

Abstract
The constitutive activation of the anoxic redox control transcriptional regulator (ArcA) in Escherichia coli during aerobic growth, with the consequent production of a strain that exhibits anaerobic physiology even in the presence of air, is reported in this work. Removal of three terminal cytochrome oxidase genes (cydAB, cyoABCD, and cbdAB) and a quinol monooxygenase gene (ygiN) from the E. coli K-12 MG1655 genome resulted in the activation of ArcA aerobically. These mutations resulted in reduction of the oxygen uptake rate by nearly 98% and production of d-lactate as a sole by-product under oxic and anoxic conditions. The knockout strain exhibited nearly identical physiological behaviors under both conditions, suggesting that the mutations resulted in significant metabolic and regulatory perturbations. In order to fully understand the physiology of this mutant and to identify underlying metabolic and regulatory reasons that prevent the transition from an aerobic to an anaerobic phenotype, we utilized whole-genome transcriptome analysis, 13C tracing experiments, and physiological characterization. Our analysis showed that the deletions resulted in the activation of anaerobic respiration under oxic conditions and a consequential shift in the content of the quinone pool from ubiquinones to menaquinones. An increase in menaquinone concentration resulted in the activation of ArcA. The activation of the ArcB/ArcA regulatory system led to a major shift in the metabolic flux distribution through the central metabolism of the mutant strain. Flux analysis indicated that the mutant strain had undetectable fluxes around the tricarboxylic acid (TCA) cycle and elevated flux through glycolysis and anaplerotic input to oxaloacetate. Flux and transcriptomics data were highly correlated and showed similar patterns.
Escherichia coli has been studied extensively with respect to its physiology, genetics, and metabolism. One of the unique features of its metabolism is the ability to support robust growth under both oxic and anoxic conditions (38). During aerobic growth, when oxygen is used as a terminal electron acceptor, E. coli divides rapidly and produces carbon dioxide and acetate as major growth by-products (38), representing an efficient form of energy metabolism. In the absence of oxygen, E. coli and other microorganisms rely on anaerobic respiration and fermentation in order to oxidize substrates, recycle electron carriers, and produce ATP (38). This metabolic versatility of E. coli allows it to survive and thrive over a wide range of conditions.
Since the ability to produce a number of reduced by-products, such as organic acids and ethanol, is of importance in the field of metabolic engineering, the majority of metabolic engineering designs rely on anaerobic conditions (15, 30, 31). It has also been shown that E. coli strains developed for overproduction of commodity chemicals can further be improved using adaptive evolution strategies (15). Adaptive evolution is often performed through a series of dilutions allowing cells to remain within the exponential phase; the environmental conditions are hereby kept similar from passage to passage, as environmental perturbations might result in an incorrect evolutionary trajectory (15). Implementation of adaptive evolution for strains requiring strict anoxic growth conditions is a challenging task; therefore, the development of a platform strain that would be insensitive to oxygen and would exhibit similar physiological behaviors under oxic and anoxic conditions would be beneficial and would significantly simplify the process of adaptation of anaerobic strain designs. It has been shown before that the simultaneous deletion of three terminal cytochrome oxidase genes (cydAB, cyoABCD, and cbdAB) and a quinol monooxygenase gene (ygiN) results in nearly complete abolition of oxygen uptake (39). The strain harboring these four mutations was named ECOM4 (Escherichia coli cytochrome oxidase mutant 4). The ECOM4 strain was unable to undergo an aerobic-anaerobic shift and exhibited similar phenotypes under both conditions, making it suitable for use as a platform strain for the implementation and adaptation of strain designs. Moreover, a strain exhibiting fermentative growth under oxic and anoxic conditions has a potential to be used for overproduction of reduced metabolic products, such as ethanol (11, 18, 24, 32), pyruvate (52), lactate (35, 43, 51, 53), and succinate (34), in industrial settings. A comprehensive understanding of the metabolism and physiology of the platform strain is important, as it provides insights for further engineering. Therefore, it is imperative to understand how the deletions reprogram the metabolic network of E. coli. To this end, we performed whole-genome transcriptome and fluxome analysis coupled with physiological characterization under both growth conditions.
MATERIALS AND METHODS
Strains and media.
The strain described in this study was generated from the cytochrome oxidase mutant strain ECOM3, presented before (39). The quinol monooxygenase gene (ygiN) (1) was removed from the unadapted ECOM3 strain; the resulting strain harbored mutations in the cydAB, cyoABCD, cbdAB, and ygiN genes and was named ECOM4 (Escherichia coli cytochrome oxidase mutant 4). The deletion of the ygiN gene was conducted by homologous recombination of a PCR-amplified linear fragment using a lambda Red recombinase system (10). In short, the gene to be deleted was replaced by a kanamycin gene flanked by FLP recombination target (FRT) sites, and the insert was removed with an FLP recombinase. In order to verify the genotype of the mutant, single colonies were isolated from solid media and tested with PCR. Primers used for deletion and verification are presented in Table S1 in the supplemental material. Wild-type (WT) E. coli colonies were tested in parallel as a negative control. The ECOM4 strain was subjected to adaptation, physiological characterization, and clonal selection (described below) in order to identify the clone with the highest lactate yield. This clone was named ECOM4LA. All physiological and high-throughput experiments were performed using the ECOM4LA strain. Bacterial strains were cultured at 37°C in M9 minimal liquid medium containing 4 g/liter glucose, except as noted.
Adaptive evolution.
The ECOM4 strain was adaptively evolved in triplicate to grow without the need of amino acid supplementation, using a technique described earlier (15, 20, 39). The method of adaptation was similar to that described for the adaptation of the parental strain (39). In short, a colony from a fresh agar plate was inoculated in 250 ml M9 medium containing amino acid supplementation provided by EZ supplements (Teknova), grown overnight, and passed into a new flask containing fresh medium. The volume of inoculum used for the transfer into the fresh medium was calculated based on the growth rate, optical density (OD), and culture volume to make sure that cultures remained in exponential phase during the next transfer in 24 h. The amount of amino acid supplementation (EZ supplements) added to the medium was reduced exponentially during the first 2 weeks of evolution. Cells were propagated aerobically for 30 days (>500 generations) by following the protocol reported by Fong et al. (15). Evolving cultures were also supplemented with 50 μg/ml kanamycin once a week and screened daily by PCR for the presence of contaminants. Samples were frozen every 2 days throughout the evolution.
Phenotype assessment.
To assess the phenotypic characteristics of the three adapted endpoint strains, growth rates and by-product secretion profiles were measured. Each strain was grown in batch culture under oxic and anoxic conditions in triplicate. Aerobic cultivation was conducted in 500-ml Erlenmeyer flasks containing 250 ml M9 medium. The temperature was controlled by a circulating water bath, and mixing and aeration were controlled with a stir bar at ∼1,000 rpm. Anaerobic cultivation was conducted in 250-ml Erlenmeyer flasks, sealed with rubber stoppers containing the necessary inlet tubing, with 200 ml medium. Anoxic conditions were achieved by continuous flushing of cultures with a 95% N2-5% CO2 gas mixture at a flow rate of 1 ml/min. The temperature was controlled by using a circulating water bath, and mixing was controlled with a stir speed of ∼200 rpm. Samples were taken from batch cultures periodically (every 30 min), filtered through a 0.2-μm filter, and stored at −20°C for by-product analysis. Glucose concentration in the media was assessed using an enzymatic assay kit (R-Biopharm, Germany), while d-lactate secretion was measured using refractive index (RI) detection by high-performance liquid chromatography (HPLC) (Waters, MA) with a Bio-Rad Aminex HPX87-H ion exclusion column (injection volume, 10 μl) and 5 mM H2SO4 as the mobile phase (0.5 ml/min, 45°C). The identities of metabolites and organic acids in the fermentation broth were further verified with enzymatic kits. The oxygen uptake rate of each aerobic culture was determined by measuring the rate of dissolved oxygen depletion in an enclosed respirometer chamber using a polarographic dissolved oxygen probe (YSI, OH).
Clonal analysis.
Three adapted ECOM4 populations were cultured overnight on solid M9 medium with 4 g/liter glucose and 50 μg/ml kanamycin. Ten random individual colonies were selected from each plate and grown overnight in M9 liquid medium. Cells were harvested by centrifugation, washed three times with medium without a carbon source, and loaded onto a Bioscreen C machine (Growth Curves USA). Cultures were inoculated into 300-μl wells containing medium; the initial OD of each well was less than 0.05. Cells were grown for 8 h with continuous shaking to ensure good mixing, and aeration and optical density at 600 nm (OD600) measurements were taken every 15 min. Once the cells reached stationary phase, the assay was stopped and the d-lactate concentration was assessed by HPLC. Strains with the highest production yield were identified and subjected to aerobic batch cultivation. Strains were grown in 500-ml Erlenmeyer flasks with 250 ml medium for 8 h with continuous agitation, as described above. Samples were taken every 30 min, filtered, and analyzed using HPLC. Growth rate (liter/h), oxygen uptake rate (mmol/g cells [dry weight]/h), sugar uptake rate (mmol/g cells [dry weight]/h), and product secretion rate (mmol/g cells [dry weight]/h) were measured as described above.
Transcriptome analysis.
Cultures were grown to the mid-log growth phase aerobically and anaerobically (OD600 of ∼0.6 for the WT and OD600 of ∼0.25 for ECOM4LA). The cultures (3 ml of the WT and 7 ml of ECOM4LA) were then added to 2 volumes of RNAprotect bacterial reagent (Qiagen, CA), and total RNA was isolated by using RNeasy columns (Qiagen, CA) with DNase I treatment. Total RNA yields were measured by using a spectrophotometer (A260), and quality was checked by visualization on agarose gels and by measurement of the sample A260/A280 ratio (>1.8). cDNA preparation was performed as described by Cho et al. (7). Affymetrix GeneChip E. coli Genome 2.0 arrays were used for genome-scale transcriptional analyses. cDNA synthesis, fragmentation, end-terminus biotin labeling, and array hybridization were performed as recommended by Affymetrix standard protocols. Differentially expressed genes were selected by using the fold change threshold and the Student t test with false discovery rate (FDR) correction as implemented in ArrayStar 3 software (DNAStar, WI). Genes with at least a 2-fold change in expression level and an FDR-adjusted P value of less than 0.05 were considered significant and were used for strain analysis. Transcriptome data were mapped to iAF1260 metabolic reconstruction of E. coli (13) by using the Simpheny software platform (Genomatica, CA).
The probability of regulon and gene ontology term enrichment among differentially expressed genes was computed using hypergeometric distribution. Regulons were obtained from RegulonDB v6.0 (16) and gene ontology terms from Ecocyc v12.0 (28, 29). Correction for multiple hypotheses was done as reported by Storey and Tibshirani (46) (FDR of 0.01). Consistency of differential expression with ArcA and FNR activity in the respective regulons was determined by comparing differential expression (up- or downregulation) with increased ArcA or FNR activity (activator or repressor) as reported by RegulonDB.
Quantitative PCR analysis.
RNA purification and cDNA synthesis were conducted following the same protocol as described for the gene expression analysis. The 50-μl quantitative PCR (qPCR) mixtures contained 25 μl of SYBR green Taq master mix (Qiagen, CA), 0.2 μM forward primer, 0.2 μM reverse primer, and cDNA as a template. Each qPCR was run in triplicate in a Bio-Rad thermocycler (Bio-Rad, CA) at 95°C for 15 min, 94°C for 15 s, 52°C for 30 s, and 72°C for 30 s; the denaturation, annealing, and extension steps were repeated for 40 cycles. Targeted gene expression of the mutant strain was analyzed under oxic and anoxic growth conditions and compared to that of the WT. By use of a standard curve for each primer set, the relative cDNA quantity for each gene was obtained by normalizing it to the quantity of acpP (acyl carrier protein) cDNA in the same sample. acpP was chosen as the internal control gene, since it is constitutively expressed in the WT and mutant strains under both aerobic and anaerobic conditions (9).
13C tracing studies. (i) Culture labeling.
Prior to labeling, single colonies were selected from stock plates and inoculated directly into 250 ml M9 medium in 500-ml Erlenmeyer flasks aerated by stirring at 1,000 rpm. Cells were grown overnight, harvested, washed twice with water, and used to inoculate 50-ml flasks containing 25 ml medium with 2 g/liter 13C-labeled d-glucose, with an initial OD600 of 0.005 to 0.01. Glucose was supplied as 100% 1-13C labeled, 100% 6-13C labeled, or a mixture of 20% uniformly (U-13C) labeled and 80% natural (which is randomly 1% 13C). Cells were grown to mid-log phase, corresponding to an OD600 of 0.6 (WT) or 0.25 (ECOM4LA). A portion (3 ml, WT; 10 ml, ECOM4LA) of each culture was harvested by centrifugation at 4°C. Media were aspirated and analyzed by HPLC to determine the remaining glucose concentration. Cell pellets were placed at −80°C prior to further analysis.
(ii) Derivatization and GC-MS analysis.
Cells were resuspended in 0.1 ml 6 M HCl and transferred to glass vials, and protein was digested into amino acids under a nitrogen atmosphere for 18 h at 105°C in an Eldex H/D work station. Digested samples were dried to remove residual HCl, resuspended with 75 μl each of tetrahydrofuran and N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide, and incubated for 1 h at 80°C to derivatize amino acids. Samples were filtered through 0.2-μm polyvinylidene difluoride (PVDF) filters and injected into a Shimadzu QP2010 Plus quadrupole gas chromatography-mass spectroscopy (GC-MS) instrument (0.5 μl, with a 1:50 split ratio). The GC injection temperature was 250°C, and the GC oven temperature was initially 130°C for 4 min, rising to 230°C at 4°C/min and to 280°C at 20°C/min, with a final hold at this temperature for 2 min. The GC flow rate with the helium carrier gas was 50 cm/s. The GC column used was a 15-m by 0.25-mm by 0.25-μm SHRXI-5ms column (Shimadzu). The GC-MS interface temperature was 300°C, and the (electron impact) ion source temperature was 200°C, with 70-eV ionization voltage. The mass spectrometer was set to scan an m/z range of 50 to 600.
(iii) Processing of GC-MS data.
Mass data were retrieved from the GC-MS instrument for fragments of 14 derivatized amino acids: cysteine and tryptophan were degraded during amino acid hydrolysis; asparagine and glutamine were converted to, respectively, aspartate and glutamate; and arginine was not stable to the derivatization procedure. For each fragment, these data comprised mass intensities for the base isotopomer (without any heavy isotopes, M+0) and isotopomers with increasing unit masses (up to M+6) relative to that of M+0. These mass distributions were normalized by dividing by the sum of M+0 to M+6 and corrected for naturally occurring heavy isotopes of the elements H, N, O, Si, S, and (in moieties from the derivatizing reagent) C, using matrix-based probabilistic methods as described previously (37, 49), implemented in Microsoft Excel. Data were also corrected for carryover of unlabeled inoculum (37).
Corrected mass distributions for amino acid fragments from [U-13C]glucose-labeled cells were used to infer the trafficking and reassortment through metabolism of linked chains of carbons derived from glucose, while mass distribution data from [1-13C]glucose- or [6-13C]glucose-labeled cells were used to track the fate of individual carbon atoms. The analysis is summarized here and is described in more detail in the supplemental methods (see the supplemental material).
Amino acid labeling data originating from [U-13C]glucose was used to estimate two aspects of pentose phosphate pathway (PPP) flux. The mass distribution data for alanine (as a marker for pyruvate) were used to calculate the fraction of alanine originating from the PPP versus that from glycolysis. Flux from glucose via glucose-6-phosphate to pentose-5-phosphates (P5P) (ribose-5-phosphate, xyulose-5-phosphate, and ribulose-5-phosphate, all assumed to be in equilibrium) in the oxidative PPP and back to glycolytic intermediates in the nonoxidative PPP ultimately yields 5 pyruvate molecules per 3 input glucose molecules. Of these 5 pyruvate molecules, 3 are composed of 3-carbon units linked as they were in glucose (same as pyruvate produced via glycolysis), and 2 are reassorted such that C-1 has a different origin from the rest of the molecule (48). The fraction of pyruvate split across the C-1-C-2 bond was calculated from the mass distributions of alanine fragments.
Second, histidine labeling from [U-13C]glucose was used to calculate the relative input to P5P from the oxidative or nonoxidative PPP. The carbon backbone of histidine is equivalent to P5P plus one carbon from the tetrahydrofolate-linked one-carbon (1-C) pool. Input to P5P from the oxidative PPP removes the C-1 carbon from glucose, but otherwise the carbon backbone remains intact (giving an M+5 P5P fraction). In contrast, inputs from the nonoxidative PPP necessarily yield reassorted P5P, with the split between different source molecules being largely across the C-2-C-3 bond (yielding M+2 or M+3 P5P).
Data from [1-13C]glucose labeling experiments were used to provide another measure of flux through the PPP versus glycolysis. As noted above, glucose routed through the oxidative branch of the PPP loses carbon from position 1 as CO2. Therefore, by measuring the degree of loss of the 13C label in alanine (pyruvate) in [1-13C]glucose-labeled cells, the relative flux through glycolysis versus the PPP was calculated.
Mass data for [U-13C]glucose labeling of aspartate, which was assumed to be in equilibrium with oxaloacetate (OAA), were used to assess the relative inputs to OAA from the tricarboxylic acid (TCA) cycle versus the anaplerotic reactions catalyzed by phosphoenolpyruvate carboxylase (PEPC) and malic enzyme. In broad terms, input from anaplerosis was apparent as +3 mass unit labeling of aspartate, indicative of incorporation of linked 13C 3-carbon units arising from PEP or pyruvate, while input from the TCA cycle appeared as +2 mass unit labeling, indicative of input of 2-carbon units originating as acetyl coenzyme A (acetyl-CoA). Data from various fragments of aspartate were used to calculate the back flux in the TCA cycle from oxaloacetate to symmetrical metabolites (e.g., fumarate) and the 13C labeling of cellular CO2/bicarbonate. The 13C labeling pattern of anaplerotic input to oxaloacetate was then modeled as the product of CO2 labeling and 13C labeling of alanine C-1-C-3 (as a surrogate for pyruvate or PEP), while the input to oxaloacetate from α-ketoglutarate in the TCA cycle was assumed to correspond to the labeling of glutamate (C-2-C-5 fragment). The relative contributions of these inputs to oxaloacetate were then calculated using the least-squares fit in MATLAB. These results were checked with alternate amino acid fragments providing the inputs (see the supplemental methods).
[1-13C]glucose or [6-13C]glucose data were used to calculate relative flux from glucose to pyruvate through the Entner-Doudoroff (ED) pathway versus glycolysis or the PPP. The ED pathway, in contrast to the other two pathways, converts [1-13C]glucose to [1-13C]pyruvate and not [3-13C]pyruvate. Flux through the ED pathway was therefore estimated by comparing labeling of C-1-C-3 and C-2-C-3 fragments of alanine (14).
Positionally labeled glucose data were also used to determine 13C labeling of the 1-C pool, utilizing methionine and aspartate labeling data, as methionine is produced from aspartate plus a 1-C unit. Furthermore, the relative contributions of serine and glycine to the 1-C pool were determined, based on the labeling of the C-3 position of serine and C-2 position of glycine.
Quinone extraction.
Ubiquinone-8 (UQ) and menaquinone (MQ) extraction was conducted according to a protocol outlined previously (4, 5, 42). In short, 2 ml of WT culture and 4 ml of ECOM4LA culture were quenched with 6 ml of ice-cold methanol. Next, 6 ml of petroleum ether was added rapidly, and the mixture was vortexed for 1 min. Following centrifugation of the mixture (900 × g for 2 min), the top phase was transferred into a new tube. Another 3 ml of petroleum ether was added, and the vortexing and centrifugation steps were repeated. The upper phases were combined and allowed to evaporate to dryness. Dried extracted quinones were resuspended in 100 μl of ethanol and analyzed using an HPLC system fitted with a Pursuit XRs (Varian, CA) C18 reverse-phase column, with methanol as a mobile phase and a flow rate of 1.0 ml/min at ambient temperature. Detection of quinones was conducted using a dual-wavelength UV detector at 290 nm for UQ and 248 nm for MQ (4, 42). Ubiquinone-10 and menaquinone-4 were used as standards. The total amount of each species was calculated using the relevant peak area, plotted against the molar absorption coefficient as described by Shestopalov et al. (42). Analytical-grade methanol, petroleum ether, and ethanol were acquired from Sigma-Aldrich.
Microarray data accession number.
Microarray data sets have been deposited in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) (3, 12) and assigned accession number GSE21839.
RESULTS
Strain engineering and adaptive evolution.
The ECOM4 strain was constructed from the unadapted ECOM3 strain previously described by us (39). In order to alleviate the need for amino acid supplementation, we adaptively evolved the ECOM4 strain in triplicate (ECOM41, ECOM42, and ECOM43), using the same technique reported earlier (39). As a result of the 30-day period of adaptation, the amino acid supplementation was successfully removed, and the lactate yield decreased slightly. The evolved oxygen uptake rate was nearly identical to that for the unadapted ECOM4 strain, at a level of 0.25 ± 0.12 mmol/g cells (dry weight)/h, and nearly 60 times lower than the oxygen uptake rate of WT E. coli (see Fig. S1 in the supplemental material). Detailed phenotypic data, such as growth rate, glucose and oxygen uptake rates, and by-product secretion rates, measured during the adaptation are presented in Table S2 in the supplemental material. In order to characterize and study heterogeneity in the adapted populations, we used clonal analysis (see Fig. S2 in the supplemental material). A single clone (ECOM4LA) was selected from one of the three adapted populations (ECOM41), based on the highest lactate yield. The following experiments were performed in triplicate using the ECOM4LA clone.
Phenotypic characterization revealed a substantial similarity between ECOM4LA strains grown aerobically and anaerobically.
The ECOM4LA strain was predicted to have similar growth rates irrespective of the oxygen supply. Growth rates were comparable for aerobic and anaerobic conditions, being 0.32 ± 0.02 liter/h and 0.27 ± 0.06 liter/h, respectively. Similarly, the conversion of glucose to d-lactate was affected only slightly by oxygen supply, with yields of 98% and 92% for aerobic and anaerobic growth conditions, respectively (Table 1). Lactate was produced with a 70% yield during the exponential phase and with a nearly 100% yield during the stationary phase (see Fig. S3 in the supplemental material), illustrating that 30% of carbon was directed toward biomass formation. Succinic acid was present in a small amount during the exponential growth phase and was metabolized in stationary phase. The highest measured concentration of succinate during the exponential phase was on the order of 30.0 ± 15.0 mg/liter; however, this measurement was highly variable due to reuptake of succinate. Based on these results, we hypothesized that the ECOM4LA strain might use the anaerobic respiratory chain in order to remove excess electrons during aerobic growth (see Discussion), while the majority of the electrons are removed by means of d-lactate production. Gene expression analysis was used to determine metabolic changes and to decipher possible regulatory alterations that underlie the inability of ECOM4LA to undergo the normal aerobic-anaerobic shift.
TABLE 1.
Phenotypic characteristics of the ECOM4LA and WT strains under oxic and anoxic conditions
| Parameter measured | Value (mean ± SD)a for: |
|||
|---|---|---|---|---|
| ECOM4LA |
MG1655 |
|||
| With O2 | Without O2 | With O2 | Without O2 | |
| Growth rate (liter/h) | 0.32 ± 0.005 | 0.27 ± 0.006 | 0.71 ± 0.01 | 0.45 ± 0.02 |
| Glucose uptake rate (mmol/g cells [dry wt]/h) | 26.4 ± 0.09 | 24.9 ± 0.72 | 9.02 ± 0.23 | 17.3 ± 0.17 |
| Lactate secretion rate (mmol/g cells [dry wt]/h) | 48.6 ± 0.76 | 41.58 ± 1.58 | 0 | 0.95 ± 0.008 |
| Acetate secretion rate (mmol/g cells [dry wt]/h) | 0 | 0 | 3.37 ± 0.9 | 10.3 ± 0.60 |
| Oxygen uptake rate (mmol/g cells [dry wt]/h) | 0.21 ± 0.16 | 0 | 16.49 ± 0.67 | 0 |
| Lactate/glucose (g/g) | 0.98 ± 0.07 | 0.92 ± 0.04 | NAb | NA |
Cells were grown in triplicate.
NA, not available.
Gene expression analysis reveals a shift to anaerobic metabolism in ECOM4LA under oxic conditions.
Genome-wide transcriptomic profiles were determined for the WT and ECOM4LA strains under aerobic and anaerobic conditions. Expressed genes were selected based on criteria described above (see Materials and Methods). The gene expression comparison between aerobic and anaerobic growth of WT E. coli revealed that 564 genes (13% of the genome, based on 4,468 total genes in the E. coli genome [28, 29]) had significant changes in expression (see Fig. S3 in the supplemental material). Comparison of mRNA transcript levels between ECOM4LA and the WT under oxic growth conditions revealed that 538 genes were significantly affected, accounting for nearly 13% of the genome, similar to the previous comparison. Interestingly, we observed that only ∼6% of the genome (250 genes) was affected by an aerobic-anaerobic shift in the ECOM4LA cell line (see Fig. S4 in the supplemental material). This observation suggested that the inability to utilize oxygen has a significant effect on global gene expression and regulation, which contributes significantly to the inability of the ECOM4LA strain to undergo an aerobic-anaerobic shift.
Gene ontology term enrichment was employed to identify biological processes that are enriched within genes differentially expressed between various experimental conditions. Interestingly, the WT aerobic-anaerobic shift and the aerobic WT/ECOM4LA comparison shared several enriched metabolic biological processes, such as “aerobic respiration,” “anaerobic respiration,” “tricarboxylic acid cycle,” “oxidation reduction,” and “glycolysis” (see Table S3 in the supplemental material). Moreover, in the comparison between the WT and ECOM4LA under oxic conditions, most of the significantly enriched gene ontology terms in the downregulated genes were similar to the enriched terms in the WT aerobic-anaerobic shift (see Table S4 in the supplemental material), and these were dominated by metabolic processes.
Since metabolic terms dominated in the differentially expressed genes, we mapped the transcriptomic data onto the E. coli metabolic network reconstruction (13). Central metabolism was analyzed in detail (i.e., glycolysis, TCA cycle, pentose phosphate pathway [PPP], and fermentative pathways). We considered the gene expression patterns acquired from aerobic and anaerobic growth in the WT as a benchmark to which we compared gene expression in the ECOM4LA strain under similar conditions. We observed that during the aerobic-anaerobic shift, the WT downregulated the TCA cycle and upregulated expression of certain enzymes involved in glycolysis and fermentative pathways, such as formate, acetate, and succinate (Fig. 1 A). When examining differences in gene expression between the ECOM4LA strain and the WT grown in an oxic environment, we noticed that the majority of genes involved in glycolysis were significantly upregulated, while genes involved in the TCA cycle were downregulated, in ECOM4LA (Table 2; Fig. 1B).
FIG. 1.

Transcriptomics analysis of the ECOM4LA and MG1655 strains under oxic and anoxic conditions. Mean gene expression data (fold change) acquired from the WT and ECOM4LA strains grown in triplicate were mapped onto the metabolic map of central metabolism. Changes in gene expression values of 2-fold and higher were considered significant (P < 0.05). Red, at least 2-fold upregulation; green, at least 2-fold downregulation; yellow, no change. The boxes in panel B enclose the branching areas of metabolism for which relative metabolic flux is illustrated in Fig. 3.
TABLE 2.
Comparison between gene expression levels in ECOM4LA and WT MG1655 cells grown aerobically and anaerobically
| Gene name | Locus tag | Gene product | Mean fold change in expression (adjusted P value)a |
|||
|---|---|---|---|---|---|---|
| E4 +O2/WT +O2 | E4 −O2/WT −O2 | WT −O2/WT +O2 | E4 +O2/E4 −O2 | |||
| pgi | b4025 | Phosphoglucose isomerase | 3.87 (0.017) | NC | NC | NC |
| pfkABb | b3916 | 6-Phosphofructose kinase | 2.04 (0.003) | NC | 2.29 (0) | NC |
| fbaA | b2925 | Fructose bisphosphate aldolase | 3.00 (0.01) | NC | NC | NC |
| fbaB | b2097 | Fructose bisphosphate aldolase | 8.77 (0) | 3.30 (0) | 3.23 (0) | NC |
| tpiA | b3919 | Triose phosphate isomerase | 3.33 (0.005) | NC | NC | NC |
| gapA | b1779 | Glyceraldehyde 3-phosphate | 4.53 (0.02) | NC | 2.53 (0.05) | NC |
| pgk | b2926 | Phosphoglycerate kinase | 2.10 (0.02) | NC | NC | NC |
| eno | b2779 | Enolase | 3.31 (0.008) | NC | NC | NC |
| ldhA | b1380 | d-Lactate dehydrogenase | 3.36 (0.005) | NC | NC | NC |
| yieF | b3713 | NADH:menaquinone oxydoreductase | 2.47 (0.014) | 2.09 (0.001) | NC | 1.60 (0.023) |
| wrbA | b1004 | NADH:menaquinone oxydoreductase | 9.10 (0.002) | 3.22 (0.001) | 5.07 (0.003) | NC |
| frdABCDb | b4151-b4154 | Fumarate reductase | 3.60 (0.02) | NC | 4.03 (0.002) | 1.90 (0.02) |
| sdhABCDb | b0721-b0724 | Succinase dehydrogenase | −25.35 (0) | NC | −33.50 (0) | NC |
| nuoA-nuoNb | b2288-b2276 | NADH:ubiquinone oxidoreductase | −2.10 (0.005) | NC | −2.30 (0.001) | NC |
Benjamini-Hochberg false-discovery rate-adjusted P value. Cells were grown in triplicate. Changes of <2-fold were considered no change (NC). E4, ECOM4LA strain; WT, wild-type MG1655.
Average expression values are presented for large operons.
Similar expression patterns were observed between the WT grown under anaerobic conditions (anaerobic WT) and ECOM4LA grown under aerobic conditions (aerobic ECOM4LA), and both were compared to the WT grown under aerobic conditions (aerobic WT) (Fig. 1A and B). These results suggest that the ECOM4LA strain relies on glycolysis under oxic growth conditions for ATP generation through substrate-level phosphorylation. This result might be attributed to deletions in respiratory chain genes and an inability to build a proton gradient sufficient to produce ATP by ATP-synthase. Also, upregulation of the anaplerotic reaction (from phosphoenolpyruvate to oxaloacetate) was observed in the ECOM4LA strain. We also noticed that the lactate dehydrogenase (ldhA) gene was upregulated over 3-fold, similarly to what was observed earlier for the parent strain (39). Expression changes observed in anaerobic ECOM4LA were comparable to but less profound than those observed in the aerobic WT (Fig. 1C). We observed significant downregulation of the TCA cycle and upregulation of some glycolytic enzymes, suggesting that similar regulatory mechanisms are active in the ECOM4LA strain under both environmental conditions.
The most interesting result was observed when we mapped the gene expression of the ECOM4LA strain during an aerobic-anaerobic shift. This comparison indicated no changes in gene expression for enzymes involved in central metabolism in ECOM4LA under oxic and anoxic conditions (Fig. 1D). The only gene that had almost a 2-fold increase in expression was the fumarate reductase gene (frdABCD). Other significantly expressed genes mapped sparsely, without any definite pattern, onto the entire metabolic map in the iAF1260 metabolic model. The lack of more-significant changes in gene expression between aerobic and anaerobic profiles for the ECOM4LA strain illustrates that functionality of the central metabolism of the respiratory chain-deficient ECOM4LA strain has been reduced to allow the strain to perform similar functions under both studied growth conditions.
Gene expression suggests that ArcA is active in aerobic ECOM4LA, while FNR is not.
Since differential expressions of central metabolic genes are similar in ECOM4LA and in anaerobically grown WT, we asked whether this anaerobic behavior in ECOM4LA extended beyond its metabolism. E. coli has two different regulators that control expression of genes involved in the aerobic-anaerobic shift, the ArcB/ArcA two-component system and FNR. FNR is a transcriptional regulator whose activity is regulated directly by oxygen (25); therefore, it is expected that under aerobic conditions, the FNR regulon would not be significantly differentially expressed between the WT and ECOM4LA. Consistent with this, the FNR regulon (from RegulonDB v6.0) does not have more differentially expressed genes in the microarray data than expected by chance (P = 0.11, hypergeometric test). Moreover, in the comparison between aerobic WT and ECOM4LA, less than 4% of the FNR regulon (excluding ArcA/FNR-coregulated genes) is differentially expressed in the direction consistent with FNR activity (Fig. 2 A). Likewise, the comparison between the WT and ECOM4LA under anoxic conditions shows little difference. However, for both strains, the shift from aerobic to anaerobic conditions clearly causes gene expression changes in the FNR regulon consistent with known FNR activity of activation or repression (Fig. 2A), suggesting that FNR activity changes in the aerobic-anaerobic shift but not between these two strains.
FIG. 2.
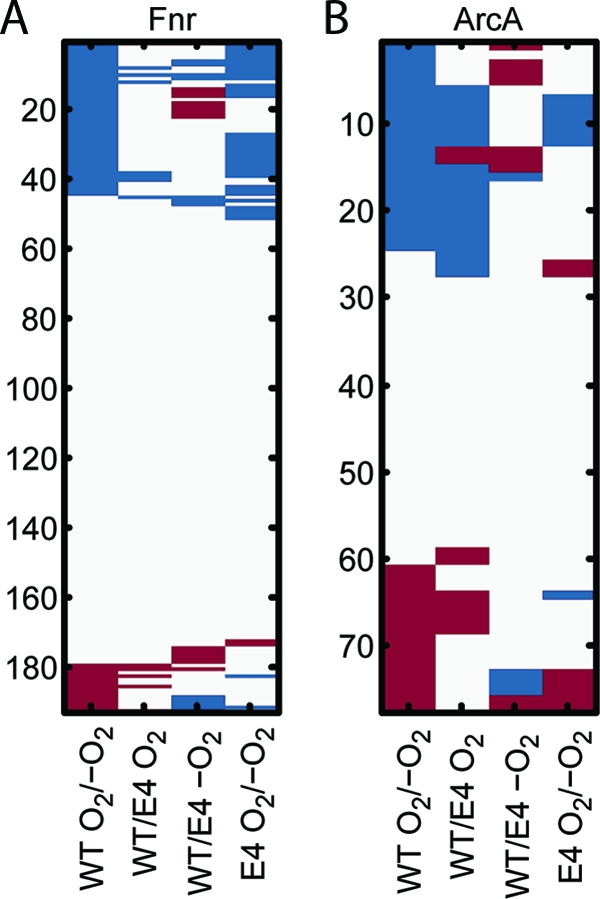
Global gene expression suggests that ArcA is active in ECOM4LA in aerobic conditions. Mean gene expression data (fold change), acquired from the WT and ECOM4LA strains grown in triplicate, for all genes that are known to be regulated by the anaerobic regulator FNR or ArcA according to RegulonDB were compared to the reported function of the regulator (activator or repressor). If a gene was differentially expressed in the direction of known regulatory activity, then the expression change is said to be consistent and is shown in blue. If the gene expression change is in the opposite direction of regulator activity, it is shown in red. Since RegulonDB regulatory logic is inferred partially from microarray data for these transcription factors, it is expected that some normal ArcA and FNR activity will be in the direction opposite from RegulonDB assignments. (A) The FNR regulon showed greater differential expression and greater consistency (blue) for the WT aerobic-anaerobic shift and the ECOM4LA (E4) aerobic-anaerobic shift. (B) The ArcA regulon shows greater differential expression and greater consistency for the WT aerobic/anaerobic shift and in the comparison between aerobic WT and aerobic ECOM4LA, thereby suggesting that ArcA is active in ECOM4LA, even under aerobic growth.
Conversely, the analysis of the ArcA regulon revealed significant differences between ECOM4LA and the WT but little ArcA-associated change in the ECOM4LA aerobic-anaerobic shift. When the WT and ECOM4LA are compared, the ArcA regulon is enriched among the differentially expressed genes (P = 4.5 × 10−12, hypergeometric test) and is the second most significantly enriched regulon for this condition (see Table S4 in the supplemental material). Moreover, in the WT aerobic-anaerobic shift, 71% of the differentially expressed ArcA regulon genes that are consistent with reported ArcA function (as an activator/repressor) also show the same consistency when the WT and ECOM4LA are compared under aerobic conditions (Fig. 2B). Furthermore, genes differentially expressed between the WT and ECOM4LA under aerobic conditions were consistent with known functions of ArcA (P = 0.03, Fischer's exact test). Conversely, few genes in the ArcA regulon are significantly differentially expressed between anaerobic WT and ECOM4LA or between aerobic and anaerobic ECOM4LA, suggesting that ArcA activities are similar in these three scenarios. Together, these results show that gene expression changes are consistent with ArcA being active in ECOM4LA under aerobic conditions.
The quinone pool is dominated by menaquinones in ECOM4LA under oxic conditions.
The activation of ArcA under aerobic conditions may be due to the fact that ArcA is a part of a two-component regulatory system that responds to the redox state of the quinone pool (4). Since the aerobic respiratory chain cannot be utilized in ECOM4LA, the ubiquinone pool is diminished and complemented by menaquinones that are involved in the anaerobic respiration mechanism (from NADH to fumarate). Relative amounts of ubiquinone and menaquinone species present in actively growing ECOM4LA and WT strains under oxic and anoxic conditions were measured (see Fig. S5 in the supplemental material). Consistent with previous reports, the quinone pool of the WT is dominated by ubiquinones during aerobic growth (800 ± 60 nmol/g cells [dry weight]) and by menaquinones during anaerobic growth (650 ± 80 nmol/g cells [dry weight]) (5). For the ECOM4LA strain, we observed that the ubiquinone contents were 150.0 and 200.0 nmol/g cells (dry weight) under anaerobic and aerobic conditions, respectively, while menaquinones were present at much higher concentrations under both conditions (500.0 ± 150.0 nmol/g cells [dry weight] under anaerobic conditions and 450.0 ± 100.0 nmol/g cells [dry weight] under aerobic conditions) (see Fig. S5 in the supplemental material). Since ECOM4LA cannot utilize molecular oxygen, the quinone pool in the mutant strain has a completely different content from that of the WT. The ECOM4LA quinone pool is dominated primarily by menaquinones under both conditions. It has been shown that the presence of ubiquinones inhibits activation of the ArcB/ArcA system, while an abundance of menaquinones alleviates this inhibition, leading to activation of ArcA (4).
These results suggest that the anaerobic phenotype of aerobically growing ECOM4LA is due to the activation of the ArcA regulon by a disruption in the cellular redox balance. Under oxic conditions, phosphorylated ArcA activates numerous operons involved in fermentative metabolism (6, 33) and represses operons involved in respiratory metabolism (19).
Targeted gene expression measurements.
In order to validate observed levels of gene expression, we used qPCR. Genes selected for qPCR analysis under oxic conditions, in ECOM4LA compared to the WT, included NADH:menaquinone oxidoreductase (yieF and wrbA), fumarate reductase (frdABCD), and succinate dehydrogenase (sdhABCD) genes. qPCR analysis confirmed that yieF was upregulated nearly 10-fold, wrbA was upregulated over 40-fold, and the frdABCD operon was upregulated over 50-fold in the aerobic ECOM4LA strain compared to levels in the aerobic WT strain. We observed a significant downregulation (over 25-fold) of the sdh operon in aerobic ECOM4LA, which is similar to levels for the WT under anoxic conditions (over 30-fold downregulation) (see Table S6 in the supplemental material). These findings, together with the observed downregulation of the TCA cycle, imply that the regulation in the aerobic ECOM4LA cell line is similar to that in anaerobic WT E. coli.
Carbon labeling experiments.
Gene expression analysis indicated major differences in metabolism of ECOM4LA and its WT E. coli parent when the strains were grown aerobically. To confirm this, metabolism was assayed directly by 13C labeling of both strains under aerobic conditions. 13C labeling was used to infer relative flux through different sections of central metabolism, particularly the pentose phosphate pathway (PPP), glycolysis, and the TCA cycle (Table 3).
TABLE 3.
Physiological parameters inferred from 13C labeling data
| Parameter measured | Glucose substrate-labeled amino acid(s)g | Mechanism or pathway | Valueh (mean % ± SD) for: |
|
|---|---|---|---|---|
| WT | ECOM4LA | |||
| Input to oxaloacetatea | [U-13C]glucose ASP, ALA, GLU | Anaplerosis | 62.4 ± 0.5 | 99.5 ± 1.4 |
| [U-13C]glucose ASP, ALA, GLU | TCA cycle | 37.6 ± 0.5 | 0.5 ± 1.4 | |
| Recycling of oxaloacetate to/from fumarate/succinateb | [U-13C]glucose ASP | 86.7 ± 4.9 | 39.2 ± 0.6 | |
| CO2 labelingc | [U-13C]glucose, [1-13C]glucose, or [6-13C]glucose ASP | [U-13C]glucose | 19.1 ± 0.2 | 14.5 ± 1.3 |
| [U-13C]glucose, [1-13C]glucose, or [6-13C]glucose ASP | [1-13C]glucose | 11.2 ± 1.8 | 9.3 ± 0.4 | |
| [U-13C]glucose, [1-13C]glucose, or [6-13C]glucose ASP | [6-13C]glucose | 1.7 ± 2.4 | −0.2 ± 0.5 | |
| Flux through PPP to pyruvate/alanined | [U-13C]glucose ALA | 13.2 ± 1.7 | −2.8 ± 1.3 | |
| Flux through oxidative PPPe | [1-13C]glucose ALA | 15.1 ± 1.2 | 1.7 ± 0.5 | |
| Flux through oxidative/nonoxidative PPP | [U-13C]glucose HIS | Oxidative PPP | 19.2 ± 1.6 | 13.2 ± 0.6 |
| into ribose | [U-13C]glucose HIS | Nonoxidative PPP | 80.8 ± 1.6 | 86.8 ± 0.6 |
| 1-C pool labeling | [1-13C]glucose or [6-13C]glucose MET, ASP | [1-13C]glucose | 37.3 ± 0.5 | 46.8 ± 0.4 |
| [1-13C]glucose or [6-13C]glucose MET, ASP | [6-13C]glucose | 46.8 ± 0.2 | 48.2 ± 0.1 | |
| Origin of 1-C pool | [1-13C]glucose or [6-13C]glucose SER, GLY, MET, ASP | From serine ([1-13C]glucose) | 93.6 ± 2.4 | 97.5 ± 1.9 |
| [1-13C]glucose or [6-13C]glucose SER, GLY, MET, ASP | From serine ([6-13C]glucose) | 91.9 ± 0.1 | 98.8 ± 0.4 | |
| [1-13C]glucose or [6-13C]glucose SER, GLY, MET, ASP | From glycine ([1-13C]glucose) | 6.4 ± 2.4 | 2.5 ± 1.9 | |
| [1-13C]glucose or [6-13C]glucose SER, GLY, MET, ASP | From glycine ([6-13C]glucose) | 8.1 ± 0.1 | 1.2 ± 0.4 | |
| Flux through Entner-Doudoroff pathwayf | [1-13C]glucose or [6-13C]glucose ALA | [1-13C]glucose | 1.4 ± 1.0 | 0.1 ± 0.7 |
| [1-13C]glucose or [6-13C]glucose ALA | [6-13C]glucose | 1.0 ± 0.2 | 0.3 ± 1.0 | |
Anaplerosis via phosphoenolpyruvate carboxylase or malic enzyme.
This is a measure of reorientation of oxaloacetate after cycling through symmetrical intermediates (succinate and fumarate) and does not include oxaloacetate derived from the TCA cycle; this portion of the oxaloacetate pool is by default randomly oriented, as it is derived from succinate and fumarate.
For [U-13C]glucose, the potential maximum is 21% (from 20% [U-13C]glucose and 1% natural label). For [1-13C]glucose or [6-13C]glucose, the potential maximum is 100%, from 100% labeled glucose as the sole carbon source.
Relative to that from glycolysis.
Measured by loss of label into alanine (pyruvate) relative to that from glycolysis.
Flux through the Entner-Doudoroff pathway relative to that via other routes from glucose to pyruvate/alanine.
ASP, aspartate; ALA, alanine; GLU, glutamate; HIS, histidine; MET, methionine; SER, serine; GLY, glycine.
From determinations on amino acids prepared from 3 separate cultures of the WT and ECOM4LA.
Pentose phosphate pathway and glycolysis.
PPP versus glycolytic flux was calculated in two ways. First, flux was estimated from labeling patterns of alanine produced from [U-13C]glucose. According to the calculation of Szyperski (48) for the reassortment of intermediates in the nonoxidative branch of the PPP (leading to a reassortment of C-1-C-2 in pyruvate and therefore alanine), in the WT, a maximum of 13% of pyruvate was formed from the PPP. In ECOM4LA, the calculated percentage was −2% (or effectively zero) (Table 3).
Similar values were found for flux through the PPP versus glycolysis by using [1-13C]glucose-generated data. Here, alanine labeling patterns were analyzed for a loss of the 13C label as a consequence of the loss of the labeled 1-carbon of glucose as CO2 during transit through the oxidative branch of the PPP. This analysis yielded values for PPP flux of 15% in the WT and 2% in ECOM4LA, relative to glycolysis flux. If it is assumed that all glucose taken up was channeled to glycolysis or the PPP, then the relative PPP flux can be converted to an absolute flux by multiplying these percentages by the measured glucose uptake rates (Table 1). This calculation gives PPP flux values of 1.4 and 0.5 mmol/g cells (dry weight)/h for the WT and ECOM4LA, respectively.
A different perspective on the PPP was provided by analyzing histidine labeling from [U-13C]glucose cultures. From histidine labeling patterns, it was possible to calculate relative input to the P5P pool (including ribose-5-phophate needed for RNA and DNA synthesis) from the oxidative or nonoxidative PPP. In the WT and ECOM4LA, respectively, 19% and 13% of input to P5P were from the oxidative PPP, with the balance from the nonoxidative PPP (Table 3). The slightly stronger preference for the nonoxidative PPP in ECOM4LA than in the WT corresponds to the generally enhanced expression of nonoxidative PPP genes in ECOM4LA versus expression in the WT, while expression of most of the nonoxidative PPP genes shows no difference (Fig. 1A and B).
Functioning of the TCA cycle.
Amino acid labeling data from [U-13C]glucose-grown E. coli were used to determine the relative input of anaplerosis (via PEPC or malic enzyme) versus the TCA cycle to OAA (aspartate) (Fig. 3C; Table 3). WT E. coli strains were found to have a split input to OAA, ∼40% from the TCA cycle and 60% from anaplerotic reactions. This result is similar to that found previously for WT E. coli strains growing in glucose minimal medium in aerated flasks (14, 48). In contrast, in the ECOM4LA strain, OAA was (within limits of error) synthesized exclusively by anaplerosis. This indicated that the TCA cycle was nonfunctional somewhere between oxoglutarate (glutamate) and OAA. The labeling patterns of aspartate fragments in ECOM4LA did indicate some recycling of oxaloacetate through the symmetrical TCA intermediate fumarate (and possibly also succinate) (Table 3), which would indicate a break in the cycle closer to oxoglutarate. These measurements corresponded to the gene expression results, which showed that in the ECOM4LA strain, expression of almost all of the TCA cycle enzymes was lower, while expression of the anaplerotic enzyme phosphoenolpyruvate carboxykinase was greater, than that in the WT (Fig. 1B).
FIG. 3.

Metabolic flux distribution through branching areas of the central metabolism of MG1655 and ECOM4LA cell lines. Mean metabolic flux distribution data acquired from WT and ECOM4LA strains grown in triplicate are presented here. (A) Overall metabolic map of central metabolism, with branching areas of metabolism boxed. The corresponding gene expression data are presented in Fig. 1. (B) Relative input to glycolysis (F6P) from glucose-6-phosphate (G6P) versus input to the pentose phosphate pathway in each strain (based on [U-13C]glucose labeling data). (C) Relative input to oxaloacetate (OA) from α-ketoglutarate (αKG) in the TCA cycle or from PEP via PEP carboxylase. Also shown, in red, is the fraction of OA formed from PEP that recycled through fumarate (FUM) in each strain.
Other pathways.
The ED pathway was evaluated as an alternate route to pyruvate from glucose. Although expression of genes encoding the ED pathway is usually weak in E. coli grown on glucose (36), it was previously shown that E. coli mutants which were disabled in components of the TCA cycle (Sdh/Mdh or FumA) produced ∼20% of their pyruvate via the ED pathway (14). Calculating the ED pathway flux (versus that from glycolysis plus the PPP) (14), we found that it was insignificant (Table 3) in the TCA-nonfunctional ECOM4LA mutant. This corresponded to a lack of enhancement in gene expression for enzymes in the ED pathway (Fig. 1B).
Using [1-13C]glucose or [6-13C]glucose data, the degree of labeling of the 1-C pool was calculated from aspartate and methionine labeling (the latter being equivalent in its origins to aspartate plus a 1-C unit). For both E. coli strains cultured with [6-13C]glucose, 1-C pool labeling was slightly below 50% (Table 3) but was lower with [1-13C]glucose, reduced (relative to the level of [6-13C]glucose labeling) by 20% for the WT and 3% for ECOM4LA. These reduced labeling levels reflected the loss of label from glucose routed through the oxidative PPP before conversion to serine and thence into the 1-C pool and corresponded roughly to the relative flux through this pathway calculated from [1-13C]glucose-labeled alanine data (Table 3). From fragment data for serine and glycine, the percent 13C labeling at serine-3 or glycine-2 was calculated, and from this (assuming that these were the only two sources for the 1-C pool), the contribution of each to the 1-C pool was calculated. In all cases, serine-3 was the predominant precursor (Table 3).
Endogenous sources of CO2.
Data from [U-13C]glucose labeling experiments indicated that most of the CO2/bicarbonate used in anaplerotic reactions was derived from glucose and not from atmospheric CO2 (Table 3). In ECOM4LA, 70% of CO2 was from glucose, versus 90% in the WT. The lesser figure in ECOM4LA is not unexpected, as the mutant was lacking in any flux through two major CO2-evolving steps in the TCA cycle, which would be expected to increase the proportion of CO2 from internal sources. Levels of CO2 labeling in both strains with 1-13C were similar (Table 3), accounting for ∼10% endogenous CO2 in both cases. The lesser PPP flux in ECOM4LA (albeit relative to that from glycolysis) suggested that in this strain the 1-carbon of glucose might be converted to CO2 via additional pathways. As the C-1 position of glucose is equivalent to the C-6 position after conversion by glycolysis to 3-carbon metabolites, labeling was also performed with [6-13C]glucose. This yielded no CO2 labeling in either the WT or ECOM4LA, demonstrating that in both strains the oxidative PPP was the only route to convert the 1-carbon of glucose into CO2.
DISCUSSION
The aim of this study was to gain insights into the physiology of the ECOM4LA strain and understand what metabolic and regulatory changes led to the inability to switch metabolism between aerobic and anaerobic growth. Three active cytochrome oxidases and a quinol monooxygenase were completely removed in order to produce a phenotype almost incapable of oxygen utilization. The oxygen uptake rate of the resultant mutant was reduced to nearly 60 times lower than that for unmutated E. coli. As a consequence of these deletions, the mutant strain was unable to undergo an aerobic-anaerobic shift and presented fermentative behavior under oxic and anoxic conditions. In order to understand metabolic changes that underlie the unique physiology of the mutant strain, we conducted whole-genome transcriptomics analysis coupled with 13C tracing experiments and physiological characterization during aerobic and anaerobic growth.
The transition between oxic and anoxic environments in E. coli has been studied extensively (9, 19, 44). In particular, the “shift” between aerobic and anaerobic modes of metabolism is regulated by two distinct systems of transcription factors: FNR and ArcB/ArcA (2, 8, 27, 40, 41, 45). It has been reported that FNR is able to sense oxygen directly (47), while the ArcB/ArcA system responds to the content of the quinone pool (4, 5, 17) and switches on the expression of fermentation genes and represses aerobic pathways when E. coli encounters low-oxygen growth conditions (21, 22).
Here, we hypothesized that oxygen uptake-mediated regulation (ArcB/ArcA) is significantly perturbed as a result of the inability to utilize oxygen, while oxygen-sensing regulation (FNR) should exhibit similarities to that in the wild type. Consistent with our hypothesis, we observed activation of the ArcA regulator under oxic conditions and consequent activation of fermentative metabolism during aerobic growth, while the regulatory action of the FNR regulator remained similar to that for the wild type.
Aerobic ECOM4LA shows anaerobic gene expression.
As expected, the deletion of the respiratory chain components had a greater effect on metabolism in an oxic environment. Comparable gene expression patterns between aerobic ECOM4LA and anaerobic WT not only indicate similar levels of regulation but also suggest similar metabolic functions. In particular, high flux (based on uptake and secretion rates) and increased expression of glycolytic enzymes suggest that glycolysis is a main ATP-producing pathway in ECOM4LA during aerobic and anaerobic growth, similar to that seen in WT anaerobic growth. It is possible that ECOM4LA is unable to build a sufficient proton gradient due to mutations in cytochrome oxidases, thus requiring the production of ATP molecules by substrate-level phosphorylation under oxic growth conditions. The similarity of growth rates between aerobically grown ECOM4LA and anaerobically grown WT E. coli (Table 1) suggests that energy requirements are similar in both strains under given conditions, unlike those of aerobically and anaerobically grown WT E. coli.
Aerobic ECOM4LA uses anaerobic respiration.
It is well known that E. coli has a highly versatile respiratory chain that allows it to adapt to conditions that vary with respect to oxygen availability and the redox state (Fig. 4 A and B) (5). It is possible that mutations introduced in the ECOM4LA strain, together with adaptive evolution, resulted in the rearrangement of the respiratory chain and a shift in the content of the quinone pool (Fig. 4C). During anaerobic growth, E. coli uses respiratory pathways different from those used during aerobic growth (38). In the respiratory chain formed by NADH:menaquinone oxidoreductase (yieF and wrbA) and fumarate reductase (frdABCD) genes, electrons are transferred from NADH to fumarate by a menaquinone pool (50), resulting in the formation of succinate (Fig. 4C) (23). Based on our gene expression results and physiological observations, we conclude that anaerobic respiration consisting of yieF, wrbA, and frdABCD is active and is used to remove excess electrons during exponential growth of the ECOM4LA strain.
FIG. 4.

Respiratory chain rearrangements in ECOM4LA compared to those in MG1655, and effect of the content of the quinone pool on ArcA activity. (A) Classical view of the aerobic respiratory chain (red) in wild-type E. coli (26, 28, 29). Electrons are transferred from NADH to oxygen via the ubiquinone pool. An alternative oxygen utilization system through quinone monooxygenase (YgiN) is presented. The high content of ubiquinones (UQs) represses the activity of the ArcA regulator. (B) Classical view of the anaerobic respiratory chain (blue) in wild-type E. coli. Electrons are transferred from NADH to succinate via the menaquinone pool. The high content of menaquinones (MQs) activates AcrA activity. (C) Rearrangement in the respiratory chain, resulting from gene deletions. A lack of significant activity of aerobic respiration (dashed line) and an increase in activity of anaerobic respiration (solid line) resulted in reformulation of the quinone pool. A shift from UQs to MQs resulted in activation of the ArcA regulator under oxic and anoxic conditions.
The anaerobic regulator ArcA is active in ECOM4LA during oxic growth.
E. coli has two distinct regulators that control expression of the many genes involved in the aerobic-anaerobic shift: the ArcB/ArcA two-component system and FNR. In the expression data, we saw that ArcA activation in ECOM4LA is likely responsible for the anaerobic phenotype under aerobic conditions. However, there was a small number of ArcA targets (19 out of 143 [see Table S5 in the supplemental material) that were further changed in anaerobic ECOM4LA, consistent with known ArcA activity. Thus, it seems that ArcA still increases its level of activity slightly in the ECOM4LA aerobic-anaerobic shift. To further validate the activation of ArcA in ECOM4LA during aerobic growth, we looked at genes previously identified as direct targets of ArcA regulation. We observed significant downregulation of the sdhABCD (succinate dehydrogenase) and frdABCD operons, known to be repressed by ArcA, under oxic conditions. These operons were also repressed significantly under anoxic conditions, indicating activity of ArcA during anaerobic growth consistent with that for WT E. coli. Our results suggest that the action mode of one of the global transcription regulators (ArcA) was altered as a result of major metabolic adjustment, which affected the gene expression in a nonintuitive way. In particular, the inability to utilize oxygen led to a decrease in ubiquinone content and an increase in menaquinone content (see Fig. S5 in the supplemental material) and subsequently to the activation of the ArcB/ArcA regulatory system (4). Thus, the change in composition of the quinone pool (Fig. 4C) leads to activation of ArcA and subsequent activation of fermentative metabolism during aerobic and anaerobic growth of the ECOM4LA cell line.
13C analysis complements gene expression data.
Metabolic flux calculations based on [13C]glucose labeling data were highly consistent with the gene expression data. Most notably, glycolysis was upregulated in ECOM4LA compared with the PPP, and flux through the TCA cycle was not detectable (Fig. 3). Flux analysis indicated reduced PPP flux between glucose and pyruvate not just relative to that from glycolysis but also with conversion to absolute flux using glucose uptake rates. This contrasted with gene expression data (Fig. 1B), which showed greater expression of genes for the nonoxidative PPP in ECOM4LA. However, the reversibility of the reactions catalyzed by the enzymes in the nonoxidative PPP should be noted. Data for input into the P5P pool via the two branches of the PPP showed a greater contribution of the nonoxidative PPP in ECOM4LA (Table 3), which might account for the increased expression of the genes for this branch of the pathway here. Producing P5P via a nonreducing route might help ECOM4LA maintain its redox balance in the absence of the ability to utilize oxygen.
Similar physiological behaviors under oxic and anoxic conditions.
Even though we observed a nearly 15% difference in growth rate of ECOM4LA between oxic and anoxic conditions, the overall physiological behaviors were similar (Table 1). The lower lactate yield observed during anaerobic growth can be attributed to a higher cell density. ECOM4LA grew to a 20% higher cell density anaerobically than aerobically (data not shown). The oxygen uptake rate measured after the gene deletions was nearly 60 times lower than that for the wild type. We were unable to identify the metabolic function accounting for the remaining oxygen uptake; however, since no major physiological differences were observed under oxic and anoxic conditions, we can conclude that oxygen does not have a significant metabolic function in the ECOM4LA strain.
The existence of similar physiological behaviors under diverse growth conditions has a significant advantage for industrial implementation, as it reduces the need for stringent control of the growth environment. The developed strain can be utilized for production of d-lactate from glucose or can be engineered further to produce reduced by-products, such as ethanol, succinate, etc.
Supplementary Material
Acknowledgments
We thank Jan Schellenberger for useful discussions regarding metabolic fluxes, and we thank Yuri Matusov, Marc Abrams, Daniel Hyduke, and Karsten Zengler for helpful discussions and critical revision of the manuscript.
This work was funded by U.S. National Institutes of Health grant GM062791 and in part by an NSF IGERT Plant Systems Biology training grant (DGE-0504645) through a fellowship to N.E.L.
We also thank the anonymous reviewers for comments and suggestions that helped us improve the manuscript.
Footnotes
Published ahead of print on 13 August 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Adams, M. A., and Z. Jia. 2005. Structural and biochemical evidence for an enzymatic quinone redox cycle in Escherichia coli: identification of a novel quinol monooxygenase. J. Biol. Chem. 280:8358-8363. [DOI] [PubMed] [Google Scholar]
- 2.Alexeeva, S., K. J. Hellingwerf, and M. J. Teixeira de Mattos. 2003. Requirement of ArcA for redox regulation in Escherichia coli under microaerobic but not anaerobic or aerobic conditions. J. Bacteriol. 185:204-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett, T., D. B. Troup, S. E. Wilhite, P. Ledoux, D. Rudnev, C. Evangelista, I. F. Kim, A. Soboleva, M. Tomashevsky, K. A. Marshall, K. H. Phillippy, P. M. Sherman, R. N. Muertter, and R. Edgar. 2009. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 37:D885-D890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bekker, M., S. Alexeeva, W. Laan, G. Sawers, J. Teixeira de Mattos, and K. Hellingwerf. 2010. The ArcBA two-component system of Escherichia coli is regulated by the redox state of both the ubiquinone and the menaquinone pool. J. Bacteriol. 192:746-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekker, M., G. Kramer, A. F. Hartog, M. J. Wagner, C. G. de Koster, K. J. Hellingwerf, and M. J. de Mattos. 2007. Changes in the redox state and composition of the quinone pool of Escherichia coli during aerobic batch-culture growth. Microbiology 153:1974-1980. [DOI] [PubMed] [Google Scholar]
- 6.Brondsted, L., and T. Atlung. 1994. Anaerobic regulation of the hydrogenase 1 (hya) operon of Escherichia coli. J. Bacteriol. 176:5423-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho, B. K., E. M. Knight, and B. O. Palsson. 2006. PCR-based tandem epitope tagging system for Escherichia coli genome engineering. Biotechniques 40:67-72. [DOI] [PubMed] [Google Scholar]
- 8.Compan, I., and D. Touati. 1994. Anaerobic activation of arcA transcription in Escherichia coli: roles of Fnr and ArcA. Mol. Microbiol. 11:955-964. [DOI] [PubMed] [Google Scholar]
- 9.Covert, M. W., E. M. Knight, J. L. Reed, M. J. Herrgard, and B. O. Palsson. 2004. Integrating high-throughput and computational data elucidates bacterial networks. Nature 429:92-96. [DOI] [PubMed] [Google Scholar]
- 10.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dellomonaco, C., C. Rivera, P. Campbell, and R. Gonzalez. 2010. Engineered respiro-fermentative metabolism for the production of biofuels and biochemicals from fatty acid-rich feedstocks. Appl. Environ. Microbiol. 76:5067-5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edgar, R., M. Domrachev, and A. E. Lash. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30:207-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feist, A. M., C. S. Henry, J. L. Reed, M. Krummenacker, A. R. Joyce, P. D. Karp, L. J. Broadbelt, V. Hatzimanikatis, and B. O. Palsson. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer, E., and U. Sauer. 2003. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 270:880-891. [DOI] [PubMed] [Google Scholar]
- 15.Fong, S. S., A. P. Burgard, C. D. Herring, E. M. Knight, F. R. Blattner, C. D. Maranas, and B. O. Palsson. 2005. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol. Bioeng. 91:643-648. [DOI] [PubMed] [Google Scholar]
- 16.Gama-Castro, S., V. Jimenez-Jacinto, M. Peralta-Gil, A. Santos-Zavaleta, M. I. Penaloza-Spinola, B. Contreras-Moreira, J. Segura-Salazar, L. Muniz-Rascado, I. Martinez-Flores, H. Salgado, C. Bonavides-Martinez, C. Abreu-Goodger, C. Rodriguez-Penagos, J. Miranda-Rios, E. Morett, E. Merino, A. M. Huerta, L. Trevino-Quintanilla, and J. Collado-Vides. 2008. RegulonDB (version 6.0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation. Nucleic Acids Res. 36:D120-D124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Georgellis, D., O. Kwon, and E. C. Lin. 2001. Quinones as the redox signal for the arc two-component system of bacteria. Science 292:2314-2316. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez, R., P. Campbell, and M. Wong. 2010. Production of ethanol from thin stillage by metabolically engineered Escherichia coli. Biotechnol. Lett. 32:405-411. [DOI] [PubMed] [Google Scholar]
- 19.Gunsalus, R. P., and S. J. Park. 1994. Aerobic-anaerobic gene regulation in Escherichia coli: control by the ArcAB and Fnr regulons. Res. Microbiol. 145:437-450. [DOI] [PubMed] [Google Scholar]
- 20.Herring, C. D., A. Raghunathan, C. Honisch, T. Patel, M. K. Applebee, A. R. Joyce, T. J. Albert, F. R. Blattner, D. van den Boom, C. R. Cantor, and B. O. Palsson. 2006. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat. Genet. 38:1406-1412. [DOI] [PubMed] [Google Scholar]
- 21.Iuchi, S., and E. C. Lin. 1993. Adaptation of Escherichia coli to redox environments by gene expression. Mol. Microbiol. 9:9-15. [DOI] [PubMed] [Google Scholar]
- 22.Iuchi, S., and E. C. Lin. 1988. arcA (dye), a global regulatory gene in Escherichia coli mediating repression of enzymes in aerobic pathways. Proc. Natl. Acad. Sci. U. S. A. 85:1888-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iverson, T. M., C. Luna-Chavez, G. Cecchini, and D. C. Rees. 1999. Structure of the Escherichia coli fumarate reductase respiratory complex. Science 284:1961-1966. [DOI] [PubMed] [Google Scholar]
- 24.Jarboe, L. R., X. Zhang, X. Wang, J. C. Moore, K. T. Shanmugam, and L. O. Ingram. 2010. Metabolic engineering for production of biorenewable fuels and chemicals: contributions of synthetic biology. J. Biomed. Biotechnol. 2010:761042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jervis, A. J., J. C. Crack, G. White, P. J. Artymiuk, M. R. Cheesman, A. J. Thomson, N. E. Le Brun, and J. Green. 2009. The O2 sensitivity of the transcription factor FNR is controlled by Ser24 modulating the kinetics of [4Fe-4S] to [2Fe-2S] conversion. Proc. Natl. Acad. Sci. U. S. A. 106:4659-4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa, M., S. Goto, M. Furumichi, M. Tanabe, and M. Hirakawa. 2010. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 38:D355-D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang, Y., K. D. Weber, Y. Qiu, P. J. Kiley, and F. R. Blattner. 2005. Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J. Bacteriol. 187:1135-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karp, P. D., I. M. Keseler, A. Shearer, M. Latendresse, M. Krummenacker, S. M. Paley, I. Paulsen, J. Collado-Vides, S. Gama-Castro, M. Peralta-Gil, A. Santos-Zavaleta, M. I. Penaloza-Spinola, C. Bonavides-Martinez, and J. Ingraham. 2007. Multidimensional annotation of the Escherichia coli K-12 genome. Nucleic Acids Res. 35:7577-7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keseler, I. M., C. Bonavides-Martinez, J. Collado-Vides, S. Gama-Castro, R. P. Gunsalus, D. A. Johnson, M. Krummenacker, L. M. Nolan, S. Paley, I. T. Paulsen, M. Peralta-Gil, A. Santos-Zavaleta, A. G. Shearer, and P. D. Karp. 2009. EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res. 37:D464-D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, K. H., J. H. Park, T. Y. Kim, H. U. Kim, and S. Y. Lee. 2007. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol. Syst. Biol. 3:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee, S. J., D. Y. Lee, T. Y. Kim, B. H. Kim, J. Lee, and S. Y. Lee. 2005. Metabolic engineering of Escherichia coli for enhanced production of succinic acid, based on genome comparison and in silico gene knockout simulation. Appl. Environ. Microbiol. 71:7880-7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li, Z. J., J. Jian, X. X. Wei, X. W. Shen, and G. Q. Chen. 2010. Microbial production of meso-2,3-butanediol by metabolically engineered Escherichia coli under low oxygen condition. Appl. Microbiol. Biotechnol. 87:2001-2009. [DOI] [PubMed] [Google Scholar]
- 33.Lynch, A. S., and E. C. Lin. 1996. Transcriptional control mediated by the ArcA two-component response regulator protein of Escherichia coli: characterization of DNA binding at target promoters. J. Bacteriol. 178:6238-6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma, J. F., M. Jiang, K. Q. Chen, B. Xu, S. W. Liu, P. Wei, and H. J. Ying. 22 May 2010, posting date. Succinic acid production with metabolically engineered Escherichia coli recovered from two-stage fermentation. Biotechnol. Lett. [Epub ahead of print.] doi: 10.1007/s10529-010-0313-x. [DOI] [PubMed]
- 35.Mazumdar, S., J. M. Clomburg, and R. Gonzalez. 2010. Escherichia coli strains engineered for homofermentative production of D-lactic acid from glycerol. Appl. Environ. Microbiol. 76:4327-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murray, E. L., and T. Conway. 2005. Multiple regulators control expression of the Entner-Doudoroff aldolase (Eda) of Escherichia coli. J. Bacteriol. 187:991-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nanchen, A., T. Fuhrer, and U. Sauer. 2007. Determination of metabolic flux ratios from 13C-experiments and gas chromatography-mass spectrometry data: protocol and principles. Methods Mol. Biol. 358:177-197. [DOI] [PubMed] [Google Scholar]
- 38.Neidhardt, F. (ed.). 1996. Escherichia coli and Salmonella, 2nd ed., vol. 1. ASM Press, Washington, DC.
- 39.Portnoy, V. A., M. J. Herrgard, and B. O. Palsson. 2008. Aerobic fermentation of D-glucose by an evolved cytochrome oxidase-deficient Escherichia coli strain. Appl. Environ. Microbiol. 74:7561-7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salmon, K., S. P. Hung, K. Mekjian, P. Baldi, G. W. Hatfield, and R. P. Gunsalus. 2003. Global gene expression profiling in Escherichia coli K12. The effects of oxygen availability and FNR. J. Biol. Chem. 278:29837-29855. [DOI] [PubMed] [Google Scholar]
- 41.Scott, C., J. D. Partridge, J. R. Stephenson, and J. Green. 2003. DNA target sequence and FNR-dependent gene expression. FEBS Lett. 541:97-101. [DOI] [PubMed] [Google Scholar]
- 42.Shestopalov, A. I., A. V. Bogachev, R. A. Murtazina, M. B. Viryasov, and V. P. Skulachev. 1997. Aeration-dependent changes in composition of the quinone pool in Escherichia coli. Evidence of post-transcriptional regulation of the quinone biosynthesis. FEBS Lett. 404:272-284. [DOI] [PubMed] [Google Scholar]
- 43.Shukla, V. B., S. Zhou, L. P. Yomano, K. T. Shanmugam, J. F. Preston, and L. O. Ingram. 2004. Production of D(-)-lactate from sucrose and molasses. Biotechnol. Lett. 26:689-693. [DOI] [PubMed] [Google Scholar]
- 44.Spiro, S., and J. R. Guest. 1991. Adaptive responses to oxygen limitation in Escherichia coli. Trends Biochem. Sci. 16:310-314. [DOI] [PubMed] [Google Scholar]
- 45.Spiro, S., and J. R. Guest. 1990. FNR and its role in oxygen-regulated gene expression in Escherichia coli. FEMS Microbiol. Rev. 6:399-428. [DOI] [PubMed] [Google Scholar]
- 46.Storey, J. D., and R. Tibshirani. 2003. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A. 100:9440-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sutton, V. R., E. L. Mettert, H. Beinert, and P. J. Kiley. 2004. Kinetic analysis of the oxidative conversion of the [4Fe-4S]2+ cluster of FNR to a [2Fe-2S]2+ cluster. J. Bacteriol. 186:8018-8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szyperski, T. 1995. Biosynthetically directed fractional 13C-labeling of proteinogenic amino acids. An efficient analytical tool to investigate intermediary metabolism. Eur. J. Biochem. 232:433-448. [DOI] [PubMed] [Google Scholar]
- 49.van Winden, W. A., C. Wittmann, E. Heinzle, and J. J. Heijnen. 2002. Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 80:477-479. [DOI] [PubMed] [Google Scholar]
- 50.Yagi, T., and A. Matsuno-Yagi. 2003. The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry 42:2266-2274. [DOI] [PubMed] [Google Scholar]
- 51.Zhou, S., T. B. Causey, A. Hasona, K. T. Shanmugam, and L. O. Ingram. 2003. Production of optically pure D-lactic acid in mineral salts medium by metabolically engineered Escherichia coli W3110. Appl. Environ. Microbiol. 69:399-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu, Y., M. A. Eiteman, R. Altman, and E. Altman. 2008. High glycolytic flux improves pyruvate production by a metabolically engineered Escherichia coli strain. Appl. Environ. Microbiol. 74:6649-6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu, Y., M. A. Eiteman, K. DeWitt, and E. Altman. 2007. Homolactate fermentation by metabolically engineered Escherichia coli strains. Appl. Environ. Microbiol. 73:456-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.