

Abstract
The ability of our cells to maintain genomic integrity is fundamental for protection from cancer development. Central to this process is the ability of cells to recognize and repair DNA damage and progress through the cell cycle in a regulated and orderly manner. In addition, protection of chromosome ends through the proper assembly of telomeres prevents loss of genetic information and aberrant chromosome fusions. Cells derived from patients with ataxia-telangiectasia (A-T) show defects in cell cycle regulation, abnormal responses to DNA breakage, and chromosomal end-to-end fusions. The identification and characterization of the ATM (ataxia-telangiectasia, mutated) gene product has provided an essential tool for researchers in elucidating cellular mechanisms involved in cell cycle control, DNA repair, and chromosomal stability.
Keywords: ATM, DNA damage and repair, ionizing radiation, chromosomal breakage and instability, cancer
INTRODUCTION
The integrity of our genome is constantly under attack from both exogenous and endogenous sources. Many thousands of DNA damaging events occur each day in every cell in our bodies [1], placing DNA damage response mechanisms in a paramount position of importance. The ability of a cell to correctly respond to and repair DNA damage and maintain genomic stability is critical for protection from cancer development. This dependence is particularly evident in human genetic disorders such as ataxia-telangiectasia (A-T), Li-Fraumeni, Nijmegen breakage syndrome (NBS), A-T-like disorder (ATLD), BRCA1/2 familial breast/ovarian cancer syndromes, and others where inherited mutations in the cellular machinery that helps cells respond to DNA damage result in a profound predisposition to cancer development[1]. This review will focus on the cancer predisposition syndrome ataxia-telangiectasia (A-T), and the gene product mutated in the disorder, ataxia-telangiectasia, mutated (ATM), which plays a central role in orchestrating molecular events involved in DNA double-strand break signaling and repair.
A-T is a rare, autosomal recessive disorder characterized by progressive cerebellar ataxia, neuro-degeneration, radiosensitivity, cell-cycle checkpoint defects, genome instability, and a predisposition to cancer[2–4]. Mutations in the ATM gene generally result in an absence of full-length, functional protein product. The gene responsible for the A-T phenotype was first cloned by Yosef Shiloh and colleagues and named ATM for A-T, mutated[5]. The human ATM gene is located at 11q22–23 and covers 160kb of genomic DNA; the gene product, ATM protein, is produced from a 13kb transcript that codes for a predicted 315-kDa protein that migrates at approximately 370kDa in SDS-PAGE.
The ATM protein is a serine/threonine protein kinase and a member of the phosphoinositide 3-kinase-related protein kinase (PIKK) family. All members of the PIKK family are large serine/threonine protein kinases involved in signaling following cellular stress. The ATM consensus phosphorylation motif is hydrophobic-X-hydrophobic-[S/T]-Q[6]. The other members of the PIKK family include ATR (ATM and Rad3 related protein kinase), DNA-PKcs (DNA dependent protein kinase catalytic subunit), mTOR (mammalian target of rapamycin), and hSMG1. These members all share common domain structures including N-terminal HEAT repeats, a FAT domain, a protein kinase domain, and a C-Terminal FAT-C domain (Figure 1).
Figure 1. PI3KK family members.
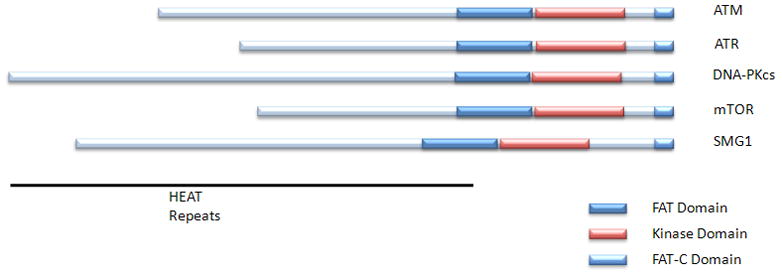
The PIKK family members have a C-terminal protein kinase doman flanked on either side by an N-terminal FAT domain and a C-terminal FAT-C domain. The N-termini are largely composed of HEAT repeats.
Another common feature of the PIKK family members is their association with proteins or protein complexes which facilitate their activation and function (reviewed in [7]). ATM binds to the MRN (Mre11, Rad50, NBS1) heterotrimer at sites of double strand breaks, allowing for ATM retention on chromatin and efficient activation of downstream signaling events and completion of DNA repair[8]. The roles of MRN in ATM activation and ATM dependent DNA damage signaling are discussed below. Germline hypomorphic mutations in mre11 and nbs1 lead to the disorders A-T-like disorder (ATLD) and Nijmegen breakage syndrome (NBS), disorders with similar phenotypes to A-T[1].
The genetic disorder A-T was first described over 50 years ago[2]. Prior to identification of the gene responsible for the A-T phenotype, many studies were carried out on cells derived from A-T patients leading to the elucidation of many cellular phenotypes. One of the earliest cellular phenotypes discovered was a defect in cell cycle control. This was first reported as a reduced inhibition of DNA synthesis during S-phase of the cell cycle following ionizing radiation and was termed “radioresistant DNA synthesis”[9]. A phenotype of radiosensitivity (increased cell death following exposure to ionizing radiation) was first noted when an A-T patient with cancer had a toxic reaction to radiation therapy and was later shown to be a characteristic feature of cells derived from A-T patients[10]. Radioresistant DNA synthesis and radiosensitivity are still considered fundamental identifying characteristics of the disease.
ATM Function I – Cell Cycle Control
The cell cycle is divided into 4 sequential phases: G1, S, G2, and M. It is important that cellular regulation for progression through each of these phases be functioning correctly in order to maintain genomic integrity. Cell cycle checkpoints are pauses in cell cycle progression that allow the cell time to deal with physiologic signals or challenges, such as DNA damage. One of the most prominent control points in the cell cycle is the entry into S-phase from G1, referred to here as the G1/S checkpoint. The tumor suppressor p53 was shown to play a critical role in the G1/S checkpoint following irradiation[11] and efficient induction of p53 after ionizing radiation requires ATM[12]. Once ATM was shown to be a protein kinase[13], both the p53 protein and proteins that interact with p53, MDM2 and Chk2, were found to be phosphorylated by ATM. The ATM-dependent induction of p53 allows p53 to trans-activate target genes, particularly the cyclin dependent kinase (CDK) inhibitor p21, induction of which results in the inhibition of the Cyclin-E/CDK2 complex and inhibition of progression from G1 into S-phase[3].
Since p53 is not required for intra S-phase arrest after irradiation, but cells lacking ATM are defective in this checkpoint, it was clear that the ATM kinase had to have other targets. The first ATM target shown to play a role in the radiation-induced intra S-phase checkpoint was the NBS1 protein, part of the MRN complex[14]. ATM mediated S-phase arrest is also dependent on signaling through the BRCA1 (Breast Cancer Associated 1) gene. Interestingly ATM was found to phosphorylate BRCA1 on multiple sites, and these different phosphorylation events elicited different effects on cell cycle progression. Phosphorylation on serine 1387 is necessary for proper S-phase arrest following ionizing radiation[15], while, phosphorylation on serine 1423 is necessary for the ATM mediated G2/M arrest[16]. Phosphorylation of SMC1 and FANCD2 proteins were also shown to be important for IR induced S-phase arrest[17;18]. However, it is not yet clear how any of these post-translational modifications affect DNA replication. In contrast, the ATM target CHK2 (cell cycle checkpoint kinase 2) does signal to the cell cycle machinery. Following the induction of DNA double strand breaks in S-phase, ATM activates CHK2 by phosphorylation on threonine 68 (T68). The activation of CHK2 leads to the phosphorylation of phosphatase CDC25A, resulting in its degradation and the inability to load Cdc45 on to replication origins necessary for the initiation of DNA replication. The Cyclin-E/CDK2 complex is also a target of CDC25A. The decrease in CDC25A following DNA damage results in an increase phosphorylation level of CDK2 resulting in degradation of CDK2 and thus delayed progression through S-phase[19], in addition to the delayed S-phase entry mentioned above.
The CHK2-mediated inhibition of the CDC25 family of phosphatases is also important for the activation of the G2/M checkpoint. In addition to the inhibition of the CDC25 family members, phosphorylation of Rad17 is critical to the induction of the G2/M checkpoints following exposure to ionizing radiation[20]. Taken together with new evidence suggesting that ATM interacts with the DNA repair enzyme Artemis[21] specifically in G2 to promote DNA repair[22], there is mounting evidence for the importance of ATM signaling during all cell cycle phases in coordinating optimal cellular responses to DNA damage.
ATM Function II – DNA repair
The linkage of specific irradiation-induced, ATM-dependent phosphorylation events to specific cell cycle checkpoints did not account for the increased radiosensitivity of cells lacking ATM [15;16]. Those observations suggested the possibility that ATM activity may also be required for optimal repair of DNA double strand breaks, the most cytotoxic lesion caused by ionizing radiation. Even prior to identification of the ATM gene, increases in ionizing radiation-induced chromosomal breakage were a reported phenotype of A-T cells[23]. However, despite the increased chromosomal breakage, no definitive defects in DNA double strand break repair kinetics or fidelity could be demonstrated.
Recently, with the advent of more sensitive techniques by which to measure repair, increasing evidence has accumulated suggesting that ATM activity has an impact on DNA double strand break repair. One such technique is the assessment of γH2AX foci disappearance. Following ionizing radiation, the histone variant H2AX becomes phosphorylated, generating γH2AX[24], in chromatin regions directly surrounding DNA strand breaks. This phosphorylation event can be visualized by immunofluorescence as foci immediately after exposure to ionizing radiation and can be used as a marker for DNA breaks[25]. It is important to note that disappearance of γH2AX foci is not a direct measure of repair (re-ligation of broken DNA ends), but rather probably represents the restoration of the local chromatin environment following DNA repair. γH2AX foci can clearly be dissociated from DNA double strand break repair since treatment of cells with phosphatase inhibitors can prolong the existence of γH2AX foci with only having a small effect on completion of repair[26]. Nevertheless, the disappearance of γH2AX foci under most conditions seems to correlate with completed DNA repair[25;27]. Cells lacking ATM show persistent γH2AX foci following ionizing radiation suggesting a direct role for ATM in DNA repair[21]. Further, it is suggested that these persistent foci are located in heterochromatin possibly providing a role for ATM in repair of a specific subset of DNA double strand breaks[28]. Supporting a role for ATM in heterochromatic DNA double strand break repair is a recent finding identifying KAP1 (KRAB-associated protein 1), a core component of heterochromatin, as a direct target of the ATM kinase[29]. Phosphorylation of KAP1 by ATM results in a relaxation of heterochromatin and may facilitate repair by providing access to the damaged DNA. Moreover, siRNA knockdown of KAP1 alleviates the heterochromatin repair defect found when ATM kinase activity is inhibited.
Since disappearance of γH2AX foci is neither a direct nor perfect assessment of DNA repair, it is desirable to have other approaches that directly measure re-ligation of DNA ends. A method which has successfully been used in the budding yeast S. cerevisiae is directly monitoring DNA re-ligation by PCR following induced expression of the sequence-specific HO endonuclease, which creates a DNA double strand break at a defined site in the genome[30;31]. A similar approach, modeled on the HO endonuclease system from yeast, was recently developed in mammalian cells using the sequence specific endonuclease I-PpoI to induce DNA double strand breaks at defined sites in the human genome, thus permitting monitored recruitment of proteins to sites of DNA double strand breaks as well as direct assessments of the repair of those DNA breaks[32;33]. These studies demonstrated a requirement for ATM kinase activity (as well as functional NBS1) for the disruption of nucleosomes surrounding a DNA double strand break. Cells lacking ATM also exhibited an increased accumulation of breaks following induction of I-PpoI compared to wild-type. While I-PpoI expression was continuous in these studies, the increased accumulation of breaks over wild-type suggests that cells lacking ATM have a defect in DNA double strand break repair. These results in combination with those found monitoring γH2AX provide evidence that ATM activity is necessary for effective DNA double strand break repair for at least a subset of DNA breaks.
ATM activation
Initial studies demonstrating that ATM was a protein kinase also showed that DNA damage caused an increase in its cellular activity [13;34]. However, the mechanisms underlying this increase in ATM activity were obscure. A key step in elucidating the mechanism by which ATM becomes active was achieved by the identification of the ATM autophosphorylation site serine 1981 (S1981). ATM exists in an inactive dimer in undamaged cells, but following induction of DNA damage, or treatment with agents that alter chromatin structure, ATM undergoes an intermolecular autophosphorylation on S1981 resulting in disassociation of the dimer into active monomers[35]. Since the initial discovery of S1981 autophosphorylation, other ATM post-translational modifications have been reported, including additional phosphorylation sites important for the complete activation of ATM downstream signaling pathways[36]. In addition, recent studies have suggested roles for one or more phosphatases in the activation or maintenance of ATM. Indeed, PP2A (protein phosphatase 2A)[37] and the WIP1 phosphatase [38] interact with ATM and modulate the phosphorylation levels of ATM and its downstream targets. In addition to phosphorylation, the histone acetyl-transferase, TIP60, was reported to acetylate ATM on Lysine 3016 (K3016) in parallel to phosphorylation on S1981 and this modification was suggested to be important for ATM activation[39].
The importance of ATM auto-phosphorylation has recently been questioned based on a BAC-transgenic mouse model where these ATM phosphorylation sites were mutated and ATM function was not detectably altered in the mouse [40;41]. Cells from these mice exhibited normal DNA damage responses (phosphorylation of p53, CHK2, and SMC1) and exhibited no evidence of increased radiosensitivity or cell cycle checkpoint abnormalities. Though human ATM protein lacking the S1981 autophosphorylation site (ATM S1981A) was not detected at sites of DNA damage induced by the I-PpoI endonuclease [32], the corresponding mouse mutant ATM protein, S1987A, did appear to accumulate at sites of UV-laser-induced damage [40;41]. These discrepancies could either result from: 1) fundamental differences between ATM activation mechanisms in mouse versus human cells; 2) from BAC reconstituted mice behaving differently than more conventional knock-in mice; or 3) differences in types of DNA damage used. Related to the second possibility, it is noted that over-expression of ATM (both wild-type and S1981A) in human cells leads to the production of active ATM monomers in the absence of DNA damage and BAC over-expression in the murine system may be a contributing factor to some of these discrepancies. Recently a study further exploring the role of S1981 phosphorylation and ATM activation in human cells reported that ATM S1981A could be recruited to breaks immediately following damage, but that the retention of ATM at breaks was defective when this phosphorylation site was lost, resulting in an inability to maintain normal levels of ATM-dependent DNA damage response pathway activation[42]. This study went on to demonstrate that the phosphorylation of S1981 was important for the interaction between ATM and MDC1 and this interaction mediated ATM retention at sites of DNA double strand breaks. If ATM S1981A is indeed recruited to break sites with similar kinetics as wild-type, but not retained, this provides a potential explanation for some of the biochemical differences reported between human ATM S1981A and mouse ATM S1987A studies. It remains unclear why the BAC transgenic S1987A knock-in mice had no discernible phenotypic abnormalities since lack of ATM retention at sites of DNA damage should have physiologic implications.
ATM dependent DNA damage response
Following DNA damage, ATM is rapidly recruited to sites of DNA double strand breaks. This rapid recruitment is visualized by focus formation assays using fluorescence microscopy and by directly measuring recruitment to DNA using chromatin immunoprecipitation (ChIP) assays. In addition to ATM, the MRN complex is also rapidly recruited to sites of DNA double strand breaks. The MRN complex is a sensor of DNA damage and does not require ATM for localization to DNA double strand breaks[43]. Studies done in cells lacking MRE11 or NBS1 demonstrated a requirement for the MRN complex in the full activation of the ATM dependent DNA damage response[18;44]. Further the MRN complex is a substrate for ATM[14,45]. Recently, it was reported that Mre11 activity at the sites of DNA double strand breaks produces small DNA fragments that can stimulate ATM activation[46]. ATM also interacts directly with MRN through the C-terminus of NBS1[8]. These studies demonstrate that ATM and MRN work in concert with one another at the sites of DNA double strand breaks to promote an effective DNA damage response and repair.
Immediately following recruitment of ATM to chromatin following DNA damage, ATM contributes to the phosphorylation of the histone variant H2AX, producing γH2AX, on Serine 139[47]. The adapter protein MDC1, which binds to γH2AX via its BRCT repeats, is then recruited to the DNA double strand break and phosphorylated by ATM[48;49]. Interestingly, MDC1 also appears to be important for the retention, but not the initial recruitment, of ATM to sites of DNA double strand breaks[42]. It is thought that the formation of γH2AX and the phosphorylation of MDC1 at the sites of DNA damage provide a docking station for many components of the DNA damage repair and signaling pathways. Proteins such as the RING-finger ubiquitin ligases RNF8 and RNF168 are recruited by binding of phosphorylated MDC1[50–53]. In turn, the ubiquitination of γH2AX by RNF8 stabilizes the recruitment of p53 binding protein 1 (53BP1) and BRCA1, both of which are also phosphorylated by ATM[54]. Thus the ATM mediated phosphorylation events that occur at the DNA double strand break are critical in orchestrating the repair and chromatin-modifying machinery surrounding the DNA double strand break (Figure 2).
Figure 2. Recruitment of DNA damage response proteins to a DNA double strand break.
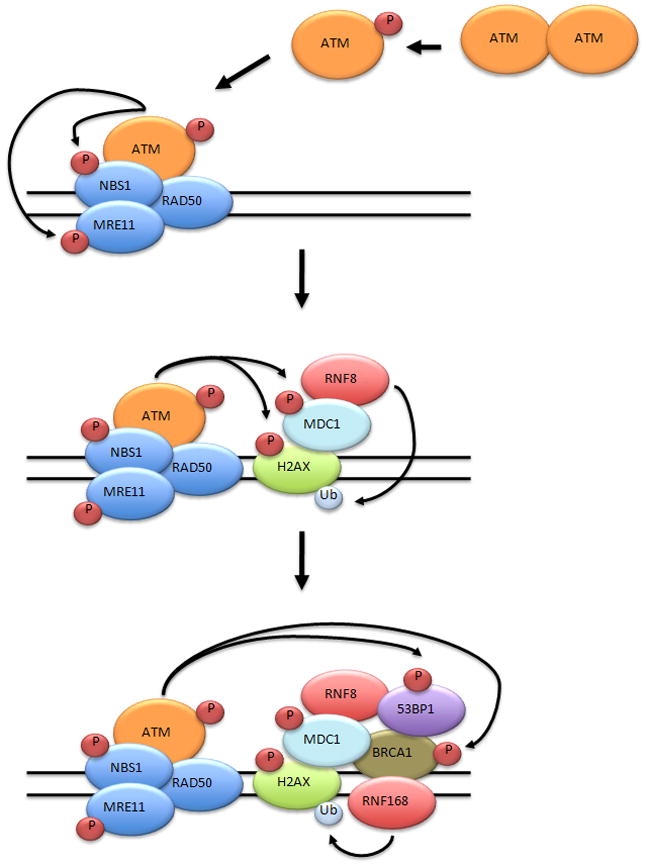
Prior to DNA damage ATM exists as an inactive dimer. Following the induction of a DNA double strand break, ATM undergoes autophosphorylation producing active ATM monomers. ATM and MRN are rapidly recruited to the site of the DNA double strand break. Upon recruitment, ATM phosphorylates MRE11, NBS1, and H2AX. The phosphorylation of H2AX leads to the recuitment of MDC1. MDC1 is phosphorylated by ATM and phosphorylated MDC1 serves as a docking site recruiting the RING-finger ubiquitin ligase RNF8. RNF8 mono-ubiquitinates γH2AX resulting in the recruitment of 53BP1, BRCA1, and RNF168. The RING-finger ubiquitin ligase RNF168 maintains the ubiquitinated status of γH2AX, aiding in the stabilization 53BP1 and BRCA1 at the break site.
ATM and telomeres
Unlike many prokaryotes, the human genome is not organized into a circular structure, but rather linear chromosomes. Each chromosome has two ends capped by telomeres. An early observation in cells isolated from A-T patients was the presence of chromosomal end-to-end fusions[55], implying a potential role for ATM function at telomeres. Telomeres are a complex structure involving DNA, RNA, and proteins designed to mask the free end of a chromosome from being recognized by the cell as the free end of a double strand break. This telomeric protection complex, known as Shelterin, is composed of multiple proteins (TIN2, TRF1, TRF2, TPP1, POT1, RAP1). It binds to the telomeric sequence and forms a t-loop structure, now known to be essential in the protection of telomeres and for their ability to elude the DNA damage sensing machinery[56].
Depletion of TRF2, or its inhibition through the use of a dominate-negative TRF2 allele, disrupts the Shelterin complex resulting in the formation of un-capped telomeres and the activation of an ATM-dependent DNA damage response[57;58]. The un-capped telomeres result in the recruitment of DNA damage response proteins and the generation of telomere dysfunction-induced foci, TIFs[56]. In addition to TRF2 depletion resulting in ATM activation, TRF2 can bind ATM and over-expression of TRF2 can inhibit the ATM dependent DNA damage response following ionizing irradiation[57]. The interaction between ATM and TRF2 may be essential for preventing telomeres from being recognized as DNA damage since TRF2 allows for ATM to be inhibited locally at the telomeres. Recently it has been shown that TRF2 also interacts with the MRN complex[59;60]. Mre11 nuclease activity has also been shown to play a role in the processing of telomeric sequences suggesting a role for MRN in the joining of uncapped telomeric sequences, and may play a role in normal telomere function[61].
These findings illustrate an important role for proper telomere assembly in preventing chromosome ends from being detected as DNA damage and activating the ATM-dependent DNA damage response. However, they do not explain the possible role for ATM at normal telomeres as suggested by the observation of aberrant telomeres in cells from A-T patients, which have all components of the Shelterin complex. Recent studies in yeast, on the ATM ortholog Tel1, provide evidence for Tel1 functioning in telomere maintenance. These studies demonstrate a role for ATM-like functions in the maintenance of yeast telomeres by measuring telomere length in strains lacking tel1 [62;63]. However, studies done in mouse cells show that ATM is not required for telomere maintenance[64], suggesting that telomere maintenance may be different in yeast versus mammalian cells. Further studies are needed to elucidate the role of ATM in telomere maintenance and the explanation for the aberrant telomeres in ATM-deficient cells.
In addition to roles for TRF2 and ATM in telomere biology, there is debate about a role for TRF2 in non-telomeric DNA repair. Studies attempting to detect TRF2 at sites of double strand breaks have produced varying results. While it has proven unsuccessful to detect TRF2 co-localizing to DNA damage induced γH2AX foci following ionizing radiation[65], localization of TRF2 to sites of DNA damage has been reported following micro-laser irradiation[66]. One possible explanation is that these types of DNA damage are fundamentally different in their severity. Ionizing radiation produces double strand breaks at the rate of ~30 DNA double-strand breaks per Gy of irradiation and individual foci formed at these breaks represent the protein localized within a defined DNA region at a single break. Micro-irradiation produces double strand breaks as well, but does so at a much greater local concentration and may result in detection of proteins recruited at low levels to DNA double strand breaks. Recently an ATM dependent phosphorylation site on TRF2 was discovered[67] and reported to be necessary for complete DNA repair and important for cell survival following treatment with ionizing radiation[68]. These studies provide biochemical evidence suggesting that TRF2 plays a role in normal DNA double strand break repair, though molecular mechanisms remain to be clarified.
Clinical Applications for modulation of ATM activity
Given the putative role of ATM in DNA repair and the increased radiosensitivity following inhibition of ATM, modulation of ATM kinase activity could prove beneficial in the treatment of cancer. A majority of currently used chemotherapeutic agents are DNA damaging agents. Therefore, agents that increase the sensitivity of tumor cells to DNA damaging agents could enhance killing of tumor cells during cancer treatment. Small molecule inhibitors of the ATM kinase have recently been identified and transient ATM inhibition has been shown to be sufficient to increase the sensitivity of cells to ionizing radiation[69;70]. Since transient inhibition of ATM in the absence of DNA damage has no discernable effect on cell survival, this approach has the potential for clinical impact in treating cancers in combination with radiation therapy, which is a “localized” treatment of tumor masses.
An intriguing phenotype of A-T patients is their propensity for insulin resistance. ATM has been shown to be stimulated by insulin signaling and in some cell types ATM loss was shown to have an adverse effect on insulin dependent signaling[71]. Metabolic syndrome, thought to be caused in part by insulin resistance, contributes to the development of atherosclerosis and obesity. The role of ATM in insulin signaling suggested the possibility that its dysfunction could contribute to the development of metabolic syndrome. In fact, the loss of ATM enhanced the onset of metabolic syndrome in ApoE −/− mice. Treatment of these mice with chloroquine, an activator of ATM, decreased all major phenotypes associated with metabolic syndrome in an ATM-dependent manner[72]. From these studies it became apparent that activation of ATM may prove to be a beneficial course of treatment for patients suffering from metabolic syndrome, a highly prevalent disorder.
CONCLUSIONS AND PERSPECTIVES
In the 50 years since A-T was first clinically described, significant advances have been made in elucidating the cellular mechanisms involved in creating the abnormal clinical phenotypes of A-T. The identification of the ATM kinase as the gene product lost in A-T paved the way for establishing A-T as a model syndrome for studying cancer predisposition and radiosensitivity. The ability of the ATM kinase to become active, bind to, and be retained on chromatin at sites of DNA double strand breaks is a key component of a complete activation of the DNA damage response. Chromatin is the site where ATM-dependent phosphorylation of H2AX, MDC1, and others serves as a scaffold for the proper recruitment and retention of a host of DNA damage signaling and repair proteins. In addition, the regulation of cell cycle checkpoints by ATM provides the cell with the time necessary to complete faithful repair. DNA alterations which can activate ATM and the DNA damage response pathway include exposure to exogenous agents that alter DNA integrity as well as those caused by endogenous process. Telomeres present a particularly challenging problem and cells go to great lengths to prevent these chromosomal ends from activating ATM and the damage response under normal physiologic conditions.
Many of the agents used today to treat malignancies directly cause DNA damage. The role of ATM in orchestrating both repair and cell cycle responses following DNA damage make it an enticing target for the development of novel therapeutics. Modulation of ATM kinase activity can be beneficial for increasing radiosensitivity of cells, and therefore may be beneficial in increasing the efficacy of currently available cancer therapeutics. This provides a promising platform for future studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ford JM, Kastan MB. Abeloff’s Clinical Oncology. Churchill Livingstone - Elsevier; 2008. DNA Damage Response Pathways and Cancer; pp. 139–152. [Google Scholar]
- 2.Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. 1958;21:526–554. [PubMed] [Google Scholar]
- 3.Kastan MB, Lim D-S. The many substrates and functions of ATM. Nature Reviews Molecular Cell Biology. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 4.Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 5.Savitsky K, Sfez S, Tagle DA, Sartiel A, Collins FS, Shiloh Y, Rotman G. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Human Molecular Genetics. 1995;4:2025–2032. doi: 10.1093/hmg/4.11.2025. [DOI] [PubMed] [Google Scholar]
- 6.Kim S-T, Lim D-S, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. Journal of Biological Chemistry. 1999;274:37538–37543. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 7.Lovejoy CA, Cortez D. Common mechanisms of PIKK regulation. DNA Repair (Amst) 2009;8:1004–1008. doi: 10.1016/j.dnarep.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 9.Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc Natl Acad Sci. 1980;77:7315–7317. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gotoff SP, Amirmokri E, Liebner EJ. Ataxia telangiectasia. Neoplasia, untoward response to x-irradiation, and tuberous sclerosis. Am J Dis Child. 1967;114:617–625. doi: 10.1001/archpedi.1967.02090270073006. [DOI] [PubMed] [Google Scholar]
- 11.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 12.Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 13.Canman CE, Lim D-S, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 14.Lim D-S, Kim S-T, Xu B, Maser RS, Lin J, Petrini JHJ, Kastan MB. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 15.Xu B, O’Donnell AM, Kim S-T, Kastan MB. Phosphorylation of serine 1387 in Brca1 is specifically required for the Atm-mediated S-phase checkpoint after ionizing irraditaion. Cancer Research. 2002;62:4588–4591. [PubMed] [Google Scholar]
- 16.Xu B, Kim S-T, Kastan MB. Involvement of Brca1 in S-phase and G2-phase checkpoints after ionizing irradiation. Molecular & Cellular Biology. 2001;21:3445–3450. doi: 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D’Andrea AD. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109:459–472. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- 18.Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 20.Garg R, Callens S, Lim DS, Canman CE, Kastan MB, Xu B. Chromatin association of rad17 is required for an ataxia telangiectasia and rad-related kinase-mediated S-phase checkpoint in response to low-dose ultraviolet radiation. Mol Cancer Res. 2004;2:362–369. [PubMed] [Google Scholar]
- 21.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 22.Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pandita TK, Hittelman WN. Initial chromosome damage but not DNA damage is greater in ataxia telangiectasia cells. Radiat Res. 1992;130:94–103. [PubMed] [Google Scholar]
- 24.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 25.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010;9:662–669. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 28.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 29.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 30.Holmes AM, Haber JE. Double-strand break repair in yeast requires both leading and lagging strand DNA polymerases. Cell. 1999;96:415–424. doi: 10.1016/s0092-8674(00)80554-1. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Ira G, Tercero JA, Holmes AM, Diffley JF, Haber JE. Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:6891–6899. doi: 10.1128/MCB.24.16.6891-6899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 33.Berkovich E, Monnat RJ, Jr, Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc. 2008;3:915–922. doi: 10.1038/nprot.2008.54. [DOI] [PubMed] [Google Scholar]
- 34.Banin S, Moyal L, Shieh S-Y, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 35.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 36.Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006;25:3504–3514. doi: 10.1038/sj.emboj.7601231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23:4451–4461. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, Minami Y, Appella E, Bulavin DV. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–764. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, Nussenzweig A. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443:222–225. doi: 10.1038/nature05112. [DOI] [PubMed] [Google Scholar]
- 41.Daniel JA, Pellegrini M, Lee JH, Paull TT, Feigenbaum L, Nussenzweig A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. The Journal of Cell Biology. 2008;183:777–783. doi: 10.1083/jcb.200805154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009;187:977–990. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26:7749–7758. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
- 44.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–7748. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- 46.Jazayeri A, Balestrini A, Garner E, Haber JE, Costanzo V. Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J. 2008 doi: 10.1038/emboj.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 48.Stucki M, Jackson SP. MDC1/NFBD1: a key regulator of the DNA damage response in higher eukaryotes. DNA Repair (Amst) 2004;3:953–957. doi: 10.1016/j.dnarep.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004;23:2674–2683. doi: 10.1038/sj.emboj.7600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, Pelletier L, Jackson SP, Durocher D. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 53.Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, Lukas J, Lukas C. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 54.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 55.Pandita TK, Pathak S, Geard CR. Chromosome end associations, telomeres and telomerase activity in ataxia telangictasia cells. Cytogent Cell Genet. 1995;71:86–93. doi: 10.1159/000134069. [DOI] [PubMed] [Google Scholar]
- 56.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 57.Karlseder J, Hoke K, Mirzoeva OK, Bakkenist C, Kastan MB, Petrini JH, de Lange T. The Telomeric Protein TRF2 Binds the ATM Kinase and Can Inhibit the ATM-Dependent DNA Damage Response. PLoS Biol. 2004;2:E240. doi: 10.1371/journal.pbio.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 59.Zhu XD, Kuster B, Mann M, Petrini JH, de Lange T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet. 2000;25:347–352. doi: 10.1038/77139. [DOI] [PubMed] [Google Scholar]
- 60.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 61.Deng Y, Guo X, Ferguson DO, Chang S. Multiple roles for MRE11 at uncapped telomeres. Nature. 2009;460:914–918. doi: 10.1038/nature08196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seidel JJ, Anderson CM, Blackburn EH. A novel Tel1/ATM N-terminal motif, TAN, is essential for telomere length maintenance and a DNA damage response. Mol Cell Biol. 2008;28:5736–5746. doi: 10.1128/MCB.00326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma Y, Greider CW. Kinase-independent functions of TEL1 in telomere maintenance. Mol Cell Biol. 2009;29:5193–5202. doi: 10.1128/MCB.01896-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feldser D, Strong MA, Greider CW. Ataxia telangiectasia mutated (Atm) is not required for telomerase-mediated elongation of short telomeres. Proc Natl Acad Sci U S A. 2006;103:2249–2251. doi: 10.1073/pnas.0511143103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams ES, Stap J, Essers J, Ponnaiya B, Luijsterburg MS, Krawczyk PM, Ullrich RL, Aten JA, Bailey SM. DNA double-strand breaks are not sufficient to initiate recruitment of TRF2. Nat Genet. 2007;39:696–698. doi: 10.1038/ng0607-696. [DOI] [PubMed] [Google Scholar]
- 66.Bradshaw PS, Stavropoulos DJ, Meyn MS. Human telomeric protein TRF2 associates with genomic double-strand breaks as an early response to DNA damage. Nat Genet. 2005;37:193–197. doi: 10.1038/ng1506. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka H, Mendonca MS, Bradshaw PS, Hoelz DJ, Malkas LH, Meyn MS, Gilley D. DNA damage-induced phosphorylation of the human telomere-associated protein TRF2. Proc Natl Acad Sci U S A. 2005;102:15539–15544. doi: 10.1073/pnas.0507915102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huda N, Tanaka H, Mendonca MS, Gilley D. DNA damage-induced phosphorylation of TRF2 is required for the fast pathway of DNA double-strand break repair. Mol Cell Biol. 2009;29:3597–3604. doi: 10.1128/MCB.00944-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008;68:7466–7474. doi: 10.1158/0008-5472.CAN-08-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.White JS, Choi S, Bakkenist CJ. Irreversible chromosome damage accumulates rapidly in the absence of ATM kinase activity. Cell Cycle. 2008;7:1277–1284. doi: 10.4161/cc.7.9.5961. [DOI] [PubMed] [Google Scholar]
- 71.Yang D, Kastan MB. Participation of ATM in insulin signalling through phosphorylation of eIF-4E binding protein 1 (4E-BP1) Nature Cell Biology. 2000;2:893–898. doi: 10.1038/35046542. [DOI] [PubMed] [Google Scholar]
- 72.Schneider JG, Finck BN, Ren J, Standley KN, Takagi M, MacLean KH, Bernal-Mizrachi C, Muslin AJ, Kastan MB, Semenkovich CF. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4:377–389. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]