and this may suggest calpain as a possible therapeutic target in IIMs.
Keywords: Apoptosis, Calpain, Inflammation, Interferon-gamma, Mitochondria, Myoblast cells, Myositis
Introduction
Idiopathic inflammatory myopathies (IIMs) are a constellation of acquired disorders of skeletal muscles, which cause weakness and inflammation in skeletal muscles. The most clearly defined IIMs are polymyositis (PM), dermatomyositis (DM), and sporadic inclusion body myositis (s-IBM) (Dalakas, 2006). Several types of immune cells have been identified in the skeletal muscles of IIMs, including T cells, macrophages, dendritic cells, and B cells (Greenberg, 2007). However, the degree of inflammation does not consistently correlate with the severity of the structural changes in the muscle fibers or with clinical severity, suggesting that non-immune processes also play a role in their pathogenesis (Nagaraju et al., 2005). The non-immune process in IIMs is comprised of muscle fiber destruction including necrosis, perifascicular atrophy in DM, vacuole formation and amyloid deposition in s-IBM, and mitochondrial abnormality in PM and s-IBM. Although it is speculated that some molecules/mechanisms induce these intracellular changes in response to the extracellular immune/inflammatory stimulation in IIMs, they have not yet been clearly identified.
In muscle, intracellular proteolysis is mediated by the proteases, including calpain and cathepsin. In muscle biopsy specimen obtained from patients with PM, sporadic but intense immunostaining for calpain and cathepsin was observed in muscle fibers, primarily near the areas of mononuclear cell infiltrates. Most of these fibers were atrophic or necrotic (Kumamoto et al., 1997). It has been hypothesized that infiltrating mononuclear cells activate the intracellular proteolytic system, resulting in the muscle fiber destruction (Kumamoto et al., 1997). Upregulation of calpain and cathepsin L are also found in perifascicular muscle fibers in DM (Gallardo et al., 2001).
Calpain is an extralysosomal, intracellular protease. Ubiquitously expressed calpain exists in two forms: μcalpain and mcalpain that require μM and mM Ca2+ concentrations, respectively (Murachi, 1984; Banik et al., 1992). Calpain activity is regulated by Ca2+, calpastatin (its endogenous inhibitor), lipids, and an activator protein (Murachi, 1984; Chakrabarti et al., 1990a, 1990b; Coolican and Hathaway, 1984). Calpain plays important roles in induction of apoptosis (Ray and Banik, 2003). Calpain cleaves caspase-3 into its active form (Blomgren et al., 2001). Caspase-3 degrades calpastatin through proteolysis and thereby increases calpain activity further (Wang et al., 1998). Calpain also cleaves caspase-12, which is an endoplasmic reticulum (ER) stress associated caspase (Nakagawa and Yuan, 2000). Calpain increases the ratio of the pro-apoptotic protein Bax to anti-apoptotic protein Bcl-2 by directly cleaving Bax to promote apoptosis through mitochondrial pathway (Wood et al., 2000) or by inactivating Bcl-2, resulting in the mitochondrial release of cytochrome c and induction of apoptosis (Gao and Dou, 2000).
Activation and expression of calpain due to increase in intracellular free Ca2+ is observed in various neurological disorders including Alzheimer's disease (Saito et al., 1993) and stroke (Bartus et al., 1994; Seubert et al., 1989). In experimental allergic encephalomyelitis (EAE), which is an animal model of multiple sclerosis (MS), axonal degeneration and loss of neurons and glial cells have been observed. Examination of the acute EAE spinal cord identified that Ca2+ influx and calpain expression correlated with axonal damage, mitochondrial damage, and loss of structural integrity of microtubules and filaments (Guyton et al., 2005). These results indicate that calpain is deeply involved in the development of intracellular degenerative changes or even cell death, and suggest possible induction of intracellular changes secondary to extracellular immune/inflammatory stimulation.
Here, we hypothesize that calpain induces intracellular changes in muscle cells in response to extracellular inflammatory stimulation. In this report, we have presented data from our studies on cell morphology and protein expression in cultured myoblast cells following interferon-gamma (IFN-γ) stimulation. We have also examined whether calpeptin (calpain inhibitor) can attenuate these intracellular changes.
Materials and methods
Cell culture and treatment with IFN-γ and calpeptin
The rat L6 myoblast cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were grown in 100-mm Petri dishes (Becton Dickinson, Franklin Lakes, NJ, USA) or in 75-cm2 cell culture flasks (Corning, Corning, NY, USA) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 μg/ml) (Sigma-Aldrich, St. Louis, MO, USA), grown to 50-60% confluency, and washed twice with phosphate-buffered saline (PBS, p 7.4). The medium was switched to 1% FBS-containing DMEM one overnight and then recombinant rat IFN-γ (R&D Systems, Minneapolis, MN, USA) at various concentrations (100, 300, or 500 units/ml) was added. To observe the effect of calpain inhibition, two different doses of calpeptin (1 and 5 μM) were added 5 minutes after IFN-γ (500 units/ml) stimulation. The treatment of myoblast cells with IFN-γ or/and calpeptin was carried out with incubation for 48 hours at 37°C with 5% CO2 and full humidity. Cells were then used for determination of morphological changes due to apoptosis and expression of specific proteins.
Wright staining for detection of morphological features of apoptosis
After the treatment with IFN-γ or/and calpeptin, attached myoblast cells were scraped and mixed with floating cells. The cells were centrifugated at 2000 rpm for 5 minutes to obtain a pellet. Then, the cells were resuspended in PBS and sedimented onto the microscopic slides and fixed with methanol. Finally, Wright staining was performed to detect at least one of the characteristic apoptotic features including chromatin condensation, cell shrinkage, membrane blebbing, and membrane-bound apoptotic bodies.
Western blotting for protein analysis
After cell pellets were obtained as above, they were homogenized on ice-cold buffer (50 mM Tris-HCl, pH 7.4, 1 mM PMSF and 5 mM EDTA). Protein content was determined using standard Lowry protein assay. Then, each sample was diluted with the same volume of sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 5 mM β-mercaptoethanol, and 10% glycerol). Samples containing 10-15 μg protein were separated on 4-20% linear gradient SDS-PAGE gels according to standard procedure (Laemmli, 1970). After electrophoresis, proteins were transferred onto nitrocellulose membranes for immunoblot analysis (Towbin et al., 1979). The blots were probed at 4°C overnight with primary IgG antibodies against calpastatin, Bax, Bcl-2, cathepsin D, caspase-3, and caspase-12 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), amyloid precursor protein (APP) (abcam, Cambridge, MA, USA), αII-spectrin (Enzo Life Sciences, Plymouth Meeting, PA, USA) and calpain. We also used primary IgG antibody against αII-spectrin to detect level of calpain-cleaved 145 kD spectrin break down product (SBDP), which was taken as a measure of calpain activity. The primary IgG antibody against glyceraldeyde-3-phosphate dedydrogenase or GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to standardize the loading of cytosolic protein. Then, the horseradish peroxidase conjugated goat anti-mouse or anti-rabbit secondary IgG antibody (MP Biomedicals, Solon, OH, USA) was applied to the blots at 37°C for 2 hours. For subsequent detection of specific proteins, the enhanced chemiluminescence (ECL) system was used (GE Health Care, Piscataway, NJ, USA). Blots were immediately processed to digitalize image using FluorChem FC2 system (AlphaInnotech, San Leandro, CA, USA). Protein bands were quantified by using Image J software program (http://rsb.info.nih.gov/ij/). Compared with control, the percent change in amount of protein was calculated.
Statistical analysis
Results were assessed using StatView software (Abacus Concepts, Berkeley, CA, USA) and compared using one-way analysis of variance (ANOVA) with Fisher's protected least significant difference (PLSD) post-hoc test at a 95% confidence interval. Data were presented as mean ± standard deviation (SD) of separate experiments (n≥3). Significant difference between control and IFN-γ treatment was indicated by *p<0.05 or **p<0.01. Significant difference between IFN-γ treatment and calpeptin pretreatment + IFN-γ was indicated by #p<0.05 or ##p<0.01.
Results
IFN-γ treatment induced apoptosis in muscle cells
To determine IFN-γ induced apoptosis in muscle cells, rat L6 myoblast cells were treated with various doses of IFN-γ (100, 300, and 500 units/ml) for 48 hours (Fig. 1). Wright staining was performed to examine morphological features of apoptosis including cell shrinkage, membrane blebbing, chromatin condensation, and formation of membrane-bound apoptotic bodies (Fig. 1A). Determination of amounts of apoptosis indicated that IFN-γ treatment significantly (p<0.05) induced apoptotic death in a dose-dependent manner in myoblast cells.
Fig. 1.
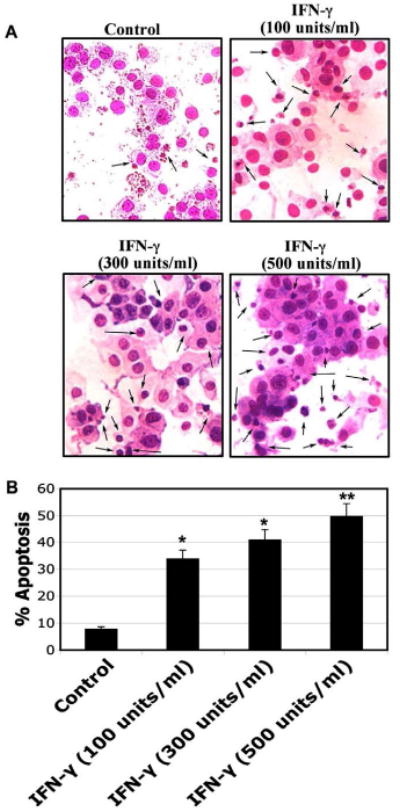
Morphological features of apoptotosis in rat L6 myoblast cells. Cells were treated with various doses of IFN-γ (100, 300, and 500 units/ml) for 48 hours. (A) Wright staining showing representative apoptotic cells after treatments. The arrows indicate apoptotic cells. (B) Determination of percentage of apoptosis based on Wright staining.
IFN-γ treatment altered expression of apoptosis-related proteins in muscle cells
We performed Western blotting following treatment of myoblast cells with IFN-γ (100, 300, and 500 units/ml) (Fig. 2) and found that IFN-γ increased Bax expression (Fig. 2A) resulting in significant increase (p<0.01) in Bax:Bcl-2 ratio, compared with control cells (Fig. 2B), to promote mitochondrial release of pro-apoptotic factors. Caspase-12 (activated due to ER stress) and caspase-3 (final executioner) belong to the cysteine protease family that plays important roles in regulating pathological cell death. We examined the expression and activation of caspase-12 and caspase-3 in myoblast cells. Our results showed significant increases (p<0.05) in active 40 kD caspase-12 and active 20 kD caspase-3 fragments in L6 myoblast cells following treatment with IFN-γ (100, 300, and 500 units/ml), compared with control cells (Fig. 2C). Expression of GAPDH was monitored to ensure that equal amounts of protein were loaded in all lanes.
Fig. 2.
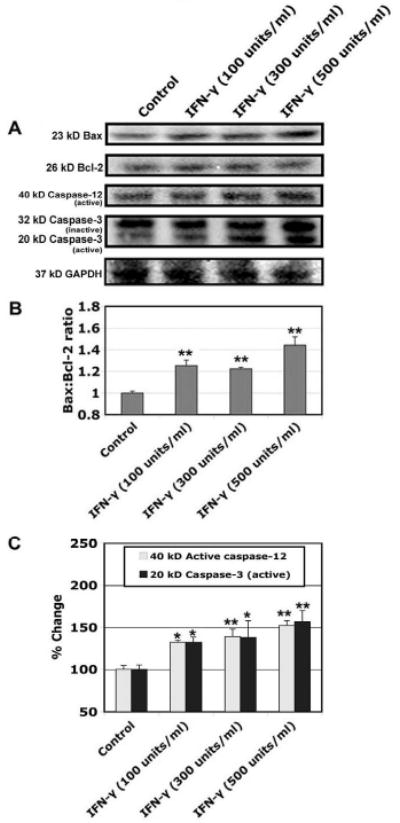
Alterations in protein expression of Bax, Bcl-2, and caspase-12, and caspase-3. Cells were treated with various doses of IFN-γ (100, 300, and 500 units/ml) for 48 hours. (A) Representative Western blots to show levels of 23 kD Bax, 26 kD Bcl-2, active 40 kD caspse-12, inactive 32 kD, active 20 kD caspase-3, and 37 kD GAPDH. (B) Determination of Bax:Bcl-2 raio. (C) Determination of changes in active caspase-12 and active caspase-3.
IFN-γ treatment altered expression of other proteases and amyloid protein in muscle cells
In order to clarify the involvement of other proteases, including extra-lysosomal calpain and intra-lysosmal cathepsin, and amyloid protein in muscle cells following inflammatory stimulation, we examined protein expression of calpain and its endogenous inhibitor calpastatin, cathepsin D, and APP (Fig. 3). We found increase in expression of calpain and decrease in expression of calpastatin leading to significantly high (p<0.05) calpain:calpastatin ratio in the cells following treatment with IFN-γ (100, 300, and 500 units/ml), compared with control cells (Figs. 3A, 3B). There was an increase in level of 145 kD SBDP, which was taken as a measure of calpain activity in the cells treated with IFN-γ (100, 300, and 500 units/ml) (p<0.05), compared with control cells (Figs. 3A, 3B). We found the increased expression of 33 kD cathepsin D (p<0.01) in IFN-γ (100, 300, and 500 units/ml) treated cells and also increased expression of 100 kD APP (p<0.01) in IFN-γ (100, 300, and 500 units/ml) treated cells, compared with control cells (Figs. 3A, 3C).
Fig. 3.
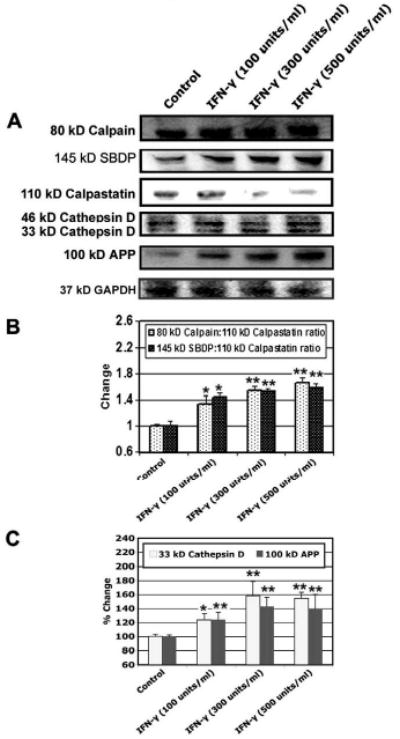
Alterations in protein expression of calpain, SBDP, calpastatin, cathepsin D, and APP. Cells were treated with various doses of IFN-γ (100, 300, and 500 units/ml) for 48 hours. (A) Representative Western blots to show levels of 80 kD calpain, 145 kD SBDP, 110 kD calpastatin, 46 kD and 33 kD cathepsin D, 100 kD APP, and 37 kD GAPDH. (B) Determination of changes in calpain:calpastatin and SBDP: calpastatin ratios. (C) Determination of percent changes in 33 kD cathepsin D and 100 kD APP.
Capain inhibition attenuated morphological features of apoptosis in IFN-γ treated muscle cells
In order to confirm our hypothesis that calpain induces intracellular changes secondary to extracellular inflammatory stimulation, we have examined the effect of calpain inhibitor on apoptotic changes following inflammatory stimulation in rat L6 myoblast cells (Fig. 4). Morphological features of apoptosis were examined following Wright staining (Fig. 4A). The post-treatment of L6 myoblast cells with calpeptin (1 and 5 μM) in the presence of IFN-γ (500 units/ml) significantly deceased (p<0.01) apoptotic cell death, compared with IFN-γ treated cells (Fig. 4B). Thus, our data demonstrated that calpain inhibitor prevented occurrence of apoptotic changes in rat L6 myoblast cells.
Fig. 4.
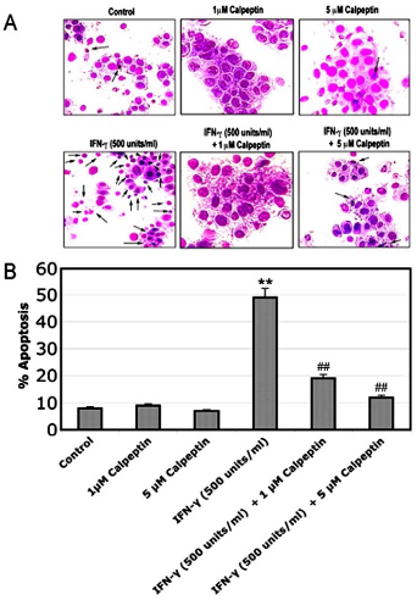
Attenuation of morphological features of apoptosis. Cells were treated with IFN-γ (500 units/ml) for 48 hours. Calpeptin (1 and 5 μM) was added 5 minutes after the IFN-γ addition. (A) Wright staining showing representative apoptotic cells. The arrows indicate apoptotic cells. (B) Determination of percentage of apoptosis based on Wright staining.
Calpain inhibition attenuated expression of apoptosis related proteins, proteases, and amyloid protein in IFN-γ treated muscle cells
We carried out Western blotting to examine whether calpain inhibition affected intracellular apoptosis related protein expression due to extracelluar inflammatory stimulation (Fig. 5). We first examined whether calpain inhibitor blocked IFN-γ induced apoptosis related intracellular proteins including Bax, Bcl-2, caspase-12, and caspase-3 (Fig. 5A). The post-treatment of rat L6 myoblast cells with calpeptin (1 and 5 μM) in the presence of IFN-γ (500 units/ml) significantly decreased (p<0.01) Bax:Bcl-2, calpain:calpastatin, and 145 kD SBDP: calpastatin ratios, compared to IFN-γ treated cells (Fig. 5B). We also observed that post-treatment with calpeptin inhibited the expression of active 40 kD caspase-12 (p<0.01) and active 20 kD caspase-3 (p<0.05) bands, compared with IFN-γ treated cells (Fig. 5C). Since calpain is activated along with wild-type APP and cathepsin D, we have also examined whether calpain inhibitor blocks IFN-γ induced activation of cathepsin D and APP. Our results demonstrated that post-treatment of rat L6 myoblast cells with calpeptin (1 and 5 μM) significantly attenuated expression of 33 kD cathepsin D (p<0.01) and 100 kD APP (p<0.05), compared with IFN-γ treated cells (Fig. 5C).
Fig. 5.
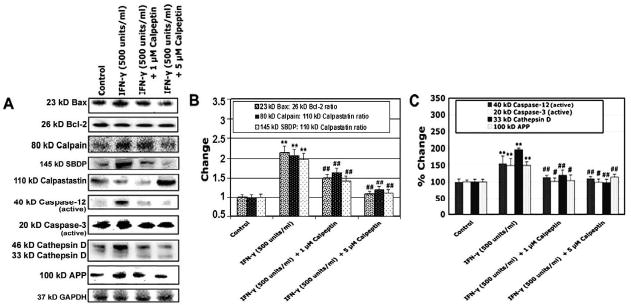
Alterations in protein expression of Bax, Bcl-2, calpain, SBDP, calpastatin, caspase-12, and caspase-3, cathepsin D, and APP. Cells were treated with IFN-γ (500 units/ml) for 48 hours. Calpeptin (1 and 5 μM) was added 5 minutes after the IFN-γ addition. (A) Representative Western blots to show levels of 23 kD Bax, 26 kD Bcl-2, 80 kD calpain, 145 kD SBDP, 110 kD calpastatin, active 40 kD caspse-12, active 20 kD caspase-3, 46 kD and 33 kD cathepsin D, 100 kD APP, and 37 kD GAPDH. (B) Determination of changes in Bax:Bcl-2, calpain:calpastatin and SBDP:calpastatin ratios. (C) Determination of percent changes in active 40 kD caspase-12, active 20 kD caspase-3, 33 kD cathepsin D, and 100 kD APP.
Discussion
Cytokines and chemokines are variably overexpressed in PM, DM, and s-IBM; and these are considered to play important roles in their pathogenesis. In DM, not only IFN-γ, IFN-α, and IFN-β but also other cytokines and chemokines facilitate an inflammatory cascade, which is mediated by complement deposition in the microvasculature. In addition, IFN-γ or IFN-α and IFN-β induce expression of major histocompatibility complex (MHC) class I molecules and various markers of regeneration, degeneration, and cytokine activation in perifascicular muscle fibers in DM. In PM and s-IBM, CD80 and ICOSL are overexpressed on MHC class I antigen-positive muscle fibers, and make contact with their respective ligands expressed on auto-invasive CD8+ T cells. Expression of CD80 and ICOSL on human myoblasts is induced by IFN-γ or TNF-α (Dalakas, 2006). Based on these observations, we used IFN-γ as an extracellular inflammatory stimulant. We treated rat L6 skeletal myoblast cells with IFN-γ and observed various intracellular changes.
We showed that IFN-γ induced apoptotic morphological changes and increased expression of caspase-12 and caspase-3 in skeletal myoblast cells. Caspases belong to cysteine protease family, which plays an essential role for induction of apoptosis. Caspase-9 mediates apoptotic signals after mitochondrial damage (Li et al., 1997), while caspase-12 mediates ER stress-specific apoptosis (Nakagawa et al., 2000). The TNF-α induced apoptosis in HL-1 cardiomyocyte is mediated by ER stress but not by mitochondrial pathway (Bajaj and Sharma, 2006). We treated rat skeletal myoblast cells with IFN-γ, instead of TNF-α, and showed increases in Bax, Bax:Bcl-2 ratio, caspase-12, caspase-3, and apoptotic morphological changes. Bcl-2 is a mitochondrial membrane protein that blocks apoptosis (Hockenbery et al., 1990), while overexpression of Bax accelerates apoptosis (Oltvai et al., 1993). Hence, our results indicate that IFN-γ induces apoptosis through both activation of mitochondrial and ER stress pathways in muscle cells. Mitochondrial changes are frequently encountered in s-IBM and impaired mitochondrial function probably contributes to muscle weakness and wasting in s-IBM (Oldfors et al., 2006). Further, mitochondrial pathology is correlated with poor response to immunosuppressive therapy in PM patients (Blume et al., 1997; Temiz et al., 2009).
We also showed that treatment with IFN-γ induced increase in expression of APP in rat skeletal myoblast cells. Amyloid-β and its precursor protein APP are abnormally accumulated in muscle fibers of s-IBM and they are considered to play important pathogenic roles in s-IBM (Askanas and Engel, 2006). In PM and s-IBM, ER stress leads to accumulation of misfolded MHC glycoproteins, including amyloid related proteins (Dalakas, 2006). It has been questioned whether those intracellular changes seen in IIMs are related to or independent from extracellular inflammatory stimulation. Studies showed significant and consistent correlation between mRNA level of APP and degree of inflammation, as well as mRNA levels of chemokines and IFN-γ in muscle specimen obtained from s-IBM patients (Schmidt et al., 2008). Also, exposure of human myotubes to IL-1 β and IFN-γ caused upregulation of APP with subsequent intracellular aggregation of β-amyloid (Schmidt et al., 2008). These authors consider that production of high amounts of pro-inflammatory mediators induces β-amyloid associated muscle degeneration in s-IBM.
Besides, we showed that treatment with IFN-γ increased expression of calpain and cathepsin D in rat skeletal myoblast cells. Calpain is considered to play important roles in muscle fiber destruction in response to inflammatory stimulation in IIMs. A previous study showed calpain staining of atrophic and necrotic muscle fibers near mononuclear infiltration in PM (Kumamoto et al., 1997). This group also showed strong Ca2+ staining in calpain-positive fibers. They hypothesize that infiltrating mononuclear cells damage sarcolemma, which allows diffusion of extracelluar Ca2+ into the muscle cells freely. The increase in intracellular free Ca2+ concentration then activates calpain, which further causes muscle fiber degradation (Kumamoto et al., 1997). Calpain is also important in muscle degradation in sepsis. Calpeptin reduced protein degradation in muscles obtained from septic rats, as well as in cultured muscle cells treated with dexamethasone (Wei et al., 2005). Calpain activation induces cleavage of myofibrillar cytoskeletal proteins, resulting in disruption of sarcomere and release of myofilaments that are subsequently ubiquitinated and degraded by the 26S proteasome (Smith et al., 2008).
In our study, we observed upregulation of calpain expression in myoblast cells after treatment with IFN-γ for 48 hours. Considering the long metabolic half-lives (approximately 5 days) of m-calpain and μ-calpain (Zhang et al., 1996), calpain upregulation found in our studies may be due to inflammatory response. Such an increase in calpain expression as well as activity has been found in MS and EAE (Shields et al., 1999; Guyton et al., 2010). Calpain activation may decrease its half-life once the influence of inflammatory response is over.
In conclusion, we demonstrated that calpain inhibition attenuated intracellular apoptosis related changes following extracellular inflammatory stimulation in skeletal muscle cells. Since mitochondrial abnormality and accumulation of amyloid protein are important intracellular non-inflammatory pathological changes seen in IIMs, our results suggest that calpain plays an essential role in the pathogenesis of IIMs. Currently, there is no treatment to prevent these intracellular, non-inflammatory changes in IIMs. Our results suggest that calpain may be a promising therapeutic target in IIMs, which may be further explored in an animal model of experimental allergic myositis.
Research Highlights.
Extracellular stimulation with interferon-gamma (IFN-γ) induced apoptosis in rat L6 myoblast cells.
In course of apoptosis, IFN-γ increased Bax:Bcl-2 and calpain:calpastatin ratios, upregulated expression of calpain, caspases, and amyloid precursor protein (APP), and also increased proteolytic activity of calpain in rat L6 myoblast cells.
These intracellular changes were significantly attenuated by treatment with calpeptin, a calpain inhibitor.
The results suggested an important role for calpain in inducing various intracellular changes including induction of apoptosis, mitochondrial dysfunction, and accumulation of APP in muscle cells in response to extracellular inflammatory stimulation. These studies also imply therapeutic potential of the calpain inhibitor in prevention of immune/inflammatory myopathies.
Acknowledgments
This work was supported in part by the NINDS grants (NS–31622, NS–38146, NS–41088, and NS–57811) and the NEI grant (EY–13520) from the National Institutes of Health (Bethesda, MD, USA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Aβ, protein misfolding, and proteasome inhibition. Neurology. 2006;66:S39–S48. doi: 10.1212/01.wnl.0000192128.13875.1e. [DOI] [PubMed] [Google Scholar]
- Bajaj G, Sharma RK. TNF-α-mediated cardiomyocyte apoptosis involves caspase-12 and calpain. Biochem Biophy Res Commun. 2006;345:1558–1564. doi: 10.1016/j.bbrc.2006.05.059. [DOI] [PubMed] [Google Scholar]
- Banik NL, Chakrabarti AK, Konat GW, Gantt-Wilford G, Hogan EL. Calcium-activated neutral proteinase (calpain) activity in C6 cell line: compartmentation of mu and m calpain. J Neurosci Res. 1992;31:708–714. doi: 10.1002/jnr.490310414. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Baker KL, Heiser AD, Sawyer SD, Dean RL, Elliott PJ, et al. Postischemic administration of AK275, a calpain inhibitor, provides substantial protection against focal ischemic brain damage. J Cereb Blood Flow Metab. 1994;14:537–544. doi: 10.1038/jcbfm.1994.67. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Zhu C, Wang X, Karlsson J, Leverin A, Bahr BA, et al. Synergistic activation of caspase 3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- Blume G, Pestronk A, Frank B, Johns DR. Polymyositis with cytochrome oxidase negative muscle fibres. Early quadriceps weakness and poor response to immunosuppressive therapy. Brain. 1997;120:39–45. doi: 10.1093/brain/120.1.39. [DOI] [PubMed] [Google Scholar]
- Chakrabarti AK, Dasgupta S, Banik NL, Hogan EL. Regulation of the calcium-activated neurtral proteinase (CANP) of bovine brain by myelin lipids. Biochem Biophys Acta. 1990a;11038:195–198. doi: 10.1016/0167-4838(90)90204-s. [DOI] [PubMed] [Google Scholar]
- Chakrabarti AK, Dasgupta S, Banik NL, Hogan EL. Ganglioside-modulated proteolysis by Ca2+- activated neutral proteinase (CANP): a role of glycoconjugates in CANP regulations. J Neurochem. 1990b;54:1816–1819. doi: 10.1111/j.1471-4159.1990.tb01241.x. [DOI] [PubMed] [Google Scholar]
- Coolican SA, Hathaway DR. Effect of L-α-phsphatidylinositol on a vascular smooth muscle Ca2+ dependent protease. Reduction of the Ca2+, requirement for autolysis. J Biol Chem. 1984;259:11627–11630. [PubMed] [Google Scholar]
- Dalakas M. Mechanism of disease: signaling pathways and immunology of inflammatory myopathies. Nat Clin Pract Rheumatol. 2006;2:219–227. doi: 10.1038/ncprheum0140. [DOI] [PubMed] [Google Scholar]
- Gallardo E, de Andres I, Illa I. Cathepsins are upregulated by IFN-γ/STAT1 in human muscle culture: a possible active factor in dermatomyositis. J Neuropath Exp Neurol. 2001;60:847–855. doi: 10.1093/jnen/60.9.847. [DOI] [PubMed] [Google Scholar]
- Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome c release and apoptotic cell death. J Cell Biochem. 2000;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::aid-jcb60>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Greenberg S. Proposed immunologic models of the inflammatory myopathies and potential therapeutic implications. Neurology. 2007;69:2008–2019. doi: 10.1212/01.WNL.0000291619.17160.b8. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Matzelle DD, Tyor WR, Ray SK, Banik NL. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res. 2005;81:53–61. doi: 10.1002/jnr.20470. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Das A, Samantaray S, Wallece GC, IV, Butter JT, Ray SK, et al. Calpeptin attenuated inflammation, cell death, and axonal damage in animal model of multiple sclerosis. J Neurosci Res. 2010;88:2398–2408. doi: 10.1002/jnr.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery D, Nunez G, Milliman C, Schreiber RD, Kosmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- Kumamoto T, Ueyama H, Sugihara R, Kominami E, Goll DE, Tsuda T. Calpain and cathepsins in the skeletal muscle of inflammatory myopathies. Eur Neurol. 1997;37:176–181. doi: 10.1159/000117430. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Murachi T. Calcium-dependent proteinases and specific inhibitors: calpain and calpastatin. Biochem Soc Symp. 1984;49:149–167. [PubMed] [Google Scholar]
- Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis. Potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–1835. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J. Cross-talk between tow cystein protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Oldfors A, Moslemi AR, Jonasson L, Ohlsson M, Kollberg G, Lindberg C. Mitochondrial abnormalities in inclusion-body myositis. Neurology. 2006;66:S49–S55. doi: 10.1212/01.wnl.0000192127.63013.8d. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Ray SK, Banik NL. Calpain and its involvement in the pathophysiology of CNS injuries and diseases: therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–89. doi: 10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J, Barthel K, Wrede A, Salajegheh M, Bahr M, Dalakas MC. Interrelation of inflammation and APP in sIBM: IL-1 β induces accumulation of β-amyloid in skeletal muscle. Bain. 2008;131:1228–1240. doi: 10.1093/brain/awn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert P, Lee K, Lynch G. Ischemia triggers NMDA receptor-linked cytoskeletal proteolysis in hippocampus. Brain Res. 1989;492:366–370. doi: 10.1016/0006-8993(89)90921-9. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci U S A. 1999;96:11486–11491. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IJ, Lecker SH, Hasselgren P. Calpain activity and muscle wasting in sepsis. Am J Physiol Endocrinol Metab. 2008;295:E762–E771. doi: 10.1152/ajpendo.90226.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temiz P, Weihl CC, Pestronk A. Inflammatory myopathies with mitochondrial pathology and protein aggregates. J Neurol Sci. 2009;278:25–29. doi: 10.1016/j.jns.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KKW, Posmantur R, Nadimappli R, Nath R, Mohan P, Nixon R, et al. Caspase-mediated Fragmentation of Calpain Inhibitor Protein Calpastatin during apoptosis. Arch Biochem Biophys. 1998;356:187–196. doi: 10.1006/abbi.1998.0748. [DOI] [PubMed] [Google Scholar]
- Wei W, Fareed MU, Evenson A, Menconi MJ, Yang H, Petkova V, et al. Sepsis stimulates calpain activity in skeletal muscle by decreasing calpastatin activity but does not activate caspase-3. Am J Physiol Regul Integr Comp Physiol. 2005;288:R580–R590. doi: 10.1152/ajpregu.00341.2004. [DOI] [PubMed] [Google Scholar]
- Wood DE, Newcomb EW. Cleavage of Bax enhances its cell death function. Exp Cell Res. 2000;256:375–382. doi: 10.1006/excr.2000.4859. [DOI] [PubMed] [Google Scholar]
- Zhang W, Lane RD, Mellgren RL. The major calpain isozymes are long-lived proteins. J Biol Chem. 1996;271:18825–18830. doi: 10.1074/jbc.271.31.18825. [DOI] [PubMed] [Google Scholar]