

Abstract
The transcription factors that regulate Aspergillus fumigatus interactions with host cells and virulence are incompletely defined. We investigated the role of the putative C2H2 transcription factor DvrA in governing these processes. Although DvrA was identified by its limited homology to Candida albicans Bcr1, a ΔdvrA mutant strain of A. fumigatus had wild-type adherence to host constituents in vitro. However, it had increased capacity to damage both endothelial cells and a pulmonary epithelial cell line compared to the ability of the wild-type strain and a ΔdvrA::dvrA-complemented strain. This increase in damage required direct contact between the mutant and host cells. The ΔdvrA mutant also stimulated greater CCL20, interleukin-8, and tumor necrosis factor mRNA expression in a pulmonary epithelial cell line compared to levels induced by the control strains. Also, it was resistant to nikkomycin Z, suggesting an altered cell wall composition. As predicted by these in vitro results, the ΔdvrA mutant had increased virulence and stimulated a greater pulmonary inflammatory response than the wild-type strain and ΔdvrA::dvrA-complemented strains in the nonneutropenic mouse model of invasive pulmonary aspergillosis. These results indicate that DvrA influences A. fumigatus virulence as well as its capacity to damage host cells and stimulate a proinflammatory response.
Aspergillus fumigatus is the leading cause of invasive pulmonary aspergillosis, a disease that is still associated with a 25 to 35% mortality rate, even with current antifungal therapy (23, 41). Invasive aspergillosis is initiated by the inhalation of conidia, which are deposited on and adhere to the epithelial cell lining of the alveoli. In susceptible hosts, these adherent conidia subsequently germinate and then invade and damage the pulmonary cells. Host cells respond by synthesizing proinflammatory mediators, which recruit phagocytic leukocytes to the foci of infection. Although these phagocytes may kill some of the invading fungi, they also can cause significant tissue destruction and thereby contribute to the pathogenesis of invasive aspergillosis (22, 38).
The molecular mechanisms that mediate the interactions of A. fumigatus with host cells during invasive aspergillosis are incompletely defined. In particular, the regulatory factors that govern the capacity of A. fumigatus to adhere to and damage host cells have not been identified. In the pathogenic yeast Candida albicans, the C2H2 transcription factor Bcr1 governs the expression of multiple cell surface proteins that mediate biofilm formation as well as adherence to host cells (20, 21). These proteins include Hwp1, Als1, and Als3 (11, 25, 39, 43). Here, we identified a putative C2H2 transcription factor in A. fumigatus that shares limited homology with Bcr1. We found that unlike Bcr1, this protein does not regulate adherence to host constituents. Instead, it appears to functions as a negative regulator of host cell damage and stimulation as well as virulence during invasive pulmonary disease. Hence, we named it DvrA (for damage and virulence regulator).
MATERIALS AND METHODS
Fungal strain and growth conditions.
A. fumigatus Af293, originally a clinical isolate, was used as the wild-type strain in these investigations. It was a generous gift from P. T. Magee, University of Minnesota, St. Paul, MN. Conidia were obtained by growing the organism on Sabouraud agar (Difco) at 37°C for 7 days. The conidia were harvested by rinsing with phosphate-buffered saline (PBS) containing 0.1% Tween 80 (Sigma-Aldrich). Germlings were prepared as described previously (17). Briefly, freshly harvested conidia were suspended in Sabouraud broth and then added to 150-mm petri dishes. The petri dishes were incubated at 37°C in 5% CO2 for 6 to 7 h until the majority of the conidia had begun to germinate and had produced germ tubes that were approximately 2 conidial diameters in length. The resulting germlings were rinsed with PBS, harvested with a cell scraper, and enumerated using a hemacytometer.
Strain construction.
A split-marker approach was used to disrupt the first 1,679 bp of the protein coding sequence of dvrA (Afu3g09820) in A. fumigatus Af293 (5). Briefly, a DNA fragment encompassing 1,578 bp upstream of dvrA was PCR amplified from genomic DNA of strain Af293 using primers BCR-F1 and BCR-F2 (see Table S1 in the supplemental material). Similarly, a fragment encompassing 1,451 bp downstream of dvrA was amplified using primers BCR-F3 and BCR-F4 (Table S1). The 5′ region of the hygromycin resistance cassette then was amplified from plasmid pAN7-1 (28) using primers HYG-F and HY, and the 3′ region of this cassette was amplified using primers HYG-R and YG. The dvrA upstream sequence was combined with the 3′ portion of the hygromycin resistance cassette by fusion PCR using primers BCR-F1 and YG. The dvrA downstream sequence was combined with the 5′ region of the resistance cassette using primers HY and BCR-F4. A. fumigatus Af293 was cotransformed with both fragments by protoplasting (4). Hygromycin-resistant clones were screened for the disruption of dvrA by PCR using the primers BCR-VER1, BCR-VER2, and HY-VER (Table S1). Southern blotting with a 412-bp fragment of the hygromycin resistance cassette was used to confirm the homologous integration of the disruption cassette and to ensure that only a single copy of a disruption cassette was integrated into the genome during transformation (data not shown).
The ΔdvrA mutant was complemented with a wild-type copy of dvrA as follows. The dvrA open reading frame plus 2,506 bp of upstream sequence and 67 bp of downstream sequence was cloned from genomic DNA of strain Af293 by high-fidelity PCR using primers BCRREV-F and BCRREV-R (Table S1). The resulting fragment was cloned into plasmid p402, which contains the phleomycin resistance gene (30). The resulting plasmid was linearized with SspI and used to transform the ΔdvrA mutant. Hygromycin and phleomycin-resistant transformants were selected, and the proper integration of the plasmid upstream of the disrupted dvrA locus was verified by PCR using primers BCR-VER1 and BCR-VER2.
Real-time PCR.
The time course of dvrA mRNA expression in the wild-type strain was analyzed by real-time PCR. This procedure also was used to verify that dvrA mRNA expression was restored to wild-type levels in the ΔdvrA::dvrA-complemented strain and to determine the transcript levels of ace2, asg3, and ecm33 in the various strains. The real-time PCR experiments were performed as described previously using the primers listed in Table S1 in the supplemental material (9). Briefly, conidia were inoculated into Sabouraud broth and incubated at 37°C in a shaking incubator. At selected time points, an aliquot of the organisms was removed for RNA extraction and subsequent cDNA synthesis. Real-time PCR amplification was monitored using SYBR green. The relative gene expression levels were determined by the 2−ΔΔCT method using tef1 as the reference gene (13, 33).
Germination rate, radial growth rate, and conidial production.
To determine the germination rate, 105 conidia of the different A. fumigatus strains were suspended in either Sabouraud broth alone or RPMI 1640 medium containing 50% fetal bovine serum. These conidia were added to wells of a 24-well tissue culture plate and incubated at 37°C in 5% CO2. At various time points, the medium above the conidia was aspirated and the organisms were fixed in 3% paraformaldehyde in PBS. The wells were examined using an inverted microscope, and 100 organisms per strain were evaluated for germination, which was defined as the production of a germ tube that was at least 1 conidial diameter in length.
The radial growth rate and conidial production of the different A. fumigatus strains were measured as described previously (9). Briefly, 5 × 103 conidia were spotted onto Sabouraud, YPD (yeast extract, peptone, and d-glucose), or Aspergillus minimal medium (27) agar and incubated at 37°C. The colony diameter was measured daily (4). After a 7-day incubation, the conidia were harvested in a standardized manner and then counted using a hemacytometer.
Susceptibility to stressors.
The susceptibility of conidia and germlings of the different strains to various stressors was determined by the microdilution method as outlined previously (9). Susceptibility to cell wall, cell membrane, and osmotic stress was determined by adding 5 × 103 conidia or germlings in Sabouraud broth to separate wells of a 96-well microtiter plate containing twofold dilutions of nikkomycin Z (range, 1 to 1,000 μg/ml; Sigma-Aldrich), Congo red (5 to 5,000 μg/ml; Sigma-Aldrich), calcofluor white (5 to 5,000 μg/ml; Sigma-Aldrich), caspofungin (0.004 to 4 μg/ml; Merck), SDS (0.0002 to 0.2%), or NaCl (0.002 to 2 M). After incubation at 37°C for 2 days, the wells were scored visually for growth. The MIC was determined as the concentration of each compound that caused at least an 80% reduction in growth compared to the growth of organisms in the absence of the compound. Susceptibility to nikkomycin Z and Congo red also were tested by the agar dilution approach. Serial 10-fold dilutions of conidia of the various strains were spotted onto YPD agar containing either 75 μg/ml nikkomycin Z or 200 μg/ml Congo red. The plates were incubated at 37°C for 24 h and then imaged. Susceptibility to each stressor was tested at least three times.
Host cells.
The A549 type II pneumocyte cell line was obtained from the American Type Culture Collection and grown in F-12K medium containing 10% fetal bovine serum (Gemini Bio-Products), penicillin, and streptomycin. Endothelial cells were harvested from the veins of human umbilical cords and maintained in M199 medium (Gibco) containing 10% fetal bovine serum, 10% bovine calf serum (Gemini Bio-Products), and 2 mM l-glutamine with penicillin and streptomycin (Irvine Scientific) as previously described (15, 17). All host cells were grown in 5% CO2 at 37°C.
Adherence assay.
The capacity of the different strains to adhere to A549 epithelial cells, endothelial cells, gelatin, and fibronectin was determined as previously described (14). When adherence to epithelial or endothelial cells was determined, the cells were grown to confluence in 6-well tissue culture plates. For adherence to extracellular matrix proteins, the 6-well plates were coated overnight with 1 ml of 0.01 mg/ml gelatin (Sigma) or fibronectin (Becton Dickinson). For adherence to all substrates, 200 conidia or germlings in 1 ml of Hanks' balanced salt solution (HBSS; Irvine Scientific) were added to each well. After a 30-min incubation, the wells were rinsed three times with HBSS in a standardized manner and then overlaid with Sabouraud agar. The number of adherent organisms in each well was determined by colony counting after an overnight incubation at 37°C. The adherence of each strain was expressed as a percentage of the original inoculum, which was measured by quantitative culture. Each experiment was performed in triplicate and repeated at least three times.
Host cell damage.
The extent of damage to the A549 cells and endothelial cells caused by the various A. fumigatus strains was determined with a 51Cr release assay as described previously (9, 17). Briefly, A549 epithelial cells or endothelial cells were grown to confluence in 24-well tissue culture plates and loaded with 51Cr. After the unincorporated 51Cr was removed by rinsing, the epithelial cells were infected with 5 × 105 conidia per well, and the endothelial cells were infected with 5 × 105 germlings per well in their respective culture media. After 24 h of incubation, the medium above the cells was collected, and the residual 51Cr remaining in the host cells was collected by lysing the wells once with 6N NaOH and rinsing twice with RadiacWash (Biodex Medical Systems, Inc.). The rinses from each well were combined, and the amount of 51Cr in the medium and rinses was determined by gamma counting. To measure the spontaneous release of 51Cr, uninfected host cells exposed to medium alone were processed in parallel. The percent specific release of 51Cr was calculated using the following formula: (experimental release − spontaneous release)/(total incorporation − spontaneous release) × 100. Each experiment was performed in triplicate three different times.
To determine the role of soluble factors produced by A. fumigatus in inducing host cell damage, the organisms were added to cell culture inserts (2-μm pore size; Nunc) that were suspended approximately 1 mm above 51Cr-loaded A549 pulmonary epithelial cells (17). The extent of epithelial cell damage was determined after 24 h, as described above.
Epithelial cell stimulation.
The capacity of each strain of A. fumigatus to stimulate epithelial cell cytokine mRNA expression was determined by real-time PCR. The A549 cells were grown to 80% confluence in 6-well tissue culture plates and infected with 5 × 105 conidia of each strain in serum-free F-12K medium. After 24 h of incubation, the epithelial cell RNA was harvested using TRIzol (Gibco-Invitrogen), followed by phenol-chloroform extraction and ethanol precipitation. Cytokine mRNA levels were analyzed by real-time reverse transcription-PCR (RT-PCR) using TaqMan probes (Applied Biosystems) for CCL20 (H00171125_m1), tumor necrosis factor (TNF; Hs00174128_m1), and interleukin-8 (IL-8; Hs00174103_m1). Relative gene expression was determined by the 2−ΔΔCT method using 18s rRNA as the reference.
Galleria melonella model of invasive aspergillosis.
The A. fumigatus strains were screened for alterations in virulence in G. melonella larvae (Magazoo) (19, 29). Larvae at the sixth instar of development were injected through the last proleg with 106 conidia suspended in 10 μl PBS using a Hamilton syringe. The larvae were kept at 37°C in a humidified chamber and monitored daily for survival. The virulence of each strain was tested twice in a total of 20 larvae, and 20 additional larvae were injected with PBS alone as a negative control.
Mouse model of invasive pulmonary aspergillosis.
The virulence of the various strains was assessed in a mouse model of invasive aspergillosis using two different immunosuppressive regimens (7, 35, 36). In the nonneutropenic regimen, male BALB/c mice (Taconics Labs) were immunosuppressed with cortisone acetate (Sigma-Aldrich) administered at 500 mg/kg subcutaneously every other day, starting on day −4 relative to infection and finishing on day +4. In the neutropenic regimen, the mice were immunosuppressed with cortisone acetate administered subcutaneously at 250 mg/kg on days −2 and +3 and cyclophosphamide (Western Medical Supply) administered intraperitoneally at 250 mg/kg on day −2 and 200 mg/kg on day +3. In both models of infection, the mice received daily subcutaneous injections of 5 mg of ceftazidime while they were immunosuppressed to prevent bacterial infections. The immunosuppressed mice were infected by placing them for 1 h in an acrylic chamber into which 1.2 × 1010 conidia were aerosolized. Control mice were immunosuppressed with either of the two regimens but were not infected.
In the survival studies, 11 to 13 mice were infected with each strain of A. fumigatus. Shortly after inoculation, three mice from each group were sacrificed, after which their lungs were homogenized and quantitatively cultured to verify conidial delivery to the lungs. In these experiments, a median of 4.1 × 103 conidia (interquartile range, 1.6 × 103 to 4.9 × 103 conidia) was delivered to the lungs of each mouse, and the pulmonary conidial delivery was similar for all strains of A. fumigatus. The remaining mice were monitored for survival. The survival experiments were repeated twice and the results were combined. The animal studies were approved by the Institutional Animal Use and Care Committee and performed according to the National Institutes of Health guidelines for animal housing and care.
Pulmonary fungal burden and inflammatory response.
To determine the pulmonary fungal burden and host response to infection, the mice were immunosuppressed with cortisone acetate alone, and 9 to 12 mice per strain were infected as described above. After 4 days of infection, the mice were sacrificed and their lungs were harvested, weighed, and then homogenized in ice-cold PBS containing protease inhibitor cocktail (Sigma-Aldrich). The homogenates were clarified by centrifugation, and aliquots of the resulting supernatants were stored at −80°C for later analysis.
The pulmonary fungal burden was assessed by measuring the pulmonary galactomannan content using the Platelia Aspergillus enzyme immunoassay (Bio-Rad). The assay was performed by following our previously described method (9, 34). Briefly, each pulmonary homogenate was diluted 1:10 in ultrapure water and processed according to the manufacturer's instructions. To determine the relative galactomannan content per gram of lung tissue, the resulting optical densities were compared to a standard curve, which was made using serial dilutions of a pool of lung homogenates from five heavily infected immunosuppressed mice (7 days after intranasal infection with strain Af293).
In preliminary experiments, we determined that the ΔdvrA mutant released an amount of galactomannan similar to that of strain Af293 when grown in vitro (see Fig. S1 in the supplemental material). Conidia from each strain were inoculated into RPMI 1640 medium containing morpholinepropanesulfonic acid (MOPS) to achieve a final concentration of 105 conidia per ml and then incubated in an orbital shaker at 37°C. After 24, 48, or 72 h, the conditioned medium was collected by filtration and stored at −80°C. The galactomannan content of each sample was determined as described above and normalized to the dry weight of the organisms in culture. The experiment was performed two times.
The pulmonary inflammatory response to the various A. fumigatus strains was assessed by measuring the myeloperoxidase (MPO) and cytokine content of the lungs. The MPO content was used to assess the accumulation of phagocytes during infection (12, 40). The MPO levels in the lung homogenates were measured by enzyme immunoassay (Cell Sciences). The pulmonary content of IL-6, IL-10, IL-12, gamma interferon (IFN-γ), TNF, and monocyte chemoattractant protein 1 (MCP-1) was measured using the mouse inflammation cytometric bead array kit (BD Biosciences).
Statistical analyses.
The results of the in vitro experiments were compared by analysis of variance followed by pair-wise comparisons using the Student's t test. Differences in survival were analyzed using the log-rank test, and the pulmonary fungal burden, MPO, and cytokine data were analyzed with the Wilcoxon rank sum test. P values of ≤0.05 were considered significant.
RESULTS
Identification and disruption of A. fumigatus dvrA.
The closest A. fumigatus homolog of C. albicans BCR1 was identified by BLAST searches of the A. fumigatus genome. Reciprocal BLAST searches verified that the A. fumigatus gene dvrA (Afu3g09820) shared the closest homology to BCR1 within the A. fumigatus genome. The products of dvrA and BCR1 share significant homology in their C2H2 zinc finger domains, whereas there are very limited regions of homology outside this region (Fig. 1). Interestingly, orthologs of dvrA are present in other species of Aspergillus as well as other filamentous fungal pathogens, such as Penicillium marneffei and Coccidioides immitis. The products of these dvrA orthologs are predicted to have much greater similarity to DvrA than to Bcr1 (Fig. 1).
Fig. 1.

Alignment of DvrA orthologs from Aspergillus spp., Penicillium marneffei, and Coccidioides immitis with Candida albicans Bcr1. Shaded areas indicate amino acids that are conserved among all of the indicated fungi. Boxed areas indicate amino acids that are conserved in ≥75% of the organisms. Thick horizontal lines indicate the predicted zinc cluster DNA binding domains.
The time course of dvrA mRNA levels in A. fumigatus Af293 was analyzed by real-time RT-PCR. When the organism was grown in Sabouraud broth, relatively low dvrA mRNA levels were detected in swollen conidia (Fig. 2A). The level of dvrA expression increased as the conidia germinated and formed mature hyphae, peaking at 40 h of incubation. After 48 h, dvrA transcript levels declined, possibly due to the depletion of nutrients from the medium.
Fig. 2.
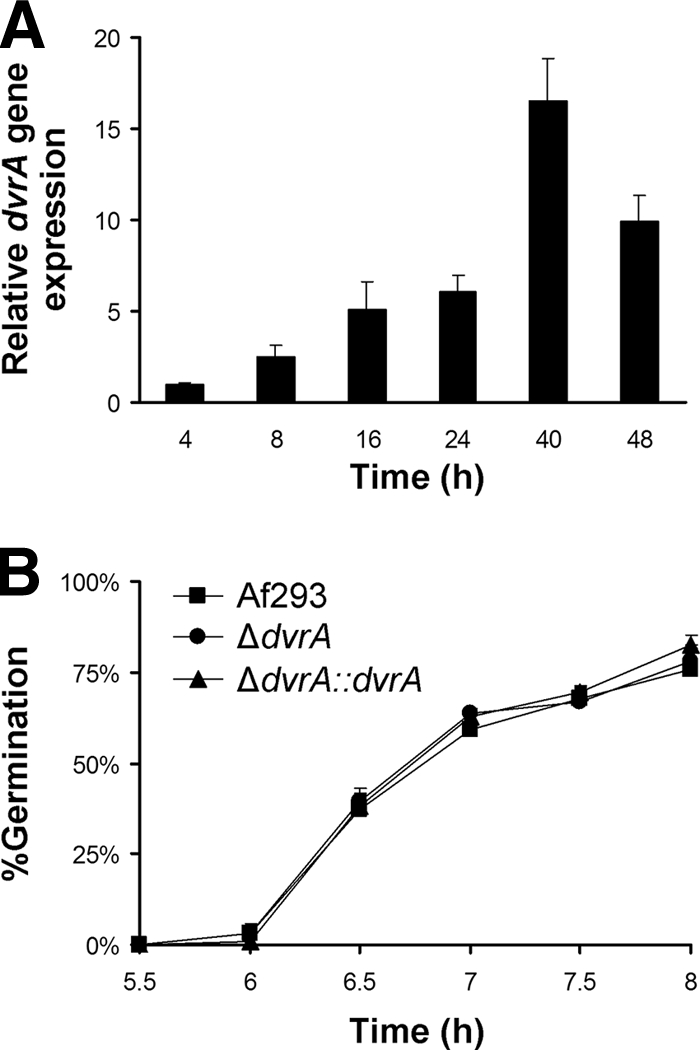
Time course of dvrA mRNA levels in wild-type A. fumigatus (A) and the effect of dvrA disruption on germination (B). (A) Conidia of strain Af293 were incubated in Sabouraud broth at 37°C in a shaking incubator. At the indicated time points, an aliquot of the organisms was removed for RNA extraction. The dvrA transcript levels were determined by real-time PCR using tef1 as the endogenous control gene. Results are means ± standard deviations (SD) from two biological replicates, each measured in duplicate. (B) Conidia of the three strains of A. fumigatus were incubated in RPMI 1640 medium containing 50% fetal bovine serum at 37°C in 5% CO2. At the indicated time points, the organisms were fixed and the percentage of cells that had germinated was determined. Results are means ± SD from three independent experiments.
To determine the function of dvrA, we constructed a ΔdvrA deletion mutant. We also constructed a ΔdvrA::dvrA-complemented mutant to verify that any phenotype of the mutant was due to the disruption of dvrA. Using real-time RT-PCR, we confirmed the absence of dvrA mRNA in the ΔdvrA mutant and the restoration of dvrA mRNA to wild-type levels in the ΔdvrA::dvrA-complemented strain (data not shown).
The ΔdvrA mutant had normal germination, growth, and conidiation.
We next investigated whether dvrA influences the growth, development, and stress resistance of A. fumigatus. The ΔdvrA conidia germinated at a frequency similar to that of wild-type conidia in both RPMI 1640 medium containing 50% fetal bovine serum (Fig. 2B) and Sabouraud broth and produced hyphae that were morphologically similar to wild-type hyphae (data not shown). Also, the ΔdvrA mutant had a normal radial growth rate on three different media, and these colonies produced numbers of conidia similar to those of the wild-type strain (data not shown).
Deletion of dvrA resulted in increased resistance to nikkomycin Z.
The susceptibility of the ΔdvrA mutant to environmental stress was examined. When this mutant was tested in a broth microdilution assay using the endpoint of at least 80% growth inhibition at 48 h, it had wild-type susceptibility to SDS, NaCl, caspofungin, calcofluor white, Congo red, and nikkomycin Z (data not shown). However, we noticed that after exposure to nikkomycin Z for 24 h, the majority of the hyphae of the ΔdvrA mutant had normal morphology in all nikkomycin Z concentrations tested (up to 1,000 μg/ml). In contrast, most of the hyphae of the wild-type strain had a ballooning of the cell walls when exposed to 63 μg/ml nikkomycin Z, and hyphae of the ΔdvrA::dvrA-complemented strain had abnormal morphology at 31 μg/ml. To verify these results, we assessed the nikkomycin Z susceptibility of the three strains in an agar dilution assay. As expected, the ΔdvrA mutant grew better in the presence of nikkomycin Z than the wild-type and ΔdvrA::dvrA-complemented strains (Fig. 3). We also tested the growth of the ΔdvrA mutant in the presence of Congo red as a control in the agar dilution assay. This strain grew similarly to the control strains in this assay, as predicted by the broth microdilution studies. The resistance to nikkomycin Z, which is a chitin synthase inhibitor, suggests that the ΔdvrA mutant has an altered cell wall and possibly increased chitin content.
Fig. 3.

Reduced susceptibility to nikkomycin Z of the ΔdvrA mutant. Serial 10-fold dilutions of conidia of the indicated strains were spotted onto YPD agar containing either 75 μg/ml nikkomycin Z or 200 μg/ml Congo red. Images are of the plates after a 24-h incubation at 37°C.
Adherence of A. fumigatus was not influenced by deletion of dvrA.
In C. albicans and Candida parapsilosis, BCR1 is required for the normal expression of multiple cell surface proteins, especially adhesins (8, 20, 21). Therefore, we investigated the adherence of the ΔdvrA mutant to biologically relevant host substrates. We found that the adherence of ΔdvrA conidia and germlings was similar to that of the wild-type strain for all substrates tested, including vascular endothelial cells, pulmonary epithelial cells, fibronectin, and gelatin (data not shown). Therefore, DvrA does not appear to govern adherence in A. fumigatus.
The ΔdvrA mutant caused increased damage to pulmonary epithelial cells and vascular endothelial cells.
To determine if DvrA influences other interactions of A. fumigatus with host cells, we measured the extent of damage to the A549 pulmonary epithelial cell line caused by conidia of the ΔdvrA mutant. We also determined the capacity of ΔdvrA germlings to damage human umbilical vein endothelial cells. Germlings were used instead of conidia in the endothelial cell experiments, because hyphae are the main form of the organism that interact with endothelial cells during invasive aspergillosis (10, 13). We found that the ΔdvrA mutant caused nearly twofold more damage to the pulmonary epithelial cells and 1.4-fold more damage to the endothelial cells than either the wild-type strain or the ΔdvrA::dvrA-complemented strain (P < 0.0001) (Fig. 4). These results suggest that DvrA negatively regulates A. fumigatus factors that mediate host cell damage.
Fig. 4.
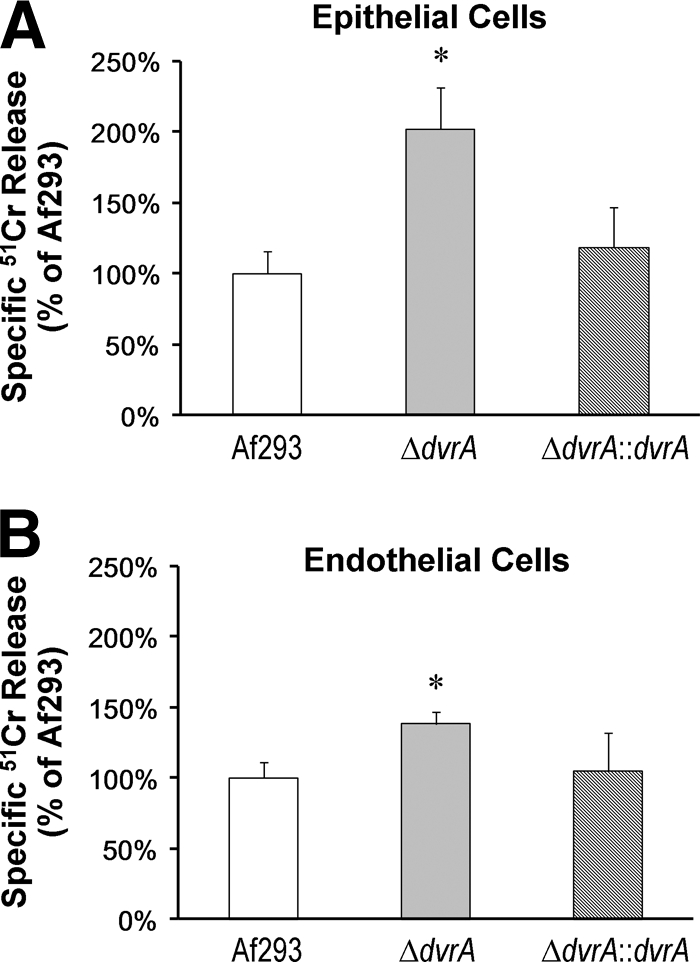
ΔdvrA mutant causes increased damage to pulmonary epithelial cells and vascular endothelial cells. (A) The A549 pulmonary epithelial cell line was infected with conidia of the indicated strains of A. fumigatus for 24 h, and the extent of epithelial cell damage was measured using a 51Cr release assay. (B) Extent of endothelial cell damage following a 24-h incubation with germlings of the indicated strains. Results are the means ± SD from three experiments, each performed in triplicate. *, P < 0.02 compared to Af293 or the ΔdvrA::dvrA-complemented strain.
In both the epithelial and endothelial cell experiments, the extent of the germination and hyphal elongation of the ΔdvrA mutant were similar to that of the wild-type strain, as determined by microscopic examination. Therefore, the increased host cell damage of the ΔdvrA mutant was not due to accelerated germination. We next investigated whether the increased host cell damage caused by the ΔdvrA mutant was due to the release of soluble factors, such as mycotoxins (2, 24). Conidia of the ΔdvrA mutant were incubated for 24 h in cell culture inserts that were suspended over the pulmonary epithelial cells. We found that when contact between the organisms and the epithelial cells was prevented, no detectable epithelial cell damage was induced even after 24 h of incubation (data not shown). Thus, the enhanced capacity of the ΔdvrA mutant to induce epithelial cell damage does not appear to be due to the increased release of toxic soluble factors.
The ΔdvrA mutant stimulated increased pulmonary epithelial cell cytokine gene expression.
Although many of the manifestations of invasive aspergillosis are due to the direct damage of host cells by A. fumigatus, it is probable that the host inflammatory response to the organism also contributes to the pathogenesis of this disease in some hosts. We found previously that A549 pulmonary epithelial cells responded to infection with wild-type A. fumigatus by increasing mRNA levels of the chemokines CCL20 and IL-8, as well as that of the proinflammatory cytokine TNF (14). In the current study, we found that the ΔdvrA mutant induced significantly greater CCL20, IL-8, and TNF transcript levels than the wild-type and ΔdvrA::dvrA-complemented strains (P < 0.001) (Fig. 5). These findings indicate that the ΔdvrA mutant induces a very strong inflammatory response in vitro.
Fig. 5.

Increased pulmonary epithelial cell stimulation by the ΔdvrA mutant. A549 pulmonary epithelial cells were incubated with conidia of the indicated A. fumigatus strains for 12 h, after which the mRNA levels of CCL20, IL-8, and TNF were determined by real-time PCR. Results are the means ± standard deviations from three experiments, each performed in duplicate. *, P < 0.001 compared to Af293 or the ΔdvrA::dvrA-complemented strain.
The ΔdvrA mutant had increased virulence during invasive aspergillosis.
The increased capacity of the ΔdvrA mutant to damage and stimulate host cells in vitro suggested that this mutant has increased virulence. To test this hypothesis, we first infected G. melonella larvae with the ΔdvrA mutant as a screening test for alterations in virulence. We found that the length of survival of larvae injected with the ΔdvrA mutant was significantly shorter than that of larvae injected with the wild-type or ΔdvrA::dvrA-complemented strain (P < 0.0001) (Fig. 6).
Fig. 6.
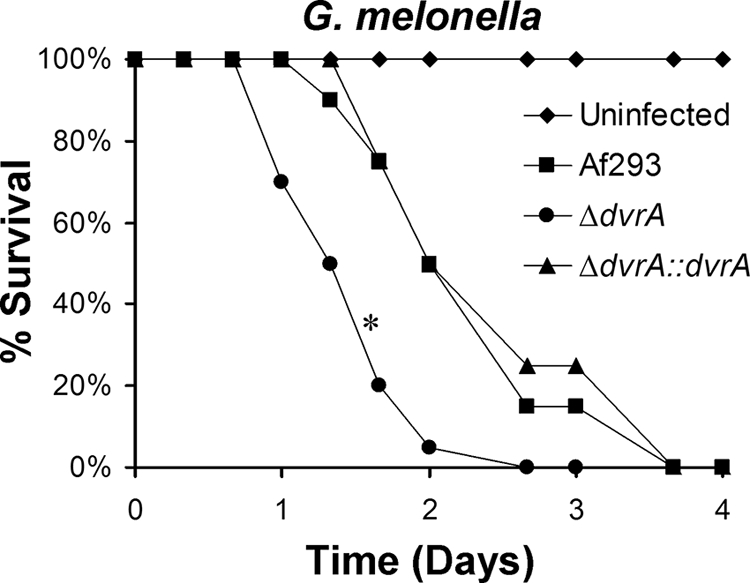
Increased virulence of the ΔdvrA mutant in G. melonella. The survival of G. melonella larvae following inoculation with conidia of each of the indicated strains of A. fumigatus is shown. Results are from 20 larvae per strain of A. fumigatus. *, P < 0.0001 compared to Af293 or the ΔdvrA::dvrA-complemented strain.
To verify that the deletion of dvrA resulted in increased virulence, we tested the ΔdvrA mutant in the mouse model of invasive pulmonary aspergillosis. The virulence of this mutant was analyzed in mice that were immunosuppressed with two different regimens. These different regimens were used because previous studies indicate that the type of immunosuppression significantly alters the pathogenesis of invasive pulmonary aspergillosis in mice (3, 7, 37). In the first regimen, the mice were given high-dose cortisone acetate, but they were not neutropenic. In the second regimen, the mice were administered cyclophosphamide to render them neutropenic, as well as a lower dose of cortisone acetate.
When the nonneutropenic mice were infected with an aerosol of conidia from the ΔdvrA mutant, the length of their survival was significantly shorter than that of mice infected with either the wild-type or ΔdvrA::dvrA-complemented strain (P < 0.001) (Fig. 7A). In contrast, when neutropenic mice were infected with the ΔdvrA mutant, their survival was similar to that of the control mice (P > 0.35) (Fig. 7B). Therefore, the effect of dvrA on virulence during invasive pulmonary aspergillosis is dependent on the immune status of the host; dvrA influences virulence in nonneutropenic mice but not in neutropenic mice.
Fig. 7.
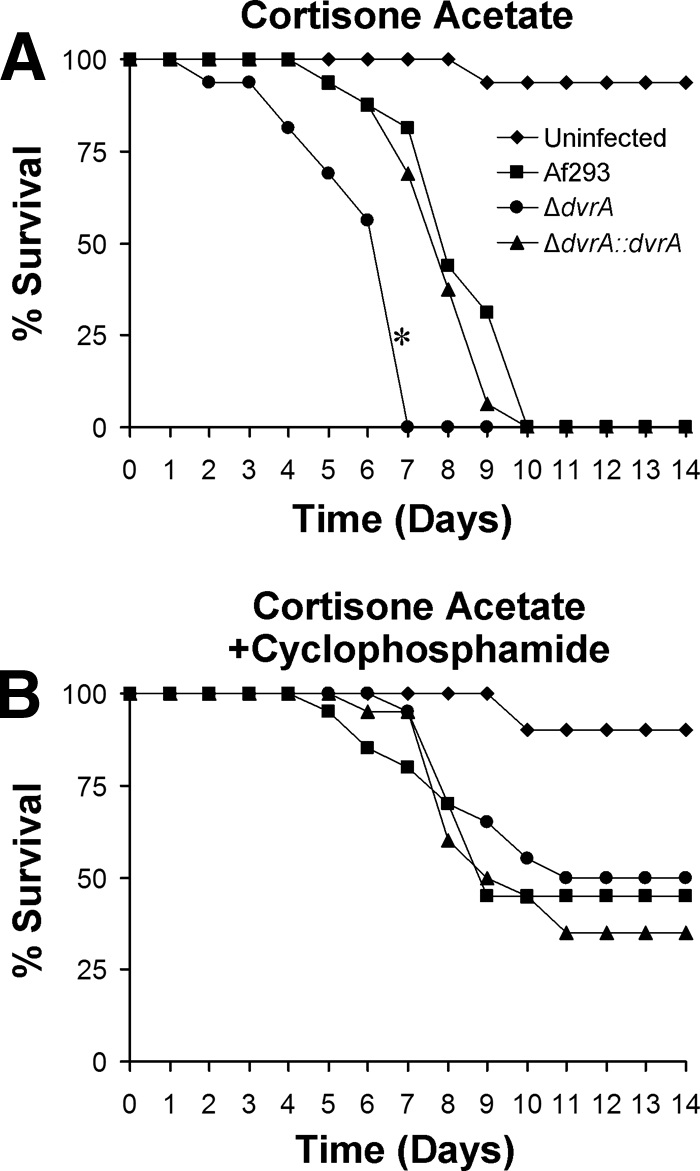
Influence of DvrA on the survival of mice with invasive pulmonary aspergillosis. Mice were immunosuppressed with either cortisone acetate alone (A) or cortisone acetate and cyclophosphamide (B) and then infected with the indicated strains of A. fumigatus in an aerosol chamber. Results are the combined data from two independent experiments for a total of 16 mice per strain (A) and 20 mice per strain (B). *, P < 0.001 compared to Af293 or the ΔdvrA::dvrA-complemented strain.
The ΔdvrA mutant induced an exaggerated inflammatory response in nonneutropenic mice.
The in vitro data indicated that the ΔdvrA mutant induced an abnormally strong inflammatory response in A549 pulmonary epithelial cells. These results suggested that the increased mortality of nonneutropenic mice infected with this mutant is due in part to an overexuberant inflammatory response. To investigate this possibility, we determined the pulmonary fungal burden, as well as pulmonary levels of MPO (a marker of phagocyte accumulation [12, 40]) and proinflammatory cytokines, in these mice. After 4 days of infection, the pulmonary galactomannan content of mice infected with the ΔdvrA mutant was significantly higher than that of mice infected with the wild-type strain (P = 0.018), but not that of mice infected with the ΔdvrA::dvrA-complemented strain (P = 0.21) (Fig. 8). Also, the pulmonary contents of MPO, IL-6, TNF, and MCP-1 were significantly greater in the mice infected with the ΔdvrA mutant than mice infected with the wild-type or ΔdvrA::dvrA-complemented strains (P < 0.03 for all comparisons) (Fig. 8). The pulmonary IL-12 levels were similar in mice infected with all three strains of A. fumigatus (Fig. 8), and the levels of IL-10 and gamma interferon were undetectable (data not shown). Collectively, these results suggest that the shortened survival of mice infected with the ΔdvrA mutant was due to both increased pulmonary fungal burden and enhanced inflammatory response.
Fig. 8.

Effects of DvrA on pulmonary fungal burden, phagocyte accumulation, and cytokine content in mice with invasive aspergillosis. Mice were immunosuppressed with cortisone acetate and then infected with the indicated A. fumigatus strains in an aerosol chamber. After 4 days of infection the mice were sacrificed, after which the amounts of galactomannan, myeloperoxidase, IL-6, TNF, MCP-1, and IL-12 in lung homogenates were measured. Results are the median ± the interquartile range of 9 to 12 mice per strain of A. fumigatus. *, P = 0.018 compared to Af293 and P = 0.21 compared to the ΔdvrA::dvrA-complemented strain; **, P < 0.03 compared to Af293 or the ΔdvrA::dvrA-complemented strain. Abbreviations: GM, galactomannan; MPO, myeloperoxidase.
The increased virulence of the ΔdvrA mutant was not due to reduced expression of genes previously known to inhibit virulence.
The deletion of ace2, ags3, and ecm33 in A. fumigatus is known to result in increased virulence in experimental models of invasive aspergillosis (9, 18, 31). Therefore, it was possible that the expression of one or more of these genes was reduced in the ΔdvrA mutant and thereby contributed to the increased virulence of this strain. To investigate this possibility, we compared the relative transcript levels of ace2, ags3, and ecm33 in the ΔdvrA mutant to those of the wild-type and ΔdvrA::dvrA-complemented strains. All three genes were expressed at similar levels in all three strains of A. fumigatus (Fig. 9), indicating that the increased virulence of the ΔdvrA mutant is due to a unique mechanism that is unrelated to the decreased expression of ace2, ags3, or ecm33.
Fig. 9.
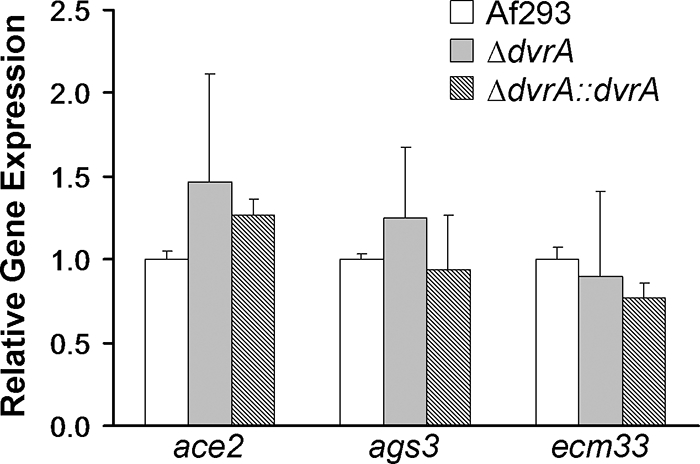
Transcript levels of ace2, ags3, and ecm33. Conidia of the indicated strains were incubated in Sabouraud broth at 37°C in a shaking incubator. After 24 h, an aliquot of the organisms was removed for RNA extraction. The transcript levels of the indicated genes were determined by real-time PCR using tef1 as the endogenous control gene. Results are means ± standard deviations from two biological replicates, each measured in triplicate.
DISCUSSION
The results of these investigations demonstrate that the deletion of A. fumigatus dvrA results in enhanced virulence in the nonneutropenic mouse model of invasive pulmonary disease. Based on the homology between A. fumigatus DvrA and C. albicans Bcr1, we had expected that the ΔdvrA mutant would have reduced adherence. However, we could find no evidence that DvrA governs the adherence properties of either conidia or germlings. While DvrA and Bcr1 share significant homology in their predicted DNA binding domains, the other regions of these proteins have only scattered areas of homology. Thus, the phenotypic data and sequence data both suggest that DvrA has a different function than that of Bcr1. Interestingly, close orthologs of DvrA are present in other Aspergillus spp. as well as other medically important filamentous fungi, including Penicillium marneffei and Coccidioides immitis. This close homology suggests that the DvrA orthologs of these organisms may have functions similar to those of A. fumigatus DvrA.
The enhanced virulence of the ΔdvrA mutant likely was due in part to its increased capacity to damage host cells. We found that this mutant caused greater damage to both endothelial and pulmonary epithelial cells in vitro. The results of experiments with cell culture inserts demonstrated that the increased capacity of the ΔdvrA mutant to damage pulmonary epithelial cells was not due to the enhanced release of soluble toxins. We have found previously that A. fumigatus must be in direct contact with host cells to damage them (16, 17). Therefore, it is probable that the enhanced capacity of the ΔdvrA mutant to damage host cells was due to an alteration in its cell surface or cell wall. Consistently with this hypothesis, we found that the ΔdvrA mutant was resistant to the chitin synthase inhibitor nikkomycin Z, which suggests that DvrA influences the composition of the A. fumigatus cell wall.
The capacity of different C. albicans strains to damage endothelial cells in vitro is a fairly good predictor of their virulence in the mouse model of disseminated candidiasis (32). Our recent data suggest that a similar relationship exists between host cell damage and virulence for A. fumigatus. Both ΔdvrA and Δace2 mutants cause increased damage to endothelial cells in vitro, and both of these mutants have increased virulence in the nonneutropenic mouse model of invasive pulmonary aspergillosis (9). Also, we have found that a ΔmedA mutant has reduced capacity to damage pulmonary epithelial cells in vitro and attenuated virulence in nonneutropenic mice (14). These results suggest that determining the capacity of an A. fumigatus mutant to damage endothelial or pulmonary epithelial cells in vitro could be used as a screening test for alterations in virulence. These data further suggest that the ability of A. fumigatus to damage host cells is an important virulence determinant of this organism.
In nonneutropenic hosts with invasive pulmonary aspergillosis, significant tissue destruction is caused not only by the fungus but also by toxic products released by activated neutrophils (3, 7, 37, 38). It thus was notable that the ΔdvrA mutant induced exaggerated proinflammatory cytokine production in the lungs of nonneutropenic mice. This excessive inflammatory response likely contributed to the accelerated mortality of mice infected with the ΔdvrA mutant. The higher pulmonary fungal burden of mice infected with the ΔdvrA mutant likely contributed to their augmented inflammatory response. However, this mutant also stimulated a stronger proinflammatory response in pulmonary epithelial cells in vitro than did the wild-type strain, even at the same inoculum. Therefore, the ΔdvrA mutant appears to have an intrinsic capacity to stimulate a supranormal inflammatory response in host cells.
The resistance of the ΔdvrA mutant to nikkomycin Z suggests that it has an increased amount of chitin in its cell wall. In C. albicans, chitin induces an inflammatory response by stimulating monocytes to synthesize IL-1β (42). Recently, it has been reported that a ΔpmrA mutant of A. fumigatus has increased cell wall chitin content but normal virulence in the mouse model of invasive pulmonary aspergillosis (26). However, the virulence of this mutant was tested only in neutropenic mice. While our ΔdvrA mutant also had normal virulence in neutropenic mice, it had increased virulence in nonneutropenic mice. Thus, it is possible that the relationship between increased chitin content and enhanced A. fumigatus virulence is detectable only in nonneutropenic hosts, where the inflammatory response induced by the fungus plays a key role in the pathogenesis of the infection.
Only a limited number of genes have been described that functioned as negative regulators of A. fumigatus virulence. The deletion of ace2, ags3, and ecm33 is associated with enhanced virulence in murine models of invasive pulmonary aspergillosis (9, 18, 31). Recently we reported that an A. fumigatus double mutant lacking the trehalose biosynthesis genes tpsA and tpsB also was hypervirulent in mice (1). This mutant had the reduced expression of ags3, suggesting that a reduction in cell wall α(1-3)glucans contributed to its hypervirulent phenotype. Our finding that the ΔdvrA mutant expressed ace2, ags3, and ecm33 at wild-type levels makes it unlikely that these genes contribute to the increased virulence of this strain. However, a common finding with the ΔdvrA, Δace2, Δags3, Δecm33, and ΔtpsAB mutants is an alteration in the fungal cell wall (1, 6, 9, 18, 31). Furthermore, the ΔdvrA, Δace2, Δags3, and ΔtpsAB mutants all induced an exaggerated host inflammatory response (1, 9, 18). Collectively, these results suggest that the composition of the cell wall plays a key role in determining the host inflammatory response to and virulence of A. fumigatus.
In summary, A. fumigatus DvrA has a negative influence on virulence during invasive pulmonary disease and functions independently of the three known virulence-modulating genes, ace2, ags3, and ecm33. Although DvrA does not govern the adherence of A. fumigatus, it likely influences virulence by governing the capacity of the organism to damage host cells and stimulate a proinflammatory response. Studies to identify DvrA target genes that are responsible for these host cell interactions are in progress.
Supplementary Material
ACKNOWLEDGMENTS
We thank the perinatal nurses at the Harbor-UCLA Medical Center Pediatric Clinical Research Center for the collection of umbilical cords and Paolo Campoli for technical assistance. The Olympus phase-contrast microscope used for these studies was generously donated by Toyota U.S.A.
This work was supported in part by grants M01RR00425 and R01AI073829, as well as contract no. N01-AI-30041, from the National Institutes of Health. D.C.S. was supported in part by a clinician-scientist award and operating grant from the Canadian Institutes of Health Research.
Footnotes
Supplemental material for this article may be found at http://ec.asm.org/.
Published ahead of print on 30 July 2010.
REFERENCES
- 1.Al-Bader N., Vanier G., Liu H., Gravelat F. N., Urb M., Hoareau C. M., Campoli P., Chabot J., Filler S. G., Sheppard D. C. 2010. Role of trehalose biosynthesis in Aspergillus fumigatus development, stress response, and virulence. Infect. Immun. 78:3007–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amitani R., Taylor G., Elezis E. N., Llewellyn-Jones C., Mitchell J., Kuze F., Cole P. J., Wilson R. 1995. Purification and characterization of factors produced by Aspergillus fumigatus which affect human ciliated respiratory epithelium. Infect. Immun. 63:3266–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balloy V., Huerre M., Latge J. P., Chignard M. 2005. Differences in patterns of infection and inflammation for corticosteroid treatment and chemotherapy in experimental invasive pulmonary aspergillosis. Infect. Immun. 73:494–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhabhra R., Miley M. D., Mylonakis E., Boettner D., Fortwendel J., Panepinto J. C., Postow M., Rhodes J. C., Askew D. S. 2004. Disruption of the Aspergillus fumigatus gene encoding nucleolar protein CgrA impairs thermotolerant growth and reduces virulence. Infect. Immun. 72:4731–4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catlett N. L., Lee B.-N., Yoder O. C., Turgeon B. G. 2002. Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet. News 50:9–11 [Google Scholar]
- 6.Chabane S., Sarfati J., Ibrahim-Granet O., Du C., Schmidt C., Mouyna I., Prevost M. C., Calderone R., Latge J. P. 2006. Glycosylphosphatidylinositol-anchored Ecm33p influences conidial cell wall biosynthesis in Aspergillus fumigatus. Appl. Environ. Microbiol. 72:3259–3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang L. Y., Sheppard D. C., Gravelat F. N., Patterson T. F., Filler S. G. 2008. Aspergillus fumigatus stimulates leukocyte adhesion molecules and cytokine production by endothelial cells in vitro and during invasive pulmonary disease. Infect. Immun. 76:3429–3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding C., Butler G. 2007. Development of a gene knockout system in Candida parapsilosis reveals a conserved role for BCR1 in biofilm formation. Eukaryot. Cell 6:1310–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ejzykowicz D. E., Cunha M. M., Rozental S., Solis N. V., Gravelat F. N., Sheppard D. C., Filler S. G. 2009. The Aspergillus fumigatus transcription factor Ace2 governs pigment production, conidiation and virulence. Mol. Microbiol. 72:155–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraser R. S. 1993. Pulmonary aspergillosis: pathologic and pathogenetic features. Pathol. Annu. 28:231–277 [PubMed] [Google Scholar]
- 11.Fu Y., Rieg G., Fonzi W. A., Belanger P. H., Edwards J. E., Jr., Filler S. G. 1998. Expression of the Candida albicans gene ALS1 in Saccharomyces cerevisiae induces adherence to endothelial and epithelial cells. Infect. Immun. 66:1783–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glasgow S. C., Ramachandran S., Blackwell T. S., Mohanakumar T., Chapman W. C. 2007. Interleukin-1beta is the primary initiator of pulmonary inflammation following liver injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 293:L491–L496 [DOI] [PubMed] [Google Scholar]
- 13.Gravelat F. N., Doedt T., Chiang L. Y., Liu H., Filler S. G., Patterson T. F., Sheppard D. C. 2008. In vivo analysis of Aspergillus fumigatus developmental gene expression determined by real-time reverse transcription-PCR. Infect. Immun. 76:3632–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gravelat F. N., Ejzykowicz D. E., Chiang L. Y., Chabot J. C., Urb M., Macdonald K. D., Al-Bader N., Filler S. G., Sheppard D. C. 2009. Aspergillus fumigatus MedA governs adherence, host cell interactions and virulence. Cell. Microbiol. 12:473–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaffe E. A., Nachman R. L., Becker C. G., Minick C. R. 1973. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Invest. 52:2745–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamai Y., Lossinsky A. S., Liu H., Sheppard D. C., Filler S. G. 2009. Polarized response of endothelial cells to invasion by Aspergillus fumigatus. Cell. Microbiol. 11:170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopes Bezerra L. M., Filler S. G. 2004. Interactions of Aspergillus fumigatus with endothelial cells: internalization, injury, and stimulation of tissue factor activity. Blood 103:2143–2149 [DOI] [PubMed] [Google Scholar]
- 18.Maubon D., Park S., Tanguy M., Huerre M., Schmitt C., Prevost M. C., Perlin D. S., Latge J. P., Beauvais A. 2006. AGS3, an alpha(1-3)glucan synthase gene family member of Aspergillus fumigatus, modulates mycelium growth in the lung of experimentally infected mice. Fungal Genet. Biol. 43:366–375 [DOI] [PubMed] [Google Scholar]
- 19.Mylonakis E. 2008. Galleria mellonella and the study of fungal pathogenesis: making the case for another genetically tractable model host. Mycopathologia 165:1–3 [DOI] [PubMed] [Google Scholar]
- 20.Nobile C. J., Andes D. R., Nett J. E., Smith F. J., Yue F., Phan Q. T., Edwards J. E., Filler S. G., Mitchell A. P. 2006. Critical role of Bcr1-dependent adhesins in C. albicans biofilm formation in vitro and in vivo. PLoS Pathog. 2:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nobile C. J., Mitchell A. P. 2005. Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Curr. Biol. 15:1150–1155 [DOI] [PubMed] [Google Scholar]
- 22.Orciuolo E., Stanzani M., Canestraro M., Galimberti S., Carulli G., Lewis R., Petrini M., Komanduri K. V. 2007. Effects of Aspergillus fumigatus gliotoxin and methylprednisolone on human neutrophils: implications for the pathogenesis of invasive aspergillosis. J. Leukoc. Biol. 82:839–848 [DOI] [PubMed] [Google Scholar]
- 23.Patterson T. F., Kirkpatrick W. R., White M., Hiemenz J. W., Wingard J. R., Dupont B., Rinaldi M. G., Stevens D. A., Graybill J. R. 2000. Invasive aspergillosis. Disease spectrum, treatment practices, and outcomes. I3 Aspergillus Study Group. Medicine (Baltimore) 79:250–260 [DOI] [PubMed] [Google Scholar]
- 24.Perrin R. M., Fedorova N. D., Bok J. W., Cramer R. A., Wortman J. R., Kim H. S., Nierman W. C., Keller N. P. 2007. Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 3:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phan Q. T., Myers C. L., Fu Y., Sheppard D. C., Yeaman M. R., Welch W. H., Ibrahim A. S., Edwards J. E., Filler S. G. 2007. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 5:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinchai N., Juvvadi P. R., Fortwendel J. R., Perfect B. Z., Rogg L. E., Asfaw Y. G., Steinbach W. J. 2010. The Aspergillus fumigatus P-type Golgi apparatus Ca2+/Mn2+ ATPase PmrA is involved in cation homeostasis and cell wall integrity but is not essential for pathogenesis. Eukaryot. Cell 9:472–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pontecorvo G., Roper J. A., Hemmons L. M., Macdonald K. D., Bufton A. W. 1953. The genetics of Aspergillus nidulans. Adv. Genet. 5:141–238 [DOI] [PubMed] [Google Scholar]
- 28.Punt P. J., Oliver R. P., Dingemanse M. A., Pouwels P. H., van den Hondel C. A. 1987. Transformation of Aspergillus based on the hygromycin B resistance marker from Escherichia coli. Gene 56:117–124 [DOI] [PubMed] [Google Scholar]
- 29.Reeves E. P., Messina C. G., Doyle S., Kavanagh K. 2004. Correlation between gliotoxin production and virulence of Aspergillus fumigatus in Galleria mellonella. Mycopathologia 158:73–79 [DOI] [PubMed] [Google Scholar]
- 30.Richie D. L., Miley M. D., Bhabhra R., Robson G. D., Rhodes J. C., Askew D. S. 2007. The Aspergillus fumigatus metacaspases CasA and CasB facilitate growth under conditions of endoplasmic reticulum stress. Mol. Microbiol. 63:591–604 [DOI] [PubMed] [Google Scholar]
- 31.Romano J., Nimrod G., Ben-Tal N., Shadkchan Y., Baruch K., Sharon H., Osherov N. 2006. Disruption of the Aspergillus fumigatus ECM33 homologue results in rapid conidial germination, antifungal resistance and hypervirulence. Microbiology 152:1919–1928 [DOI] [PubMed] [Google Scholar]
- 32.Sanchez A. A., Johnston D. A., Myers C., Edwards J. E., Mitchell A. P., Jr., Filler S. G. 2004. Relationship between Candida albicans virulence during experimental hematogenously disseminated infection and endothelial cell damage in vitro. Infect. Immun. 72:598–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheppard D. C., Doedt T., Chiang L. Y., Kim H. S., Chen D., Nierman W. C., Filler S. G. 2005. The Aspergillus fumigatus StuA protein governs the up-regulation of a discrete transcriptional program during the acquisition of developmental competence. Mol. Biol. Cell 16:5866–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheppard D. C., Marr K. A., Fredricks D. N., Chiang L. Y., Doedt T., Filler S. G. 2006. Comparison of three methodologies for the determination of pulmonary fungal burden in experimental murine aspergillosis. Clin. Microbiol. Infect. 12:376–380 [DOI] [PubMed] [Google Scholar]
- 35.Sheppard D. C., Rieg G., Chiang L. Y., Filler S. G., Edwards J. E., Jr., Ibrahim A. S. 2004. Novel inhalational murine model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 48:1908–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spikes S., Xu R., Nguyen C. K., Chamilos G., Kontoyiannis D. P., Jacobson R. H., Ejzykowicz D. E., Chiang L. Y., Filler S. G., May G. S. 2008. Gliotoxin production in Aspergillus fumigatus contributes to host-specific differences in virulence. J. Infect. Dis. 197:479–486 [DOI] [PubMed] [Google Scholar]
- 37.Stephens-Romero S. D., Mednick A. J., Feldmesser M. 2005. The pathogenesis of fatal outcome in murine pulmonary aspergillosis depends on the neutrophil depletion strategy. Infect. Immun. 73:114–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stergiopoulou T., Meletiadis J., Roilides E., Kleiner D. E., Schaufele R., Roden M., Harrington S., Dad L., Segal B., Walsh T. J. 2007. Host-dependent patterns of tissue injury in invasive pulmonary aspergillosis. Am. J. Clin. Pathol. 127:349–355 [DOI] [PubMed] [Google Scholar]
- 39.Sundstrom P., Balish E., Allen C. M. 2002. Essential role of the Candida albicans transglutaminase substrate, hyphal wall protein 1, in lethal oroesophageal candidiasis in immunodeficient mice. J. Infect. Dis. 185:521–530 [DOI] [PubMed] [Google Scholar]
- 40.Tsuruta Y., Park Y. J., Siegal G. P., Liu G., Abraham E. 2007. Involvement of vitronectin in lipopolysaccaride-induced acute lung injury. J. Immunol. 179:7079–7086 [DOI] [PubMed] [Google Scholar]
- 41.Upton A., Kirby K. A., Carpenter P., Boeckh M., Marr K. A. 2007. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin. Infect. Dis. 44:531–540 [DOI] [PubMed] [Google Scholar]
- 42.van de Veerdonk F. L., Joosten L. A., Devesa I., Mora-Montes H. M., Kanneganti T. D., Dinarello C. A., van der Meer J. W., Gow N. A., Kullberg B. J., Netea M. G. 2009. Bypassing pathogen-induced inflammasome activation for the regulation of interleukin-1beta production by the fungal pathogen Candida albicans. J. Infect. Dis. 199:1087–1096 [DOI] [PubMed] [Google Scholar]
- 43.Zhao X., Oh S. H., Cheng G., Green C. B., Nuessen J. A., Yeater K., Leng R. P., Brown A. J., Hoyer L. L. 2004. ALS3 and ALS8 represent a single locus that encodes a Candida albicans adhesin; functional comparisons between Als3p and Als1p. Microbiology 150:2415–2428 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.