

Abstract
Inherited Long QT Syndrome, a cardiac arrhythmia that predisposes to the often lethal ventricular fibrillation, is commonly linked to mutations in KCNQ1. The KCNQ1 voltage-gated K+ channel α subunit passes ventricular myocyte K+ current that helps bring a timely end to each heart-beat. KCNQ1, like many K+ channel α subunits, is regulated by KCNE β subunits, inherited mutations in which also associate with Long QT Syndrome. KCNQ1 and KCNE mutations are also associated with atrial fibrillation. It has long been known that thyroid status strongly influences cardiac function, and that thyroid dysfunction causes abnormal cardiac structure and rhythm. We recently discovered that KCNQ1 and KCNE2 form a thyroid-stimulating hormone-stimulated K+ channel in the thyroid that is required for normal thyroid hormone biosynthesis. Here, we review this novel genetic link between cardiac and thyroid physiology and pathology, and its potential influence upon future therapeutic strategies in cardiac and thyroid disease.
Keywords: atrial fibrillation, hypothyroidism, hyperthyroidism, KCNE2, KCNQ1, Long QT Syndrome, MiRP1
Introduction
Excitable cells facilitate dynamic processes such as electrical signaling in the brain and rhythmic beating of the heart. Excitability, defined in this context as the ability to sustain action potentials, requires voltage-gated sodium (NaV) channels for the depolarization phase (upstroke) and voltage-gated potassium (KV) channels for the repolarization phase (downstroke) (Figure 1 A). The ventricles of the human heart provide most of the contractile force for pumping blood to the lungs and around the rest of the body, and the integrity of the ventricular myocyte action potential is therefore crucial for life. Influx of Na+ ions through NaV1.5 (encoded by SCN5A) depolarizes human ventricular myocytes, and K+ efflux through a variety of K+ channel types, primarily KV channels, facilitates myocyte repolarization. The most prominent repolarization phase, Phase 3, is coordinated primarily by two KV α subunits: hERG and KCNQ1, which respectively generate the IKr and IKs repolarization currents (Figure 1 B). The culmination of ventricular repolarization manifests on a surface electrocardiogram (ECG) as the end of the T wave (Figure 1 C). KCNQ1 and hERG, as with all other KV α subunits, each contain six transmembrane helices within which is a voltage sensor that moves upon membrane depolarization - a conformational shift transmitted to the channel pore, or gate, which then opens to permit ion flux; four Kv α subunits are necessary and sufficient to form a functional (tetrarmeric) pore. However, in vivo, hERG and KCNQ1 each form complexes with KCNE β subunits, also referred to as MinK-related peptides (MiRPs). KCNE subunits are single-transmembrane-segment (TMS) proteins that do not pass current alone, but co-assemble with pore-forming KV α subunits to regulate their trafficking, gating, conductance, regulation by other proteins, and pharmacology (Figure 2). Importantly, both the KCNE β subunits and the KV α subunits are promiscuous, helping to create K+ current diversity but also hampering efforts to determine molecular correlates of native currents. Thus, the KCNQ1-KCNE1 channel activates more slowly and at more positive membrane potentials than homomeric KCNQ1, and generates the IKs human ventricular repolarization current (with possible contributions from other KCNQ1-KCNE complexes). hERG, the α subunit that generates human ventricular IKr, may be regulated by KCNE1, KCNE2 and potentially other KCNEs in vivo; for review, see (McCrossan and Abbott 2004).
Figure 1. Action potentials and the surface electrocardiogram.
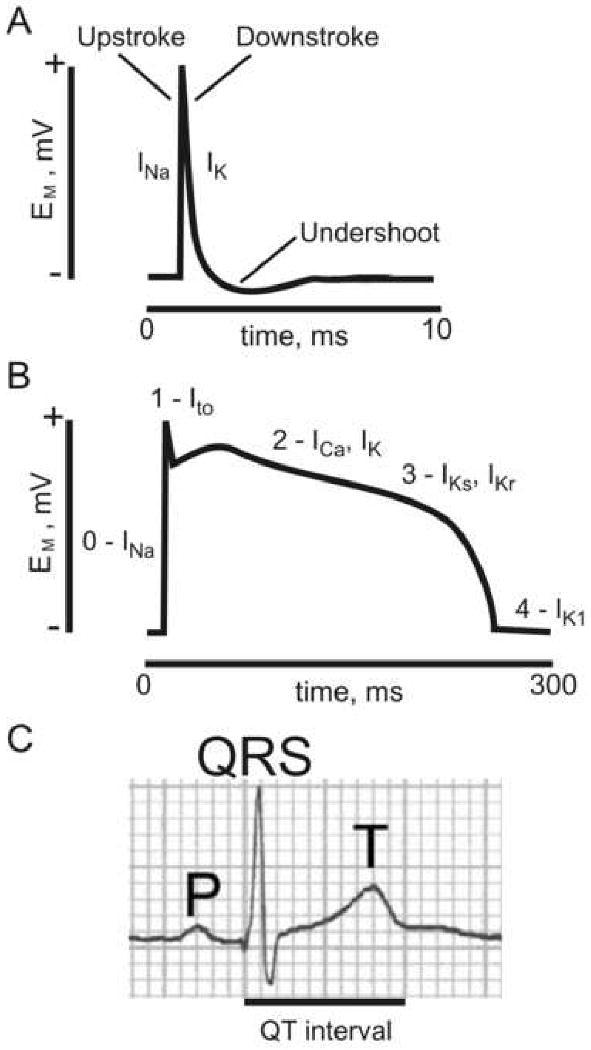
A. An idealized neuronal action potential. INa, sodium current; IK, potassium current; EM, membrane potential.
B. An idealized human ventricular myocyte action potential. Phases 0-4 are indicated, together with prominent currents during these phases: INa, sodium current; Ito, transient outward KV current (generated by KV4.3); ICa, voltage-gated calcium current (generated by CACNA1C, also termed CaV1.2); IK, potassium current (generated by various K+ channels); IKs, slowly activating K+ current (generated by KCNQ1-KCNE1); IKr, rapidly activating K+ current (generated by hERG-KCNE2); IK1, inward rectifier K+ current (generated by Kir2.x subfamily α subunits); EM, membrane potential.
C. A surface electrocardiogram showing the P and T waves, the QRS complex, and the QT interval.
Figure 2. KCNE2 and KCNQ1.
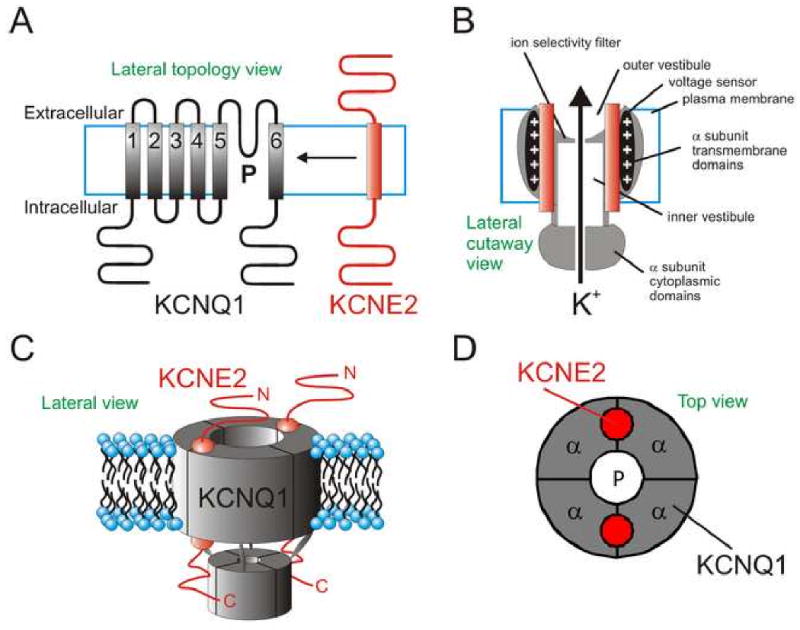
A. Topology of KCNQ1 and KCNE2 with respect to the cell membrane.
B. Idealized cutaway lateral view of a KCNQ1-KCNE2 complex (KCNQ1, grey; KCNE2, red).
C. Cartoon lateral view of a KCNQ1-KCNE2 channel complex, stoichiometry as determined for KCNQ1-KCNE1 (Chen, Kim et al. 2003).
D. Cartoon top view of a KCNQ1-KCNE2 complex suggesting a possible juxtaposition of the two subunit types.
KCNQ1 has a property unique among KV α subunits: it can be converted to a constitutively open K+ leak channel (i.e., one that does not require membrane depolarization to open) by co-assembly with the KCNE2 or KCNE3 ancillary subunits (Schroeder, Waldegger et al. 2000; Tinel, Diochot et al. 2000). While it is not yet known whether KCNQ1 forms leak channels in human heart with KCNE2 or KCNE3, the ability to open constitutively has been shown to facilitate functional roles for KCNQ1-KCNE complexes in non-excitable, polarized epithelia in vivo. In gastric parietal cells, KCNQ1-KCNE2 channels provide an apical K+ recycling pathway required for gastric acidification by the apical gastric H+/K+ATPase (Lee, Ravenel et al. 2000; Heitzmann, Grahammer et al. 2004; Roepke, Anantharam et al. 2006). In the colon, basolateral KCNQ1-KCNE3 channels help provide a driving force for cAMP-stimulated Cl- secretion (Schroeder, Waldegger et al. 2000). Recently, we also discovered that KCNQ1 and KCNE2 form a constitutively-active K+ channel in thyrocytes, and that the KCNQ1-KCNE2 channel is required for normal thyroid hormone (TH) biosynthesis (Roepke, King et al. 2009). Because the heart is strongly influenced by the thyroid, the finding suggests that these two K+ channel subunits could influence cardiac function both directly, due to their roles in cardiac myocytes, and indirectly, by virtue of their role in thyroid physiology.
Pathogenesis
Mutations in human KCNQ1 and KCNE2 are associated with cardiac arrhythmias, thought to be primarily due to disruption of their function in the heart (Wang, Curran et al. 1996; Abbott, Sesti et al. 1999). KCNQ1 mutations underlie Long QT Syndrome (LQTS) Type 1 (LQT1), sub-classified as Romano Ward Syndrome (RWS; autosomal dominant) and Jervell Lange-Nielsen Syndrome (JLNS; typically autosomal recessive). Loss-of-function mutations in KCNQ1 reduce ventricular repolarization capacity, and therefore extend the QT interval on the surface electrocardiogram, hence LQTS. JLNS manifests as both LQTS and profound sensorineural deafness – because KCNQ1-KCNE1 channels are important not only for ventricular repolarization, but also for K+ secretion into the endolymph of the inner ear. Accordingly, loss-of-function KCNE1 mutations, which underlie LQT5, also cause either RWS or JLNS; for review, see (Keating and Sanguinetti 2001). Interestingly, gain-of-function mutations in KCNQ1 (and perhaps in some KCNE genes) appear to underlie some cases of lone atrial fibrillation (AF), probably because they shorten the atrial effective refractory period (Chen, Xu et al. 2003; Yang, Xia et al. 2004).
We originally identified KCNE2 as a partner for hERG (KCNH2) and found that KCNE2 mutations associate with inherited and drug-induced LQTS, a cardiac arrhythmia that predisposes to the often lethal ventricular fibrillation. KCNE2-associated LQTS is classified LQT6. The presumed mechanism is disruption of ventricular myocyte hERG-KCNE2 channels (Abbott, Sesti et al. 1999). Despite initial controversy surrounding these findings, KCNE2 has also since been found by others to regulate IKr in vivo in canine ventricles (Jiang, Zhang et al. 2004) and mouse sinoatrial node (Hesketh, Qi et al. 2005). We also found that 3-month-old Kcne2-/- mice (which are euthyroid at this age, when bred from Kcne2+/- dams) have impaired ventricular myocyte repolarization due to loss of KCNE2 from myocyte channel complexes with the KV4.2 and KV1.5 α subunits (Roepke, Kontogeorgis et al. 2008). After these cardiac roles for KCNE2 were identified, we discovered that breeding of Kcne2-/- dams results in hypothyroidism in both the pregnant dams and their pups, due to a previously unreported role for the KCNQ1-KCNE2 K+ channel in thyrocytes (Roepke, King et al. 2009). By 3 weeks of age, Kcne2-/- mice born to Kcne2-/- dams show marked cardiac abnormalities including cardiomegaly, hypertrophy and impaired contractility. These mice also exhibit alopecia, defective skeletal development, 50% embryonic lethality, and delayed growth resulting in dwarfism (Roepke, King et al. 2009); these are all symptoms observed in human congenital hypothyroidism (Lafranchi 2010). While Kcne2-/- pups born to Kcne2+/- dams appear to develop normally, and by three months are euthyroid (indicating a strong influence of maternal genotype on phenotype), they exhibit latent hypothyroidism with cardiomegaly, alopecia, diminished T4 and elevated TSH levels by 1 year of age (Roepke, King et al. 2009) (Figure 3 A-D). As previously observed in hypothyroid rats (Hapon, Simoncini et al. 2003), Kcne2-/- dams have impaired milk ejection - alleviated by oxytocin injection - partially explaining the influence of maternal genotype in Kcne2-/- mouse phenotype severity (Roepke, King et al. 2009).
Figure 3. Manifestations of thyroid pathology in the Kcne2-/- mouse.
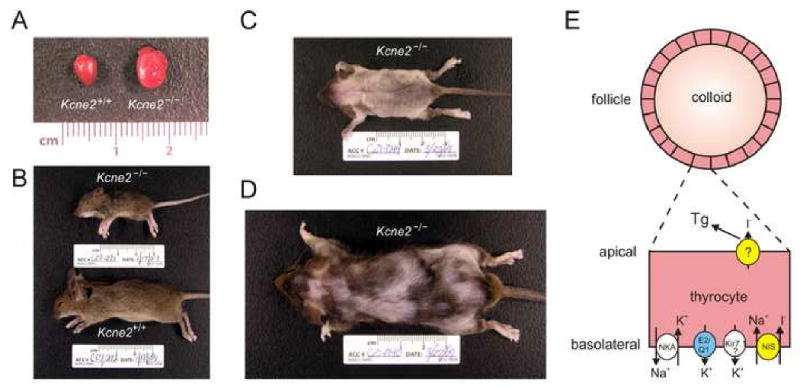
Kcne2-/- mice have a complex pathophysiology. The primary hypothyroidism due to dysfunction of thyrocyte KCNQ1-KCNE2 causes, among other defects:
A. Cardiomegaly; (B) retarded growth; and alopecia in (C) Kcne2-/- pups from Kcne2-/- dams, and (D) 1-year-old Kcne2-/- mice originally bred from Kcne2+/- dams.
E. The putative role for KCNQ1-KCNE2 (blue) in thyrocytes is to facilitate K+ efflux basolaterally. A subset of other channels and transporters are shown for context: Kir7.1, inward rectifier K+ channel; NKA, Na+/K+/ATPase; NIS, Na+/I- symporter (yellow). At the apical membrane, I- passes from the thyrocyte to the colloid for organification, through pendrin or another, unspecified protein (yellow), resulting in formation of thyroglobulin (Tg).
The thyroid hormones (THs), thyroxine (T4) and triiodothyronine (T3), are crucial for proper growth and development, and play an integral role throughout adulthood in maintenance of cognitive function, metabolism, and cardiac function. As iodide (I-) is an essential component of TH, the scarcity of dietary I- is a universal health concern monitored by the WHO, UNICEF, and the International Council for Control of Iodine Deficiency (ICCID) (Zimmermann, Jooste et al. 2008). I- deficiency manifests most severely during pregnancy, when the demand for TH is increased as the early fetus is unable to synthesize TH on its own and must rely on a supply of maternal T4 for proper development. Inability to supply the growing fetus with T4 can result in fetal mortality, defective neuronal myelination and synapse formation, retarded growth and a form of mental retardation known as cretinism (Zimmermann, Jooste et al. 2008). Though severe, defects resulting from hypothyroidism are highly preventable with proper intervention. However, controversies surround research aimed at determining the population at risk, effective treatment dosage, and the crucial point in gestation at which to implement treatment, necessitating further study to elucidate the etiology of hypothyroidism, and safe, effective means of its prevention (Loh, Wartofsky et al. 2009). The Na+/I- symporter (NIS) is responsible for the accumulation of I- in thyrocytes in the first step of TH biosynthesis. I- is then transported apically into the colloid in the thyroid lumen where it is organified into thyroglobin (Tg) and subsequently incorporated into T3 and T4. NIS uses the movement of Na+ down its concentration gradient to accumulate I- into thyrocytes. While it has previously been established that the Na+/K+ATPase, which co-localizes with NIS at the basolateral membrane, generates this gradient by pumping Na+ out in exchange for moving K+ in (Dohan, De la Vieja et al. 2003), the pathway responsible for moving K+ back out of the cell has remained enigmatic. Our recent findings show that KCNE2, probably primarily in complexes with KCNQ1, is required for normal I- accumulation in the thyroid, suggesting that the KCNQ1-KCNE2 channel may form the aforementioned thyrocyte K+ efflux pathway (Figure 3 E).
Therapy
Despite there being a long-recognized link between thyroid dysfunction and cardiovascular risk, and an awareness that THs regulate expression of K+ channels in the heart (Klein and Ojamaa 2001) (Tribulova, Knezl et al. 2010), our recent discovery of a crucial role for KCNE2 and KCNQ1 in TH biosynthesis presents a novel and unexpected genetic link between thyroid dysfunction and cardiac arrhythmias. Mutations in KCNE2 and/or KCNQ1 have previously been associated with LQTS, AF, and even early-onset myocardial infarction (Wang, Curran et al. 1996; Abbott, Sesti et al. 1999; Chen, Xu et al. 2003; Yang, Xia et al. 2004; Kathiresan, Voight et al. 2009), each of which is also predisposed to by thyroid dysfunction in the general population (Forfar, Miller et al. 1979; Hak, Pols et al. 2000; Bakiner, Ertorer et al. 2008), suggesting the intriguing possibility of an endocrine component to some KCNE2- and KCNQ1-associated human cardiac disease. Whether or not the discovery of KCNQ1-KCNE2 in the thyroid and its role in TH biosynthesis leads to use of KCNQ1-KCNE2 modulators to treat thyroid dysfunction remains to be seen, but these findings should at least be a consideration in future studies of thyroid-related cardiac disease, its molecular etiology and therapy.
It is noteworthy that subclinical hypothyroidism is associated with prolongation of the QTc (QT interval corrected for heart rate) (Tribulova, Knezl et al. 2010), especially given that loss-of-function mutations in KCNQ1 are the joint most commonly identified cause of inherited LQTS (Keating and Sanguinetti 2001), and would also be predicted to impair TH biosynthesis by loss of function of thyroid KCNQ1-KCNE2 channels, as is achieved by genetic disruption of Kcne2 (Roepke, King et al. 2009). Conversely, ∼10% of AF patients show biochemical evidence of hyperthyroidism, and in two-thirds of patients diagnosed with both idiopathic AF and hyperthyroidism, successful therapeutic conversion to a euthyroid state is accompanied by a return to normal sinus rhythm (Klein and Ojamaa 2001). One genetic cause of AF is gain of function of KCNQ1, which shortens the atrial effective refractory period, predisposing to AF (Chen, Xu et al. 2003). This raises the interesting question of whether increased KCNQ1 function in the thyroid might actually increase TH production, something which can initially be tested in mouse models, using existing KCNQ1 openers such as the benzodiazepine RL-3.
In summary, the KCNQ1-KCNE2 K+ channel appears important for normal TH biosynthesis in mice, by an as yet incompletely-defined mechanism. KCNQ1 and KCNE2 are also expressed in human thyroid but we do not yet know their function there, or the consequences of their disruption. Disruption of thyrocyte KCNQ1-KCNE2 in mice is most deleterious to health in pregnant and lactating dams and their offspring, and in older mice. A possible role for thyroid dysfunction in KCNQ1- and KCNE2-associated cardiac arrhythmias and myocardial infarction warrants consideration in future anti-arrhythmic regimes involving pharmacologic modulation of KCNQ1.
Acknowledgments
G.W.A. is supported by the National Heart, Lung and Blood Institute, National Institutes of Health (R01 HL079275; R01HL101190), the American Heart Association (0855756D), and an Irma T. Hirschl Career Scientist Award. K.P. is supported by a National Institutes of Health Predoctoral Training Grant (T32GM073546).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott GW, Sesti F, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97(2):175–87. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Bakiner O, Ertorer ME, et al. Subclinical hypothyroidism is characterized by increased QT interval dispersion among women. Med Princ Pract. 2008;17(5):390–4. doi: 10.1159/000141503. [DOI] [PubMed] [Google Scholar]
- Chen H, Kim LA, et al. Charybdotoxin binding in the I(Ks) pore demonstrates two MinK subunits in each channel complex. Neuron. 2003;40(1):15–23. doi: 10.1016/s0896-6273(03)00570-1. [DOI] [PubMed] [Google Scholar]
- Chen YH, Xu SJ, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299(5604):251–4. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- Dohan O, De la Vieja A, et al. The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr Rev. 2003;24(1):48–77. doi: 10.1210/er.2001-0029. [DOI] [PubMed] [Google Scholar]
- Forfar JC, Miller HC, et al. Occult thyrotoxicosis: a correctable cause of “idiopathic” atrial fibrillation. Am J Cardiol. 1979;44(1):9–12. doi: 10.1016/0002-9149(79)90243-1. [DOI] [PubMed] [Google Scholar]
- Hak AE, Pols HA, et al. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam Study. Ann Intern Med. 2000;132(4):270–8. doi: 10.7326/0003-4819-132-4-200002150-00004. [DOI] [PubMed] [Google Scholar]
- Hapon MB, Simoncini M, et al. Effect of hypothyroidism on hormone profiles in virgin, pregnant and lactating rats, and on lactation. Reproduction. 2003;126(3):371–82. doi: 10.1530/rep.0.1260371. [DOI] [PubMed] [Google Scholar]
- Heitzmann D, Grahammer F, et al. Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J Physiol. 2004;561(Pt 2):547–57. doi: 10.1113/jphysiol.2004.075168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh JC, Qi X, et al. Cardiac function of KCNE2 subunits revealed by targeted deletion in mice. American Heart Assocation Scientific Sessions. 2005 Session Number AOP.16.2a(Presentation Number 278) [Google Scholar]
- Jiang M, Zhang M, et al. KCNE2 protein is expressed in ventricles of different species, and changes in its expression contribute to electrical remodeling in diseased hearts. Circulation. 2004;109(14):1783–8. doi: 10.1161/01.CIR.0000124225.43852.50. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Voight BF, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41(3):334–41. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104(4):569–80. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344(7):501–9. doi: 10.1056/NEJM200102153440707. [DOI] [PubMed] [Google Scholar]
- Lafranchi SF. Newborn screening strategies for congenital hypothyroidism: an update. J Inherit Metab Dis. 2010 doi: 10.1007/s10545-010-9062-1. epub ahead of print(March 2nd) [DOI] [PubMed] [Google Scholar]
- Lee MP, Ravenel JD, et al. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest. 2000;106(12):1447–55. doi: 10.1172/JCI10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh JA, Wartofsky L, et al. The magnitude of increased levothyroxine requirements in hypothyroid pregnant women depends upon the etiology of the hypothyroidism. Thyroid. 2009;19(3):269–75. doi: 10.1089/thy.2008.0413. [DOI] [PubMed] [Google Scholar]
- McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology. 2004;47(6):787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Roepke TK, Anantharam A, et al. The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J Biol Chem. 2006;281(33):23740–7. doi: 10.1074/jbc.M604155200. [DOI] [PubMed] [Google Scholar]
- Roepke TK, King EC, et al. Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat Med. 2009;15(10):1186–94. doi: 10.1038/nm.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TK, Kontogeorgis A, et al. Targeted deletion of kcne2 impairs ventricular repolarization via disruption of I(K,slow1) and I(to,f) Faseb J. 2008;22(10):3648–60. doi: 10.1096/fj.08-110171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Waldegger S, et al. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403(6766):196–9. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- Tinel N, Diochot S, et al. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. Embo J. 2000;19(23):6326–30. doi: 10.1093/emboj/19.23.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribulova N, Knezl V, et al. Thyroid hormones and cardiac arrhythmias. Vascul Pharmacol. 2010;52(3-4):102–12. doi: 10.1016/j.vph.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Yang Y, Xia M, et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75(5):899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann MB, Jooste PL, et al. Iodine-deficiency disorders. Lancet. 2008;372(9645):1251–62. doi: 10.1016/S0140-6736(08)61005-3. [DOI] [PubMed] [Google Scholar]