

Abstract
Adaptive responses to physical and inflammatory stressors are mediated by transcription factors and molecular chaperones. The transcription factor heat shock factor 1 (HSF1) has been implicated in extending lifespan in part by increasing expression of heat shock response genes. Pyrrolidine dithiocarbamate (PDTC) is a small thiol compound that exerts in vivo and in vitro anti-inflammatory properties through mechanisms that remain unclear. Here we report that PDTC induced the release of monomeric HSF1 from the molecular chaperone heat shock protein 90 (Hsp90), with concomitant increase in HSF1 trimer formation, translocation to the nucleus, and binding to promoter of target genes in human HepG2 cells. siRNA-mediated silencing of HSF1 blocked BAG3 gene expression by PDTC. The protein levels of the co-chaperone BAG3 and its interaction partner Hsp72 were stimulated by PDTC in a dose-dependent fashion, peaking at 6 hours. Inhibition of Hsp90 function by geldanamycin derivatives and novobiocin elicited a pattern of HSF1 activation and BAG3 expression that was similar to PDTC. Chromatin immunoprecipitation studies showed that PDTC and the inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin enhanced the binding of HSF1 to the promoter of several target genes, including BAG3, HSPA1A, HSPA1B, FKBP4, STIP1 and UBB. Cell treatment with PDTC increased significantly the level of Hsp90α thiol oxidation, a posttranslational modification known to inhibit its chaperone function. These results unravel a previously unrecognized mechanism by which PDTC and related compounds could confer cellular protection against inflammation through HSF1-induced expression of heat shock response genes.
Keywords: Hsp90, pyrrolidine dithiocarbamate, transcriptional regulation, protein thiols, chromatin immunoprecipitation
1. Introduction
Pyrrolidine dithiocarbamate (PDTC) is a low-molecular mass thiol compound that has been reported to act either as an antioxidant, pro-oxidant, metal chelator and/or thiol group modulator depending on the cell types and their exposure to different stimuli. There is accumulating evidence for the induction of heat shock protein 70 (Hsp70) and several other genes, including heme oxygenase-1, in response to PDTC, which provides adaptive protection of stressed cells and tissues from various pathophysiological conditions and inflammatory responses (Long et al., 2003; Mallick et al., 2005; Tian et al., 2006). PDTC has been demonstrated to interfere with the upregulation of proinflammatory genes by inhibiting the activation of the redox-sensitive transcription factor NF-κB (Schreck et al., 1992; Zhang et al., 2008). However, other potential mechanisms may also contribute to the widely reported beneficial effects of PDTC. Indeed, PDTC attenuates interleukin-6-mediated activation of the transcription factor STAT3 and expression of acute-phase plasma proteins in hepatocytes (He et al., 2006), and STAT3 has been shown to be susceptible to S-glutathionylation after cell treatment with PDTC (Xie et al., 2009). Earlier work by Kim et al. (2001) established that PDTC induces the binding of the transcription factor heat shock factor 1 (HSF1) to DNA through a mechanism that requires protein thiol modification.
HSF1 has been shown as the master regulator of the heat shock response, one of the most fundamental biological mechanisms that confer cellular protection from environmental insults. In normal growth conditions, HSF1 is found held in the cytoplasm bound to the Hsp90-containing multichaperone complexes and, upon activation, it is released from this complex to form a homotrimer, which translocates to the nucleus and binds to the heat shock element (HSE) within target gene promoter regions (Anckar and Sistonen, 2007; Voellmy and Boellmann, 2007). Recent genetic evidence shows that HSF1 is involved in constitutive gene expression in unstressed cells and tissues (Yan et al., 2002; Takaki et al., 2006), and participates in cellular differentiation and extra-embryonic development (McArdle et al., 2006). The ability of HSF1 to confer resistance to stress-induced programmed cell death correlates with diminished expression of proapoptotic genes, which, in turn, could contribute to the initiation and maintenance of tumors in a variety of cancer models (Dai et al., 2007).
In addition to heat shock protein genes, there are many non-classical genes that have been recently described as transcriptional targets of HSF1. One such target is the anti-apoptotic Bcl2-associated athanogene domain 3 (BAG3), also known as CAIR-1 or Bis (Franceschelli et al., 2008). BAG-3 is a 74-kDa cytoplasmic co-chaperone protein involved in cell stress response, whose up-regulation protects cancer cells from apoptosis by stabilizing the level of Bcl-2 family members (Jacobs and Marnett, 2009). BAG3 and other BAG family members bind to the serine/threonine kinase Raf-1, the ATPase domain of Hsc70/Hsp70 and with TNF receptor 1 (Antoku et al., 2002; Song et al., 2001; Takayama and Reed, 2001). Induction of BAG3 confers protection against apoptosis while decrease in its expression promotes cell death in various human cell models, such as myeloid U937 cells, primary mononuclear cells, lymphocytes, and HEK 293 cells (Rosati et al., 2007). These results have several implications, whereby up-regulation in HSF1-mediated BAG3 expression may be a contributing factor in cellular protection against inflammatory stressors and resistance of cancer cells to apoptosis.
The purpose of this study was to investigate in detail the molecular events by which PDTC affects HSF1 activation and to examine the possibility that such activation might result in BAG3 expression in response to PDTC. We found that PDTC stimulation induced a rapid and dose-dependent increase in critical biochemical steps leading to HSF1 activation and induction of BAG3 expression in human tumor cell lines in vitro. Additional investigations indicate that inhibitors of Hsp90 mimic the action of PDTC on HSF1-BAG3 pathway activation.
2. Materials and methods
2.1. Cell culture and treatment
The human hepatoma HepG2, the human pancreatic carcinoma PANC-1, and the human astrocytoma 1321N1 cells (American Type Culture Collection, Manassas, VA) were cultured in Minimum Eagle's medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum, 50 units/ml penicillin and 50 mg/ml streptomycin. Cells were serum starved for 4 h in MEM:F12 (1:1) followed by the addition of vehicle, PDTC (50 μM) or a potent Hsp90 inhibitor for the indicated times. The Hsp90 inhibitors used include novobiocin and the geldanamycin analogs, 17-allylamino-17-demethoxygeldanamycin (17-AAG) and DMAG, a water-soluble analog of 17-AAG. PDTC, novobiocin, 17-AAG and DMAG were purchased from EMD-Calbiochem (San Diego, CA).
2.2 Protein crosslinking in intact cells with ethylene glycol bis(succinimidyl succinate) (EGS)
Cells were washed twice with phosphate-buffered saline and incubated with 1 mM EGS (Thermo Scientific Pierce, Rockford, IL) for 30 min at 25ºC, followed by quenching of the cross-linking reaction with the addition of 10 mM glycine. Cells were lysed in SDS sample buffer supplemented with 7.5% 2-mercaptoethanol, and the samples were processed for Western blot analysis.
2.3. Western blot analysis
Unless otherwise indicated, cells were lysed in RIPA buffer (He et al., 2006) and proteins were separated by SDS-polyacrylamide gel electrophoresis under reducing conditions and then transferred onto polyvinylidene difluoride membranes. Western blots were performed according to standard methods, which involved the visualization of immunoreactive bands by enhanced chemiluminescence, their quantitation by volume densitomety using ImageQuant software (Molecular Dynamics, Piscataway, NJ), and normalization to GAPDH. In this study, the primary antibodies were directed against HSF1 (StressGen Biotechnologies, Victoria, Canada), BAG3 (Abcam, Cambridge, MA), Hsp90α (BD Biosciences, San Jose, CA), Hsp72, BRG1, p65Rel, IκBα, Mcl-1, and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA) and were used generally at a dilution of 1:1000.
2.4. Preparation of nuclear extracts
Nuclear extracts from HepG2 cells were prepared using the NE-PER extraction kit (Thermo Scientific Pierce) and quantified using the bicinchoninic acid protein assay reagent.
2.5. RNA interference
Transfection of 21-nucleotide siRNA duplexes (Qiagen) for targeting endogenous HSF1 was carried out using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA) and 20 nM small interfering RNA (siRNA) duplex per 35-mm plate according to the supplier s instructions. Transfected HepG2 cells were assayed 3 days after reverse transfection. The sequences of HSF1 siRNA used were: r(GGUUGUUCAUAGUCAGAAU)dTdT (sense) and r(AUUCUGACUAUGAACAACC)dTdG (anti-sense), whereas the negative control siRNA was the “AllStars Neg. Control siRNA” (Qiagen) that has no known target gene. Specific target gene silencing was confirmed by real-time PCR and immunoblotting.
2.6. Reverse transcription and real-time PCR analysis
Total RNA was isolated and first strand cDNA was synthesized using the Omniscript Reverse Transcript kit (Qiagen). Real-time PCR reactions were carried out with the TaqMan® Gene Expression Assay system method on an ABI Prism 7300 sequence detection system (Applied Biosystems, Foster City, CA). Primer pairs used for the reactions are listed in Table 1. Relative quantitation of gene expression was performed using the threshold cycle. The mRNA levels were compared to standard curves (generated using serial dilutions of human HepG2 RNA) and differences in mRNA expression were calculated by the ΔΔCT method after normalizing to GAPDH mRNA.
Table 1.
List of primer identification number (ID) and size of the PCR products for real time PCR
| Gene | Assay ID (Applied Biosystems) | Size of PCR products (bp) |
|---|---|---|
| HSPA1A | Hs00359163_s1 | 124 |
| BAG3 | Hs00188713_m1 | 83 |
| HSF1 | Hs00232134_m1 | 102 |
| GAPDH | Hs99999905_m1 | 122 |
2.7. Chromatin immunoprecipitation (ChIP) assay
HepG2 cells were treated with vehicle, PDTC (50 μM) or DMAG (5 μM) for 1 h, and ChIP assays were then performed essentially as described (Wurster and Pazin, 2008). In brief, cells were crosslinked with 1% formaldehyde at room temperature for 10 min. After harvesting cells with SDS lysis buffer, DNA was sheared to 200–1000 bp by sonication, and the cell lysates were precleared by centrifugation. Polyclonal antibody developed against HSF1 (StressGen) was used to recover HSF1-bound DNA complexes. In addition, precleared lysates were also incubated with rabbit IgG as a control for potential non-specific coprecipitations. DNA was quantified by real-time PCR using promoter-specific primers for select genes (Table 2). Each experiment was repeated 3-5 times.
Table 2.
Promoter-specific primer sequences used for ChIP assays
| Gene Name | Promoter-specific primers |
|---|---|
| BAG3 | 5’-GGCCAACTTCTCTGGACTGG-3’ (forward) |
| 5’-GGTAGCAACTGGCCGGCTA-3’ (reverse) | |
| HSPA1A | 5’-GAATATTCCCGACCTGGCAG-3’ (forward) |
| 5’-CCTTCTGAGCCAATCACCGA-3’ (reverse) | |
| HSPA1B | 5’-AATATTCCCGACCTGGCAGC-3’ (forward) |
| 5’-CCCTTCTGAGCCAATCACCA-3’ (reverse) | |
| FKBP4 | 5’-CAGAAGGAGGCAAGAACCAAA-3’ (forward) |
| 5’-GGCGCTTCGGAATTTTTTTT-3’ (reverse) | |
| STIP1 | 5’-GGCTACGATTGGCAGTGCA-3’ (forward) |
| 5’-GGAGCGAACTTCTGCGACAC-3’ (reverse) | |
| UBB | 5’-AAGTTTCCAGAGCTTTCGAGGA-3’ (forward) |
| 5’-AGGCGGATGAATTTGAGTTGA-3’ (reverse) | |
| PPP1R15A | 5’-GTGGTCACGCTCGGAAACTC-3’ (forward) |
| 5’-CGGAGATTCCAGCTTTTGCA-3’ (reverse) | |
| NFKBIA | 5’-TACTTCCCTGCAGCCTGCAC-3’ (forward) |
| 5’-AGGCTCACTTGCAGAGGGAC-3’ (reverse) |
2.8. Labeling of protein reactive thiols in cellular extracts
Cells were lysed in RIPA buffer supplemented with 200 μM maleimidobutyrylbiocytin (MBB, EMD-Calbiochem), an irreversible thiol-specific biotinylating agent, for 30 min on ice followed by the addition of an excess of L-cysteine (5 mM) to quench the reaction. An aliquot of the clarified lysates was subjected to Hsp90 immunoprecipitation and Western blotting. Polyvinylidene difluoride membranes were blocked with 1% polyvinylpyrrolidone (Sigma-Aldrich, St-Louis, MO) in TBS-T and incubated with horseradish peroxidase-conjugated streptavidin (Vector Laboratories, Burlingame, CA) for the detection of biotin-labeled protein thiols. A second aliquot was incubated with captavin-agarose beads (Invitrogen) for 1 h at 4°C to immobilize thiol-biotinylated proteins, which were then eluted with 50 mM NaHCO3 (pH 10.0). After neutralization, Hsp90 immunoprecipitation and Western blot analysis were carried out.
2.9. Statistical analysis
All results are expressed as relative to the control value. Experiments were performed in at least two to three different culture preparations, and two dishes for each experimental condition were plated in each preparation. Results are expressed as means ± SD, where indicated, with n reflecting the number of observations. Statistical comparisons between groups were made by unpaired Student s t-test. Analyses were performed using the software package Kaleidagraph v4.01 (Synergy Software, Reading, PA) with values ≤ 0.05 considered significant.
3. Results
3.1. Dissociation from Hsp90α is required for PDTC-mediated activation, homotrimerization and nuclear translocation of HSF1
Given that Hsp90-containing multichaperone complexes are thought to be the most relevant repressors of HSF1 activity (Voellmy and Boellmann, 2007), we reasoned that PDTC might alter HSF1 interaction with Hsp90-containing complexes. In the first series of experiments, HSF1 immunoprecipitates were analyzed for co-sedimentation of Hsp90α by Western blotting. The results indicated a 31.4 ± 3.1% reduction in the constitutive HSF1-Hsp90α interaction at 30 min after PDTC treatment, reaching 82.0 ± 9.1% inhibition after 1 h (P < 0.05, n=3, Fig. 1A, upper panel). The blots exhibited equivalent HSF1 protein levels in all samples (Fig. 1A, bottom panel). Reciprocal immunoprecipitation with Hsp90α antibodies confirmed dissociation of the HSF1-Hsp90 complex by PDTC (Fig. 1B).
Fig. 1.
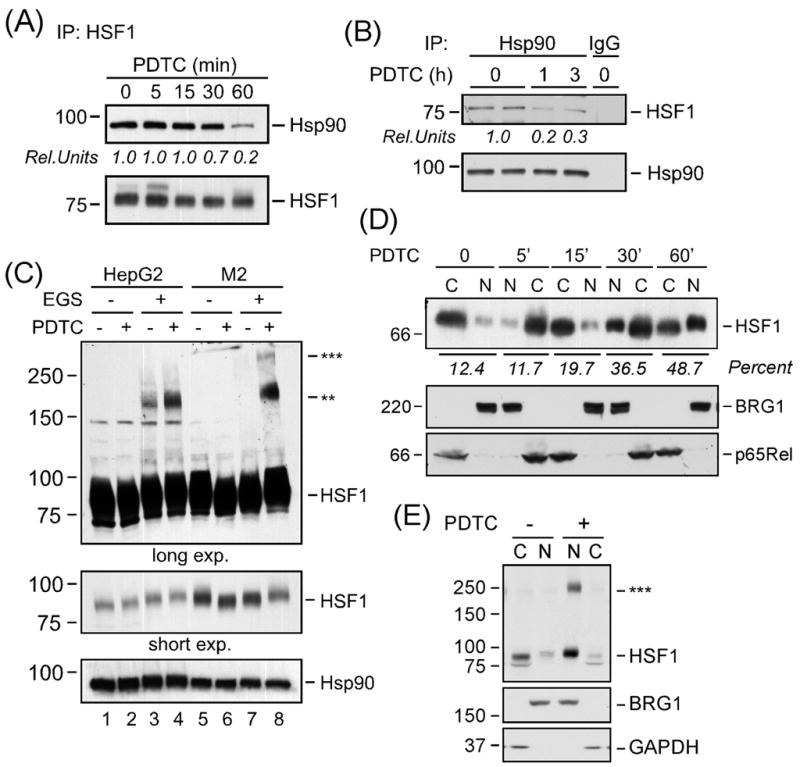
Cellular activation of HSF1 by PDTC. (A) Following exposure of HepG2 cells to PDTC (50 μM) for 0-60 min, cells were lysed and the HSF1 immunoprecipitates were analyzed by Western blot for the cosedimentation of Hsp90α (upper panel) and HSF1 (bottom panel). Rel. Units, ratio of Hsp90 over HSF1. (B) HepG2 cells were exposed to 50 μM PDTC for 1 or 3 h, after which cells were lysed and Hsp90α immunoprecipitates were analyzed by Western blot for the cosedimentation of HSF1 (upper panel) and Hsp90α (lower panel). IgG, isotype-specific mouse IgG used for control immunoprecipitation. Rel. Units, ratio of HSF1 over Hsp90. (C) Serum-starved HepG2 and M2 cells were exposed to PDTC (50 μM) for 1 h, washed and subjected to crosslinking with EGS as outlined in “Materials and Methods”. Total cell extracts were separated on 6% SDS-PAGE gels under reducing conditions and analyzed by Western blot for HSF1 (upper and middle panels) and Hsp90α, which was included as a loading control (bottom panel). The position of the molecular weight makers (in kDa) is shown on the left. ** and *** denote the formation of HSF1 oligomers. (D) The level of HSF1 in the nuclear (N) and cytosolic (C) fractions of HepG2 cells incubated with PDTC (50 μM) for 5, 15, 30 and 60 min was assessed by Western blot analysis (top panel). The membranes were reprobed for BRG1 and p65Rel to demonstrate the quality of our nuclear fractionation. Percent, ratio of [nuclear HSF1 over cytosol+nuclear pool of HSF1] x 100. (E) Serum-starved HepG2 cells were exposed to vehicle or PDTC (50 μM) for 1 h, washed and subjected to crosslinking with EGS. The nuclear (N) and cytosolic (C) fractions were prepared and analyzed by Western blot for HSF1, BRG1 and the cytosolic marker, GAPDH. ***, trimers of HSF1. For each experiment depicted in A-E, similar results were obtained in 2–3 independent experiments.
The activation of HSF1 protein requires its conversion from monomeric form to dimers and trimers in response to heat shock. To evaluate whether PDTC would elicit the oligomerization of HSF1, HepG2 cells were left untreated or treated with PDTC followed by the stabilization of oligomeric HSF1 by the addition of the crosslinker EGS in intact cells. Western blot analysis using total cell lysates showed no high molecular HSF1 species in the absence of EGS, while the in vivo crosslinking procedure yielded significant HSF1 oligomerization (P < 0.005, Fig. 1C, lanes 3-4 versus 1–2). PDTC enhanced also HSF1 oligomer formation in the human M2 melanoma cells (Fig. 1C, lane 8). Our results suggest that PDTC promotes formation of high molecular weight HSF1 complexes in these tumor cell models.
Because HSF1 translocates and functions in the nucleus, it was of interest to examine its nucleocytoplasmic localization in response to PDTC. HepG2 cells were left untreated or treated with PDTC for 5 to 60 min, and the cytosolic and nuclear fractions were separated and analyzed by Western blotting. The data in Fig 1D (upper panel) showed a time-dependent increase in the level of nuclear HSF1 in PDTC-treated cells. Exposure to PDTC for 1 h resulted in a 3.00 ± 0.36-fold increase in HSF1 nuclear accumulation over control (P < 0.0001, n=6). The blots were reprobed with the nuclear marker, BRG1, and cytosolic marker, p65Rel, to confirm the quality of our cell fractionation (Fig. 1D, bottom two panels). Subcellular fractionation of EGS-crosslinked cells confirmed the nuclear accumulation of HSF1 trimers after a 1-h treatment with PDTC (P < 0.005 vs. control n=3; Fig. 1E). Taken together, these results provide a first comprehensive demonstration that PDTC elicits HSF1 recruitment to the nucleus via the classical pathway.
3.2. HSF1 mediates PDTC-induced BAG3 expression
One of the critical aspects of HSF1 signaling is its ability to induce expression of heat shock response genes. We first examined the effect of PDTC on the expression of the co-chaperone BAG3 both at the mRNA and protein levels. Quantitative real-time PCR confirmed the time- and dose-dependent induction of BAG3 mRNA in PDTC-treated HepG2 cells (Fig. 2A, B). The maximum response was achieved at 4 h with an ED50 of 8.1 ± 0.6 μM. HSPA1A is a classical HSF1 target gene that encodes the molecular chaperone Hsp72 whose activity is controlled by its interaction with BAG3 (Jacobs and Marnett, 2009). Real-time PCR results showed a pattern of HSPA1A mRNA expression that was comparable to BAG3 (Fig. 2A, B). Human PANC-1 cells were also stimulated with PDTC, and expression of BAG3 and HSPA1A was assessed by real-time PCR. Compared to controls, exposure of PANC-1 cells to PDTC for 4 h resulted in a 4.2 ± 1.3 – and 7.9 ± 2.1 – fold induction of BAG3 and HSPA1A mRNAs, respectively (Fig. 2C). BAG3 and Hsp72 protein levels were also analyzed by immunoblotting. The expression of Hsp72 (HSPA1A transcript) and BAG3 proteins increased significantly as early as 6 h after treatment of HepG2 cells with PDTC, resulting in a 2.1 ± 0.3 – (P<0.01, n=4) and 1.8 ± 0.3 – fold increase (P<0.05, n=4) over control, respectively. Hsp72 levels remained elevated up to 18 h (P<0.05), but those of BAG3 progressively declined after 18 h of treatment (Fig. 2D). Under these conditions, PDTC had minimal effect on the expression of HSF1, Hsp90α and GAPDH, the latter serving as a loading control (Fig. 2D, bottom panel). The role of protein synthesis in the observed upregulation of BAG3 by PDTC was then investigated by preincubating HepG2 cells with cycloheximide. As anticipated, inhibition of protein synthesis resulted in a significant decrease in both the constitutive and inducible expression of BAG3 to 25 ± 1% and 32 ± 1% (P<0.01, n=3) of control (Supplemental Fig. 1). Exposure of HepG2 cells to PDTC for 4 h dose-dependently increased Hsp72 and BAG3 protein levels, resulting in a 3.8 ± 0.2 – and 2.6 ± 0.4 – fold increase with 50 μM PDTC compared to vehicle-treated cells (P< 0.01, Fig. 2E).
Fig. 2.

Effect of PDTC on BAG3 and Hsp72 (HSPA1A) expression. Total RNA was extracted from HepG2 cells treated either with 50 μM PDTC for 1, 2, 4 and 8 h (panel A) or with the indicated concentrations of PDTC for 4 h (panel B) and then analyzed by real time PCR. Data are represented as fold increase relative to vehicle-treated groups. Error bars indicate standard deviation and may be smaller than the symbols. These experiments were repeated three times with comparable results. (C) Real time PCR analysis of the two HSF1 target genes in human PANC-1 cells treated with 50 μM PDTC for periods up to 4 h. Relative mRNA levels for HSPA1A (●) and BAG3 (❍) were normalized to GAPDH mRNA expression. Data are representative of two independent experiments, each performed in duplicate dishes. (D) Western blot analysis was performed using lysates from HepG2 cells treated with 50 μM PDTC for the indicated times. GAPDH was included as a loading control (bottom panel). Graphical representation for protein blots (BAG3 and Hsp72 over GAPDH). *P<0.05 and **P<0.01 vs. control. n =4. (E) HepG2 cells treated with 0–50 μM PDTC for 4 h were lysed, and immunoblotting was performed using antibodies to BAG3 and Hsp72. GAPDH was included as a loading control. Graphical representation for protein blots (BAG3 and Hsp72 over GAPDH). **P<0.01 vs. control. n =3.
In order to clarify the role of HSF1 in the regulation of BAG3 and HSPA1A gene expression, HepG2 cells were incubated with HSF1 siRNA and non-silencing control for 72 h, after which PDTC was added for 2 and 4 h. Under conditions where more than 60-65% of HSF1 mRNA was knocked down, the silencing of HSF1 significantly abrogated BAG3 and HSPA1A mRNA levels to 32.7 ± 5.1% (P<0.001) and 10 ± 2% (P<0.0001) of control siRNA-treated cells at 4 h (Fig. 3). Control experiments indicated the depletion of HSF1 protein level in response to HSF1 siRNA (data not shown).
Fig. 3.

Effect of HSF1 siRNA on PDTC-induced expression of HSPA1A and BAG3. HepG2 cells were transfected with either negative control (❍) or HSF1 (●) siRNA for 72 h, after which they were serum-starved and then incubated with PDTC (50 μM) for 0, 2 and 4 h. Total RNA was extracted and analyzed by real time PCR. Data represent relative mRNA expression (normalized to GAPDH) and expressed as mean values ± S.D. of triplicates. Similar results were obtained in a second independent experiment. Error bars indicate standard deviation and may be smaller than the symbols
3.3 Hsp90-binding drugs mimic PDTC-mediated activation of HSF1
Given that the ATPase function of Hsp90 is required for maintaining HSF1 in an inactive conformation, we sought to investigate whether inhibition of the Hsp90 ATPase activity with geldanamycin analogs elicits responses similar to PDTC. Western blot analysis indicated that 17-AAG time-dependently induced oligomerization of HSF1 in HepG2 cells (Fig. 4A). The 1321N1 astrocytoma cells were also responsive to 17-AAG (Fig. 4B). Likewise, incubation of HepG2 cells with the structurally unrelated Hsp90 inhibitor, novobiocin, led to a dose-dependent increase in HSF1 oligomerization with concomitant decrease in monomeric HSF1 (Fig. 4C). Subcellular fractionation experiments revealed that HSF1 migration to the nucleus was increased in cells treated with either DMAG (a water-soluble analog of 17-AAG) or novobiocin, as compared to vehicle-treated cells (Fig. 4D). BRG1 and IκBα are shown as nuclear and cytosolic markers, respectively. Treatment of HepG2 cells with DMAG for 4 h resulted in increased expression of BAG3 and HSPA1A mRNA levels (Fig. 4E). Similar to the results with PDTC, the expression of Hsp72 and BAG3 proteins increased significantly by 6-h treatment of HepG2 cells with DMAG, resulting in a 2.5 ± 0.2 (P<0.01, n=6) and 1.9 ± 0.2 – fold increase (P<0.01, n=5) over control, respectively (Fig. 4F).
Fig. 4.

Hsp90-binding drugs mimic PDTC effects on HSF1 activation. (A) Serum-starved HepG2 cells were incubated with 17-AAG (5 μM) for 0-3 h, followed by crosslinking reaction with EGS as outlined in the legend of Fig. 1C. Western blot analysis using total cell lysates was performed with anti-HSF1 antibody. Note the time-dependent increase in the formation of EGS-stabilized HSF1 trimers (upper panel) with concomitant reduction in HSF1 monomers (bottom panel). (B) Serum-starved 1321N1 cells were treated with vehicle, PDTC (50 μM) or 17-AAG (5 μM) for 1 h followed by EGS crosslinking reaction. (C) Serum-starved HepG2 cells were incubated for 1 h with novobiocin (0.2 and 0.6 mM), an Hsp90 inhibitor structurally unrelated to geldanamycin. Total cell lysates were analyzed by Western blot using antibodies to HSF1 and Hsp90α. ** denotes HSF1 trimers. (D) Serum-starved HepG2 cells were treated with vehicle (Veh.), PDTC (50 μM), DMAG (5 μM), or novobiocin (0.6 mM) for 1 h followed by the preparation of nuclear (N) and cytosolic (C) extracts. HSF1 levels were detected by Western blot analysis (upper panel). The membranes were reprobed with BRG1 (middle panel) and IκBα (bottom panel) antibodies. (E) Total RNA from HepG2 cells treated with vehicle or DMAG (5 μM) for 4 h was extracted and analyzed by real time PCR. Data represent relative HSF1, HSPA1A and BAG3 mRNA levels (normalized to GAPDH) and expressed as mean value ± S.D. of four replicates. (F) HepG2 cells treated with 5 μM DMAG for 0–24 h were lysed, and immunoblotting was performed using antibodies to BAG3 and Hsp72. GAPDH was included as a loading control (bottom panel). Graphical representation for protein blots (BAG3 and Hsp72 over GAPDH). For each experiment depicted in A-F, similar results were obtained in 2-3 independent experiments.
3.4. HSF1 binds directly to the HSE element of the BAG3 promoter
ChIP assay was used to get insight into the in vivo interactions between HSF1 and target gene promoters in HepG2 cells. In addition to BAG3 and HSPA1A, a number of classical and nonclassical HSF1 target genes (e.g., HSPA1B, FKBP4, STIP1 and UBB) contain canonical HSE in their promoter regions (Trinklein et al., 2004). The results showed that cell treatment with PDTC for 1 h led to the binding of HSF1 onto the promoter regions of BAG3 and HSPA1A (Fig. 5A and 5B, left panel) as well as HSPA1B, FKBP4, STIP1 and UBB, but not PPP1R15A or NFKBIA (Fig. 5B, right panel). The same pattern of HSF1-inducible binding to HSE elements was observed in DMAG-treated cells (Fig. 5B). HSF1 was not detected upstream or downstream of several target genes, suggesting promoter specificity (data not shown). When chromatin samples were immunoprecipitated with a control rabbit IgG, there was a complete absence of HSF1 recruitment onto these promoter regions (Supplemental Fig. 2).
Fig. 5.
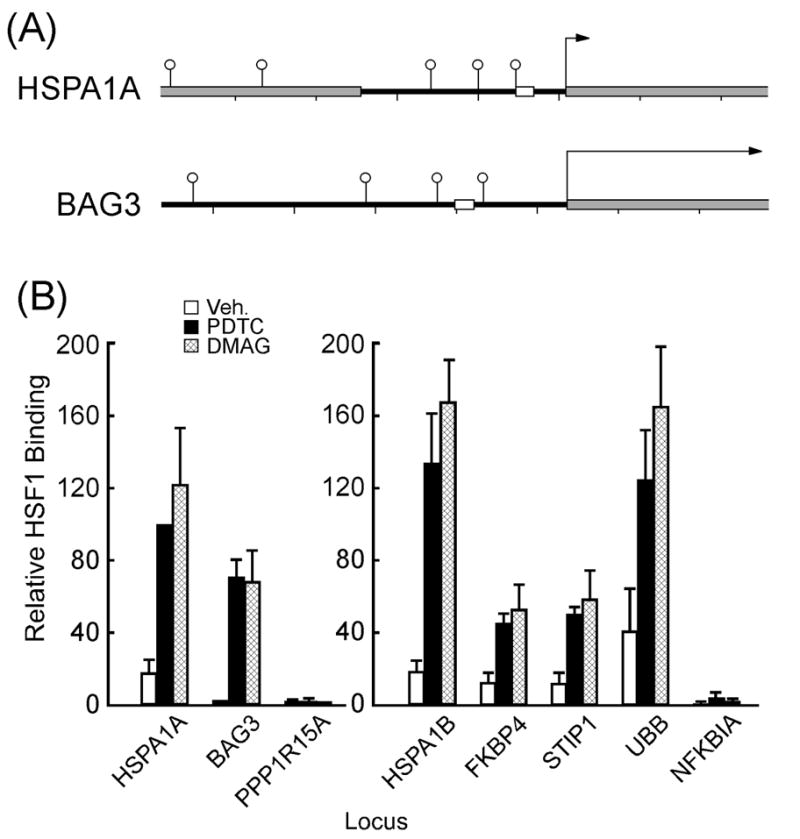
Binding of HSF1 to HSE elements in target gene promoters. (A) All loci are shown at the same scale, from 1000 bases upstream to 500 bases downstream of the annotated transcriptional start site. Open circles indicate predicted HSEs, gray rectangles indicated transcribed regions, white rectangles indicate PCR products for ChIP, tick marks are every 200 bp. The gray box upstream of the HSPA1A is the HSPA1L pseudogene. (B) ChIP assays using agarose-bound HSF1 antibody were performed in serum-starved HepG2 cells treated either with vehicle (open bars), PDTC (50 μM, filled bars) or DMAG (5 μM, hatched bars) for 60 min. HSF1 binding to HSPA1A promoter in PDTC-treated cells was set at 100. Bars indicate mean values ± SEM from 3-5 independent experiments.
3.5. PDTC and redox modulation of Hsp90α protein
Exposure of HepG2 cells to PDTC significantly reduced incorporation of the thiol-alkylating agent, maleimidobutyrylbiocytin (MBB), into accessible Hsp90α cysteines to 28.5 ± 8.5% of control (P < 0.01, n=3; Fig. 6A). Equivalent amount of Hsp90 protein was present in all samples (Fig. 6A, bottom panel). The role of PDTC in the observed reduction in Hsp90 reactive thiols was also investigated by enriching thiol-biotinylated proteins by captavidin-agarose chromatography. Western blot analysis showed a 34% recovery of captavidin-bound Hsp90 from PDTC-treated cells as compared to untreated controls (Fig. 6B). These results suggest that PDTC-mediated Hsp90α thiol oxidation may contribute to cellular activation of HSF1.
Fig. 6.
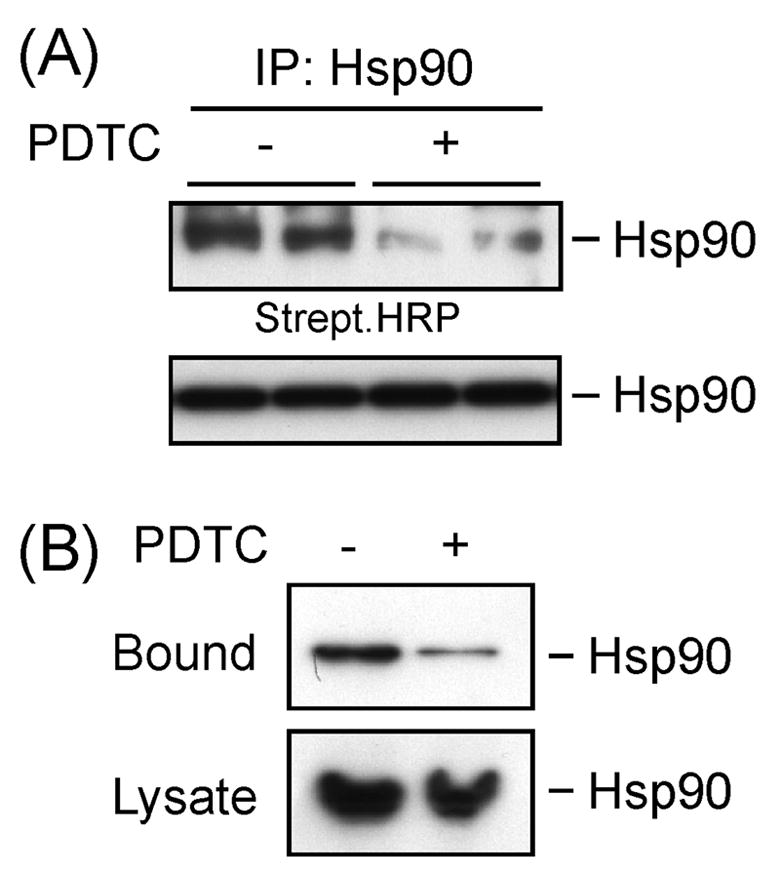
Hsp90α contains cysteine residues that are S-thiolation targets. Serum-starved HepG2 cells were treated with vehicle or PDTC (50 μM, 2 h) after which cells were homogeneized in RIPA buffer supplemented with MBB (200 μM) for 30 min on ice. The alkylation reaction was quenched with excess L-cysteine. (A) The clarified cell lysates were subjected to immunoprecipitation with Hsp90 antibody and immunoblotted either with streptavidin-conjugated HRP (upper panel) or Hsp90 to confirm equal loading (bottom panel). (B) The clarified cell lysates were incubated with captavidin-linked agarose for 1 h at 4 ºC, and the immobilized proteins were eluted with biotin, followed by Western blot analysis with anti-Hsp90 (upper panel). Input controls are shown in the bottom panel. Similar results were obtained in a second independent experiment.
4. Discussion
The present study demonstrates that PDTC and Hsp90-binding drugs promote BAG3 expression by inducing the release of HSF1 from the inhibitory Hsp90 chaperone complex and hence contribute to the increase in HSF1 transcriptional activity. Once released from the Hsp90 multichaperone complex, trimers of HSF1 were found to translocate into the nucleus in response to PDTC and Hsp90 inhibitors. This accumulation of HSF1 trimers in the nuclear compartment of PDTC-treated cells is consistent with the involvement of p62 and/or related nucleoporins in the shuttling of HSF1. Indeed, nucleoporin p62, a major component of the nuclear pore complex, has been shown to interact with the heat shock factor trimerization domain (Yoshima et al., 1997).
Our results indicate that nuclear HSF1 can bind to HSE element of target gene promoters, which included several Hsps and co-chaperones such as BAG3 (Bag3), HSPA1A (Hsp72), HSPA1B (Hsp71), FKBP4 (FKBP52), STIP1 (p60Hop), and UBB (ubiquitin B). Inducible expression of BAG3 has a critical role in proliferation and mitigating apoptosis. While elevated BAG3 expression correlates with growth inhibition (Seo et al., 2005) and tumor cell survival (Chiappetta et al., 2007; Romano et al., 2003), its silencing sensitizes leukemia cell lines to bortezomib-induced apoptosis (Liu et al., 2009). In the present study, we observed that the PDTC-mediated increase in BAG3 protein expression was rapid and had plateaued by 6-h. Under these conditions, Hsp72 (HSPA1A), another HSF1-regulated gene transcript and an interaction partner of BAG3 (Jacobs and Marnett, 2009), was also induced in response to PDTC. Morover, our time-course and dose-response experiments have demonstrated the co-expression of BAG3 with Mcl-1, a member of the anti-apoptotic Bcl-2 family (Song and Bernier, unpublished results). It is interesting that the proteasome inhibitor MG132 increases BAG3 expression (Du et al., 2009) while bortezomib up-regulates BAG3 levels in human leukemic cells to confer limited cytoprotection (Liu et al., 2009). These data suggest that proper chaperoning and stabilization of anti-apoptotic proteins by BAG3 is a key component of cancer cell viability.
An earlier report showed that PDTC activates the transcription factor HSF1 to induce heat shock protein expression by virtue of its activity as thiol group modulator (Kim et al., 2001); however, it was unclear how PDTC causes nuclear translocation of HSF1 to control the expression of inducible classical and nonclassical target genes. PDTC and other dithiocarbamates are known inhibitors of the NF-κB pathway by virtue of their ability to block the release and degradation of the small IκBα inhibitor (Cvek and Dvorak, 2007). An increase in the intracellular levels of oxidized glutathione by dithiocarbamates (Wild and Mulcahy, 1999) enables formation of thiol-modified protein adducts that could affect a number of redox-sensitive signaling pathways involved in inflammation and cancer (Chu et al., 2005; Xie et al., 2009). This process requires PDTC and its derivatives to form a complex with transition metals (e.g., Zn2+, Co2+, Cu2+), which are present in varying concentrations especially in cancer cells (Goodman et al., 2005). Thiol oxidation inactivates the Hsp90 chaperone function in response to oxidative stress (Carbone et al., 2005; Martinez-Ruiz et al., 2005) and therefore it is possible that PDTC may inhibit Hsp90 activity with concomitant release of HSF1 through formation of mixed protein disulfides between small thiols (e.g., glutathione) and the reactive cysteines of Hsp90α. Our data indicate a role of thiol oxidation in PDTC-mediated dysregulation of Hsp90 signaling (Fig. 6). Interestingly, cell exposure to pharmacological inhibitors of Hsp90 elicited a pattern of HSF1 activation and transcriptional activity similar to PDTC. It has been established that geldanamycin, and its structurally related benzoquinone derivatives, not only bind directly to Hsp90, but also participate in redox cycling via superoxide generation (Dikalov et al., 2002). Hence, future studies are needed to examine the role of redox cycling of Hsp90 in the control of HSF1 activation and induction of gene expression.
In conclusion, inhibition of the chaperone function of Hsp90α either via PDTC-mediated thiol oxidation or Hsp90-binding drugs induces the disassembly of the cytosolic HSF1-Hsp90 complex, enabling HSF1 trimer formation with concomitant increase in HSF1-mediated expression of the co-chaperone BAG3 and antiapoptotic proteins. Future studies should determine whether tumor cell dependence on normal cellular functions (Solimini et al., 2007) renders them vulnerable to therapeutic interventions aimed at targeting Hsp90-HSF1 association.
Supplementary Material
Acknowledgments
This research was supported entirely by the Intramural Research Program of the NIH, National Institute on Aging.
Abbreviations
- PDTC
pyrrolidine dithiocarbamate
- HSF1
heat shock factor 1
- HSE
heat shock element
- Hsp
heat shock protein
- BAG3
Bcl2 associated athanogene domain 3
- MEM
Minimum Eagle’s medium
- 17-AAG
17-allylamino-17-demethoxygeldanamycin
- DMAG
17-dimethylaminoethylamino-17-demethoxygeldanamycin
- EGS
ethylene glycol bis(succinimidyl succinate)
- ChIP
chromatin immunoprecipitation
- MBB
maleimidobutyrylbiocytin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Shaoming Song, Email: songs2@mail.nih.gov.
Sutapa Kole, Email: KoleS@grc.nia.nih.gov.
Patricia Precht, Email: prechtp@mail.nih.gov.
Michael J. Pazin, Email: pazinm@mail.nih.gov.
Michel Bernier, Email: Bernierm@mail.nih.gov.
References
- Anckar J, Sistonen L. Heat shock factor 1 as a coordinator of stress and developmental pathways. Adv Exp Med Biol. 2007;594:78–88. doi: 10.1007/978-0-387-39975-1_8. [DOI] [PubMed] [Google Scholar]
- Antoku K, Maser RS, Scully WJ, Jr, Delach SM, Johnson DE. Isolation of Bcl-2 binding proteins that exhibit homology with BAG-1 and suppressor of death domains protein. Biochem Biophys Res Commun. 2002;286:1003–10. doi: 10.1006/bbrc.2001.5512. [DOI] [PubMed] [Google Scholar]
- Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- Chen WY, Chang FR, Huang ZY, Chen JH, Wu YC, Wu CC. Tubocapsenolide A, a novel withanolide, inhibits proliferation and induces apoptosis in MDA-MB-231 cells by thiol oxidation of heat shock proteins. J Biol Chem. 2008;283:17184–93. doi: 10.1074/jbc.M709447200. [DOI] [PubMed] [Google Scholar]
- Chiappetta G, Ammirante M, Basile A, Rosati A, Festa M, Monaco M, et al. The antiapoptotic protein BAG3 is expressed in thyroid carcinomas and modulates apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J Clin Endocrinol Metab. 2007;92:1159–63. doi: 10.1210/jc.2006-1712. [DOI] [PubMed] [Google Scholar]
- Chu F, O'Brian CA. PKC sulfhydryl targeting by disulfiram produces divergent isozymic regulatory responses that accord with the cancer preventive activity of the thiuram disulfide. Antioxid Redox Signal. 2005;7:855–62. doi: 10.1089/ars.2005.7.855. [DOI] [PubMed] [Google Scholar]
- Cvek B, Dvorak Z. Targeting of nuclear factor-kB and proteasome by dithiocarbamate complexes with metals. Curr Pharm Des. 2007;13:3155–67. doi: 10.2174/138161207782110390. [DOI] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–18. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du ZX, Zhang HY, Meng X, Gao YY, Zou RL, Liu BQ, et al. Proteasome inhibitor MG132 induces BAG3 expression through activation of heat shock factor 1. J Cell Physiol. 2009;218:631–7. doi: 10.1002/jcp.21634. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Landmesser U, Harrison DG. Geldanamycin leads to superoxide formation by enzymatic and non-enzymatic redox cycling. Implications for studies of Hsp90 and endothelial cell nitric-oxide synthase. J Biol Chem. 2002;277:25480–5. doi: 10.1074/jbc.M203271200. [DOI] [PubMed] [Google Scholar]
- Franceschelli S, Rosati A, Lerose R, De Nicola S, Turco MC, Pascale M. Bag3 gene expression is regulated by heat shock factor 1. J Cell Physiol. 2008;215:575–7. doi: 10.1002/jcp.21397. [DOI] [PubMed] [Google Scholar]
- Goodman VL, Brewer GJ, Merajver SD. Control of copper status for cancer therapy. Curr Cancer Drug Targets. 2005;5:543–9. doi: 10.2174/156800905774574066. [DOI] [PubMed] [Google Scholar]
- He HJ, Zhu TN, Xie Y, Fan J, Kole S, Saxena S, Bernier M. Pyrrolidine dithiocarbamate inhibits interleukin-6 signaling through impaired STAT3 activation and association with transcriptional coactivators in hepatocytes. J Biol Chem. 2006;281:31369–79. doi: 10.1074/jbc.M603762200. [DOI] [PubMed] [Google Scholar]
- Jacobs AT, Marnett LJ. HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J Biol Chem. 2009;284:9176–83. doi: 10.1074/jbc.M808656200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Han SI, Oh SY, Chung HY, Kim HD, Kang HS. Activation of heat shock factor 1 by pyrrolidine dithiocarbamate is mediated by its activities as pro-oxidant and thiol modulator. Biochem Biophys Res Commun. 2001;281:367–72. doi: 10.1006/bbrc.2001.4376. [DOI] [PubMed] [Google Scholar]
- Liu P, Xu B, Li J, Lu H. BAG3 gene silencing sensitizes leukemic cells to Bortezomib-induced apoptosis. FEBS Lett. 2009;583:401–6. doi: 10.1016/j.febslet.2008.12.032. [DOI] [PubMed] [Google Scholar]
- Long SM, Laubach VE, Tribble CG, Kaza AK, Fiser SM, Cassada DC, et al. Pyrrolidine dithiocarbamate reduces lung reperfusion injury. J Surg Res. 2003;112:12–8. doi: 10.1016/s0022-4804(03)00139-2. [DOI] [PubMed] [Google Scholar]
- Mallick IH, Yang WX, Winslet MC, Seifalian AM. Pyrrolidine dithiocarbamate reduces ischemia-reperfusion injury of the small intestine. World J Gastroenterol. 2005;11:7308–13. doi: 10.3748/wjg.v11.i46.7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Ruiz A, Villanueva L, González de Orduña C, López-Ferrer D, Higueras MA, Tarín C, et al. S-Nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci USA. 2005;102:8525–30. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle A, Broome CS, Kayani AC, Tully MD, Close GL, Vasilaki A, et al. HSF expression in skeletal muscle during myogenesis: implications for failed regeneration in old mice. Exp Gerontol. 2006;41:497–500. doi: 10.1016/j.exger.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Romano MF, Festa M, Petrella A, Rosati A, Pascale M, Bisogni R, et al. BAG3 protein regulates cell survival in childhood acute lymphoblastic leukemia cells. Cancer Biol Ther. 2003;2:508–10. doi: 10.4161/cbt.2.5.524. [DOI] [PubMed] [Google Scholar]
- Rosati A, Ammirante M, Gentilella A, Basile A, Festa M, Pascale M, et al. Apoptosis inhibition in cancer cells: a novel molecular pathway that involves BAG3 protein. Int J Biochem Cell Biol. 2007;39:1337–42. doi: 10.1016/j.biocel.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Schreck R, Meier B, Männel DN, Dröge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med. 1992;175:1181–94. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo YJ, Jeon MH, Lee JH, Lee YJ, Youn HJ, Ko JH, et al. Bis induces growth inhibition and differentiation of HL-60 cells via up-regulation of p27. Exp Mol Med. 2005;37:624–30. doi: 10.1038/emm.2005.76. [DOI] [PubMed] [Google Scholar]
- Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986–8. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Song J, Takeda M, Morimoto RI. Bag1-Hsp70 mediates a physiological stress signalling pathway that regulates Raf-1/ERK and cell growth. Nat Cell Biol. 2001;3:276–82. doi: 10.1038/35060068. [DOI] [PubMed] [Google Scholar]
- Takaki E, Fujimoto M, Sugahara K, Nakahari T, Yonemura S, Tanaka Y, et al. Maintenance of olfactory neurogenesis requires HSF1, a major heat shock transcription factor in mice. J Biol Chem. 2006;281:4931–7. doi: 10.1074/jbc.M506911200. [DOI] [PubMed] [Google Scholar]
- Takayama S, Reed JC. Molecular chaperone targeting and regulation by BAG family proteins. Nat Cell Biol. 2001;3:237–41. doi: 10.1038/ncb1001-e237. [DOI] [PubMed] [Google Scholar]
- Tian XF, Yao JH, Li YH, Gao HF, Wang ZZ, Yang CM, et al. Protective effect of pyrrolidine dithiocarbamate on liver injury induced by intestinal ischemia-reperfusion in rats. Hepatobiliary Pancreat Dis Int. 2006;5:90–5. [PubMed] [Google Scholar]
- Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell. 2004;15:1254–61. doi: 10.1091/mbc.E03-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voellmy R, Boellmann F. Chaperone regulation of the heat shock protein response. Adv Exp Med Biol. 2007;594:89–99. doi: 10.1007/978-0-387-39975-1_9. [DOI] [PubMed] [Google Scholar]
- Wild AC, Mulcahy RT. Pyrrolidine dithiocarbamate up-regulates the expression of the genes encoding the catalytic and regulatory subunits of γ-glutamylcysteine synthetase and increases intracellular glutathione levels. Biochem J. 1999;338:659–65. [PMC free article] [PubMed] [Google Scholar]
- Wurster AL, Pazin MJ. BRG1-mediated chromatin remodeling regulates differentiation and gene expression of T helper cells. Mol Cell Biol. 2008;28:7274–85. doi: 10.1128/MCB.00835-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Kole S, Precht P, Pazin MJ, Bernier M. S-Glutathionylation impairs STAT3 activation and signaling. Endocrinology. 2009;150:1122–31. doi: 10.1210/en.2008-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002;21:5164–72. doi: 10.1093/emboj/cdf528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshima T, Yura T, Yanagi H. The trimerization domain of human heat shock factor 2 is able to interact with nucleoporin p62. Biochem Biophys Res Commun. 1997;240:228–33. doi: 10.1006/bbrc.1997.7662. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Wei EQ, Hu X, Qiao WL, Shi Y, Xu M, et al. The role of nuclear factor-κB in the effect of angiotensin II in the paraventricular nucleus in protecting the gastric mucosa from ischemia-reperfusion injury in rats. J Gastroenterol. 2008;43:687–98. doi: 10.1007/s00535-008-2217-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.