

Abstract
Lipoteichoic acid (LTA) is an important cell wall component of Gram-positive bacteria. The key enzyme responsible for polyglycerolphosphate lipoteichoic acid synthesis in the Gram-positive pathogen Staphylococcus aureus is the membrane-embedded lipoteichoic acid synthase enzyme, LtaS. It is presumed that LtaS hydrolyzes the glycerolphosphate head group of the membrane lipid phosphatidylglycerol (PG) and catalyzes the formation of the polyglycerolphosphate LTA backbone chain. Here we describe an in vitro assay for this new class of enzyme using PG with a fluorescently labeled fatty acid chain (NBD-PG) as the substrate and the recombinant soluble C-terminal enzymatic domain of LtaS (eLtaS). Thin-layer chromatography and mass spectrometry analysis of the lipid reaction products revealed that eLtaS is sufficient to cleave the glycerolphosphate head group from NBD-PG, resulting in the formation of NBD-diacylglycerol. An excess of soluble glycerolphosphate could not compete with the hydrolysis of the fluorescently labeled PG lipid substrate, in contrast to the addition of unlabeled PG. This indicates that the enzyme recognizes and binds other parts of the lipid substrate, besides the glycerolphosphate head group. Furthermore, eLtaS activity was Mn2+ ion dependent; Mg2+ and Ca2+ supported only weak enzyme activity. Addition of Zn2+ or EDTA inhibited enzyme activity even in the presence of Mn2+. The pH optimum of the enzyme was 6.5, characteristic for an enzyme that functions extracellularly. Lastly, we show that the in vitro assay can be used to study the enzyme activities of other members of the lipoteichoic acid synthase enzyme family.
Lipoteichoic acid (LTA) is a crucial component of the cell wall envelope in Gram-positive bacteria. Diverse functions have been ascribed to LTA, including regulation of the activity of hydrolytic enzymes (4), an essential role in divalent cation homeostasis (2, 26, 37), and retention of noncovalently attached proteins within the cell wall envelope (20, 41). In addition, functions of LTA in host-pathogen interactions have been reported (44). d-Alanine modifications on LTA protect bacteria from killing by cationic antimicrobial peptides (36, 43) and are critical during the infection and colonization processes (1, 5, 10). On the other hand, LTA may also play a positive role for the host in wound healing, by preventing excessive inflammation (25).
In the Gram-positive bacterial pathogen Staphylococcus aureus and in many other bacteria belonging to the Firmicutes, including Bacillus, Listeria, Streptococcus, Enterococcus, and Lactococcus spp., LTA is composed of a linear 1,3-linked polyglycerolphosphate backbone chain that is tethered via a glycolipid anchor to the bacterial membrane (6, 9). Recently, the staphylococcal protein LtaS was identified and shown to be responsible for polyglycerolphosphate LTA synthesis in vivo (14). An S. aureus strain depleted of LtaS is unable to synthesize LTA and shows severe growth and morphological defects (14); an S. aureus ltaS deletion strain is viable at 30°C only in a growth medium containing at least 1% NaCl or at higher temperatures at high salt (7.5%) or high sucrose (40%) concentrations (35). Taken together, these findings provide further evidence for the importance of this abundant cell envelope component for normal cell morphology and physiology.
Pulse-chase experiments have provided strong biochemical evidence that the glycerolphosphate subunits of LTA are derived from the head group of the membrane lipid phosphatidylglycerol (PG) (7, 8, 12). A rapid and almost complete turnover of the nonacylated glycerolphosphate group of PG into LTA is observed in S. aureus and other Gram-positive bacteria that synthesize polyglycerolphosphate LTA (23, 24). It is assumed that the LtaS enzyme cleaves the head group of PG and uses this glycerolphosphate subunit to polymerize the LTA backbone chain.
One or more LtaS-like enzymes are encoded in the genomes of Gram-positive bacteria that synthesize polyglycerolphosphate LTA (14). S. aureus LtaS and all other members of this enzyme family are predicted to contain five N-terminal transmembrane helices followed by an extracellular C-terminal enzymatic domain (eLtaS) (14, 29). The LtaS enzyme is processed in S. aureus, and the eLtaS domain is released into the culture supernatant as well as partially retained within the cell wall envelope (11, 29, 45). The crystal structure of the S. aureus eLtaS domain, alone and in a complex with soluble glycerolphosphate and the soluble domain of the Bacillus subtilis LtaS (LtaSBs) enzyme (YflE), identified a threonine as the catalytic residue. This is based on the location of the glycerolphosphate head group in the active site for S. aureus LtaS and on threonine phosphorylation in the B. subtilis enzyme structure (29, 37). Replacement of this threonine residue with an alanine renders the S. aureus enzyme inactive and unable to synthesize LTA in vivo (29). In addition, a Mn2+ ion was detected in the active center of the S. aureus LtaS structure, while the B. subtilis enzyme contained a Mg2+ ion.
To provide insight into the enzymatic activity of the S. aureus lipoteichoic acid synthase enzyme, we developed an in vitro assay for this enzyme using purified recombinant eLtaS and fluorescently labeled PG as a substrate. Using thin-layer chromatography (TLC) and mass spectrometry analysis of the lipid reaction products, we show that eLtaS protein is sufficient to cleave the glycerolphosphate head group from NBD-PG, resulting in the formation of NBD-diacylglycerol (NBD-DAG). Furthermore, we provide experimental evidence that LtaS requires Mn2+ for enzyme activity, while Zn2+ inhibits enzyme function. Our results suggest that LtaS has a narrow substrate specificity, with PG serving as a substrate while phosphatidylethanolamine (PE), phosphatidylcholine (PC), and phosphatidylserine (PS) do not. Lastly, we show that this in vitro assay can be used to study the enzyme functions of other members of this protein family, such as the Listeria monocytogenes LTA synthase (LtaSLm) and LTA primase (LtaPLm) enzymes. This study is the first in vitro characterization of lipoteichoic acid synthase enzymes and an important first step towards the development of an assay to screen and identify enzyme-specific inhibitors for this new and important class of bacterial enzymes.
MATERIALS AND METHODS
Growth conditions and construction of plasmids and strains.
Escherichia coli strains were grown at 37°C in Luria-Bertani (LB) medium with 100 μg/ml ampicillin for plasmid selection when appropriate. E. coli Rosetta strains (Novagen) containing plasmid pProEX-eLtaS (strain ANG571) or pProEX-eLtaS-T300A (strain ANG575) were used for the expression and purification of N-terminally His-tagged versions of the extracellular enzymatic domain of the S. aureus LTA synthase enzyme (eLtaS) or the active-site variant eLtaS-T300A, respectively. The construction of these strains has been described previously (29). Plasmids pProEX-eLtaPLm (Lmo0644) and pProEX-eLtaSLm (Lmo0927) were constructed for the expression and purification of N-terminally His-tagged versions of the extracellular enzymatic domains of the Listeria monocytogenes LTA primase enzyme (eLtaPLm; Lmo0644) and LTA synthase enzyme (eLtaSLm; Lmo0927). Gene fragments were amplified from L. monocytogenes 10403S chromosomal DNA using primer pairs 5-BamHI-LMO0644-C-term (CGGGATCCGCCGCAGATATTACTGCCAAAAACGTG)/3-XbaI-LMO0644-with-stop (CGTCTAGACCAGCAAATCTGGATACTTCTTTTGTTTA) and 5-EcoRI-LMO0927-Cterm (CGGAATTCAAGATAGTAGTGATGTTACTGAAG)/3-XbaI-LMO0927-with-stop (CGTCTAGATTATTTATCGGATGAATCAGTTGATTTTTTC), containing BamHI/XbaI and EcoRI/XbaI, respectively, and were cloned into plasmid pProEX-HTb (Invitrogen), which had been cut with the same enzymes. The resulting plasmids, pProEX-eLtaPLm and pProEX-eLtaSLm, were initially transformed into E. coli strain DH5α, resulting in strains ANG1448 (pProEX-eLtaPLm) and ANG1449 (pProEX-eLtaSLm). For protein expression and purification, these plasmids were introduced into the E. coli Rosetta strain, yielding strains ANG1478 (pProEX-eLtaPLm) and ANG1479 (pProEX-eLtaSLm). The sequences of inserts were verified by fluorescence automated sequencing at the MRC Clinical Science Center Sequencing Facility at Imperial College London.
Protein purification.
Proteins were expressed and purified by Ni affinity and size exclusion chromatography as described previously, with the modification that protein expression was induced at an optical density at 600 nm (OD600) of 0.4 by the addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) (29). Fractions containing the purified protein were pooled and concentrated using Amicon Centricon concentrators (cutoff, 10 kDa), and the protein concentration was determined using the bicinchoninic acid (BCA) kit from Pierce. The purity of proteins was confirmed by analysis on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and Coomassie staining.
Standard eLtaS enzyme assay.
eLtaS enzyme activity was measured by following the hydrolysis of a fluorescently labeled phosphatidylglycerol lipid substrate (NBD-PG) (see Fig. 1A). For enzyme assays, the commercially available 16:0-6:0 NBD-PG lipid (Avanti 810163) was further purified on TLC plates. Typically, 100 μg NBD-PG was spotted (five 20-μg spots) onto 250-μm-thick 60-Å silica TLC plates 250 μm thick (Macherey-Nagel) and was run for 15 min in a 60:20:2.7 CHCl3-MeOH-water solvent system. The major yellow fluorescent band clearly visible to the eye was excised from TLC plates, silica gel was scraped into appropriate tubes; and lipids were extracted for 15 min at room temperature (RT) with 8 ml of a 1:1 methanol-chloroform mixture with frequent vortexing. Next, 3.6 ml double-distilled H2O (ddH2O) was added; samples were vortexed; and the mixture was subsequently centrifuged for 10 min at 7,000 × g for phase separation. The bottom chloroform phase containing the purified NBD-PG lipid was transferred to a glass tube, and the silica matrix was extracted a second time as described above. Chloroform phases containing purified NBD-PG lipid were dried under a stream of nitrogen and were stored at −20°C until further use.
FIG. 1.

eLtaS in vitro assay. (A) Chemical structures of the fluorescently labeled phosphatidylglycerolphosphate lipids NBD-PG and NBD-DAG. The B. cereus phospholipase C (PLC) cleavage site is indicated on the scheme, as is the mass reduction following the release of the glycerolphosphate lipid head group. (B) TLC analysis of eLtaS in vitro reaction products. NBD-PG lipid suspensions were incubated with either eLtaS, the eLtaS-T300A protein, or no enzyme (negative control). Reaction products were separated by TLC, and plates were scanned using a fluorescence imager. As a positive control, a reaction was set up with B. cereus PLC. Note that only 10% of the PLC reaction mixture was run on the TLC. Arrows on the left indicate the positions of the NBD-PG input lipid, the main hydrolysis product, and a lipid of unknown structure (lipid X). The proteins added to each reaction mixture are shown at the top. (C) NBD-DAG standard curve. Twofold dilutions of PLC control reaction mixtures were separated by TLC (inset); the signal for the hydrolysis product was quantified; and the average values and standard deviations from two plates were plotted.
For our standard enzyme assay, 1.8 ml of 10 mM sodium succinate buffer (pH 6.0; ionic strength, 20 mM or 50 mM [adjusted with NaCl]) was added to approximately 25 μg of purified NBD-PG lipid to give a final lipid concentration of 17.8 μM (assuming that 100% of the lipid was recovered in the purification step). Lipids were solubilized by sonication for 35 to 45 s at an amplitude of 11 using a Sanyo Soniprep sonicator. Unless otherwise stated, 300 μl of these vesicles (∼ 4.166 ng lipid) was used per enzyme assay. Next, MnCl2 from a 1 M stock was added to give a final concentration of 10 mM, and reactions were initiated by the addition of 1.52 μM (25 μg) enzyme. The assay mixtures were incubated at RT on a rotator wheel or in a 37°C water bath for 3 h unless otherwise stated. Reactions were stopped by the addition of CHCl3-methanol (MeOH) to give a final assay volume/CHCl3/MeOH ratio of 0.9:1:1. After vigorous vortexing, the reaction products were centrifuged at 17,000 × g for 5 min. The chloroform phase or fractions of the chloroform phase were transferred to a new tube and dried under a stream of nitrogen. Dried lipids were suspended in 10 μl chloroform, spotted onto 60-Å silica TLC plates, and separated using a CHCl3-MeOH-water (60:20:2.7) solvent system. Plates were subsequently dried at RT; fluorescent bands were visualized using a Fujifilm FLA-5000 imager equipped with a 473-nm excitation laser and a fluorescein isothiocyanate (FITC) emission filter; and signals were quantified using Aida software.
If other buffer systems were used (10 mM sodium succinate [pH 6.0] buffer adjusted with NaCl to the ionic strengths indicated in Fig. 4B, 10 mM sodium succinate [pH 4.5 to pH 6.5]; or 20 mM HEPES [pH 7 to 8] adjusted to an ionic strength of 20 mM with NaCl), lipid vesicles were prepared in each buffer separately. Also, where indicated, assays were set up either in the absence of metal ions; in the presence of 1 to 250 mM MnCl2 or of 10 mM MgCl2, CaCl2, or ZnSO4, or combinations thereof; in the presence of 16 mM EDTA; or in the presence of glycerolphosphate. As a positive control, phospholipase C (PLC) from Bacillus cereus (Sigma) (EC 3.1.4.3) was used. Twenty-five micrograms of purified NBD-PG was brought into suspension in 1.8 ml phosphate-buffered saline (PBS) (pH 7.4) by sonication. Next, CaCl2 was added to give a final concentration of 10 mM; reactions were initiated by the addition of 2.5 U of PLC per 300 μl of lipid solution (∼ 4.166 ng lipids); and samples were incubated for 3 to 4 h on a rotator at RT. Lipids were subsequently extracted as described above. Another lipid used in this study was egg PG (Avanti 841138 or Sigma P8318). This unlabeled lipid was used in competition experiments in a 5-fold excess over the amount of NBD-PG (5:1 ratio; 20.8 ng PG to 4.166 ng NBD-PG).
To determine the enzyme substrate specificity, the fluorescently labeled lipids 16:0-6:0 NBD-PC (Avanti 810130), 16:0-6:0 NBD-PE (Avanti 810153), and 16:0-6:0 NBD-PS (Avanti 810192) were purified on TLC plates as described above for NBD-PG. Reactions with these lipids were set up in 10 mM sodium succinate (pH 6.0; ionic strength, 50 mM) containing twice the amount of lipid as in the standard reactions, and samples were incubated for 3 h at 37°C in a water bath. Two to six independent experiments with duplicate or triplicate samples were performed for all data presented. Variations between different enzyme preparations and lipid purifications were observed, and therefore, absolute fluorescence values for the hydrolysis products could be directly compared only within one experiment, which was performed with the same enzyme preparation and purified lipid substrate. To compare results between different independent experiments and for final data plotting and analysis, the average fluorescence reading for one condition (usually 10 mM MnCl2) was arbitrarily set to 1 in each independent experiment, and all other values from the same experiment were adjusted accordingly. Unless specifically stated otherwise, all normalized data points from the two to six independent experiments were combined for final data presentation and statistical analysis, and the average values and standard deviations were plotted. The two-tailed, two-sample equal-variance Student t test was used to determine statistically significant differences between the enzyme activity under standard reaction conditions (highlighted in dark grey in each graph) and that under each of the other enzyme reaction conditions shown in the same plot. Statistically significant differences with P values below 0.001 or between 0.001 and 0.05 are indicated in the figures.
Mass spectrometry analysis of lipid enzyme reaction products.
Twelve and a half micrograms of NBD-PG lipid was dried under a stream of nitrogen and was used for subsequent mass spectrometry analysis. As a positive control, PLC reactions were set up as described above; however, assays were scaled up 2.5-fold, and reaction mixtures were incubated for 3 h at RT. For the analysis of eLtaS, eLtaS-T300A, and no-enzyme (negative control) lipid reaction products, assays were scaled up 8-fold and were set up in 10 mM sodium succinate (pH 6.0; ionic strength, 50 mM) containing 10 mM MnCl2, and reaction mixtures were incubated for 3 h at 37°C. Lipids were extracted and dried as described above. For mass spectrometry analysis, dried lipids were suspended in 6 to 10 μl of a 0.5 M 2,5-dihydroxybenzoic acid (DHB) matrix-assisted laser desorption ionization (MALDI) matrix dissolved in 1:1 methanol-chloroform, and 1 μl was either spotted directly onto a MALDI plate or diluted 1:10 using a 0.5 M DHB matrix, after which 1 μl was spotted. Spotted MALDI plates were run on a MALDI micro MX machine (Waters, United Kingdom), available at the Proteomics Facility at Imperial College London. Using an automated program, 20 spectra were recorded for each spot in the reflector positive-ion mode. The machine was calibrated with polyethylene glycol (PEG) standards, and as an additional calibration standard, 25 to 50 pmol of a bradykinin peptide standard (Sigma) with an absolute mass of 757.3997 (M + H+) was spotted in an α-cyano-4-hydroxycinnamic acid (CHCA) matrix, which was suspended at 10 mg/ml in 75% acetonitrile-0.1% trifluoroacetic acid (TFA). Mass signals for lipids were manually corrected for the observed mass difference of the internal peptide standard. Experiments were performed twice, and representative results are shown in the figures.
RESULTS
In vitro enzyme assay using purified eLtaS protein and a fluorescently labeled PG lipid substrate.
To gain insight into the LtaS enzyme mechanism, we set out to develop an in vitro assay for the S. aureus eLtaS enzyme. The fluorescently labeled NBD-PG lipid substrate was used in previous studies to follow phospholipase C-type degradation reactions (39), reactions that are similar to that proposed for LtaS. Phospholipase C (PLC)-dependent cleavage of NBD-PG results in the production of fluorescently labeled diacylglycerol (NBD-DAG) (Fig. 1A). To determine if the purified soluble enzymatic domain of the S. aureus lipoteichoic acid synthase enzyme (eLtaS) is able to hydrolyze the glycerolphosphate head group of PG, we purified this enzyme as an N-terminally His-tagged version from E. coli extracts (29). The fluorescently labeled NBD-PG lipid substrate was brought into suspension by sonication, and enzyme reactions were initiated by the addition of purified eLtaS and MnCl2, the metal ion observed in the eLtaS crystal structure. Reaction mixtures were incubated for 3 h at RT; lipids were subsequently extracted and separated by TLC; and plates were scanned using a fluorescence imager. Two major fluorescent lipid bands were detected: one with the mobility of the NBD-PG input lipid and a second band for a lipid with faster mobility (Fig. 1B). The faster-migrating lipid had the same mobility as the lipid obtained in positive-control reactions, which were set up with the B. cereus PLC enzyme. This enzyme is known to hydrolyze the glycerolphosphate head group of PG (38), and the top lipid band therefore presumably corresponds to NBD-DAG (Fig. 1B) (see also the mass spectrometry analysis below). This faster-migrating lipid was not detected in the products of reactions set up with the active-site variant eLtaS-T300A or with no enzyme. Taken together, these results show that NBD-PG is hydrolyzed by eLtaS in an enzyme-specific manner and that the released hydrolysis product is presumably NBD-DAG (see also below). Besides the two main fluorescent bands (Fig. 1B, NBD-PG and hydrolysis product), a few additional minor fluorescent bands were observed. One of these lipids (Fig. 1B, lipid X) appears to be an eLtaS-specific reaction product, since this lipid is not produced in the eLtaS-T300A or the PLC control reaction. However, because of the small amounts, we were not able to obtain any additional structural information on this lipid or on any of the other minor lipid bands.
It has been reported that the intensity of the fluorescence signal of NBD-labeled lipids increases linearly with the concentration of the lipid over a wide range, allowing the quantification of enzyme reaction products (28). To test if our experimental setup would allow us to quantify enzyme reaction products, we ran 2-fold dilutions of the PLC control reaction mixture on TLC plates (16× to 1×) and quantified the NBD-DAG signal using Aida software (Fig. 1C). A linear relation between the amount of lipid and the fluorescence signal was observed over the range tested, allowing us to use this assay to quantify enzyme activities.
eLtaS-dependent NBD-PG hydrolysis results in the production of NBD-DAG.
The eLtaS reaction product runs on TLC plates with a retention time identical to that observed for the B. cereus PLC reaction product, indicating that NBD-DAG is formed upon the hydrolysis of NBD-PG. To provide further experimental evidence for this notion, we subjected the lipid products of scaled-up eLtaS, eLtaS-T300A, no-enzyme (negative control), and PLC (positive-control) reactions to MALDI mass spectrometry analysis. Dried lipids were suspended in a DHB matrix and spotted onto MALDI plates, and mass spectra were recorded in the positive reflector ion mode (Fig. 2). The sodium adduct of the NBD-PG input lipid (C34H56N4O13PNa) has a calculated absolute mass of 782.25. The mass we observed for the NBD-PG input lipid was 718.4 (Fig. 2B), considerably smaller than the calculated mass. This is presumably due to fragmentation at an unknown position within the NBD fluorescence group, since MALDI mass spectrometry analysis of other NBD-labeled lipids gave a similar mass reduction, while unlabeled lipids showed the expected absolute mass (data not shown). The peak at 718.4 is clearly NBD-PG lipid specific, since it was not observed in the sample containing the DHB matrix only (Fig. 2A). Cleavage of the glycerolphosphate head group results in the production of NBD-DAG and a calculated absolute mass reduction of 154.0 units (minus C3H7O5P) (Fig. 1A). In agreement with this predicted mass reduction, the observed mass of the B. cereus PLC reaction product was 564.2 (Fig. 2C), 154.2 mass units smaller than the 718.4 signal for the NBD-PG input lipid. A similar mass signal of 564.3 was observed for the eLtaS reaction product, in addition to the mass signal of 718.4 for the NBD-PG input lipid (Fig. 2F). This lower mass signal was absent from samples obtained from no-enzyme (Fig. 2D) and eLtaS-T300A (Fig. 2E) reaction products; and only signals at 718.3 and 718.4, respectively, identical to the mass signal of the NBD-PG input lipid, were obtained for these samples. In sum, these mass spectrometry data provide further experimental evidence that the S. aureus eLtaS enzyme cleaves the phosphodiester bond of NBD-PG, resulting in the production of NBD-DAG, a reaction similar to that of phospholipase C-type enzymes.
FIG. 2.

MALDI-TOF mass spectra of eLtaS lipid reaction products. For mass spectrometry analysis, PLC (positive control), eLtaS, eLtaS-T300A, and no-enzyme reactions were set up, and lipids were extracted and dried as described in Materials and Methods. Dried lipids, as well as 12.5 μg of the NBD-PG input lipid, were suspended in a 0.5 M DHB MALDI matrix and were spotted onto a MALDI plate. Spectra were recorded in the reflector positive-ion mode on a MALDI micro MX machine (Waters, United Kingdom), and m/z signals were plotted for the DHB matrix only (A), the NBD-PG input lipid (mass signal, 718.4) (B), the PLC reaction (mass signal, 564.2) (C), no enzyme (mass signal, 718.3) (D), the eLtaS-T300A reaction (mass signal, 718.4) (E), and the eLtaS reaction (mass signals, 564.3 and 718.4) (F). Presumed NBD-lipid-specific signals are shown in red.
eLtaS is a Mn2+-dependent metal enzyme.
Structural analysis of the S. aureus eLtaS domain and the soluble enzymatic domain of the B. subtilis LtaS (YflE) enzyme revealed the presence of a Mn2+ and a Mg2+ ion, respectively, in their active centers (29, 37). Since the ion was present in the crystallization buffer in both cases, these may not represent the relevant metal ions for enzyme activity. To test if a metal ion is required for enzyme activity and, if so, which one is required, we set up in vitro assays in the absence and presence of different divalent cations. As can be seen in Fig. 3A, addition of Mn2+ resulted in the highest enzyme activity. Addition of MgCl2 or CaCl2 resulted in low enzyme activity, while no enzyme activity was observed in the presence of ZnSO4. Increasing the concentration of MnCl2 revealed that the highest in vitro enzyme activity was observed in the presence of 50 to 100 mM Mn2+ ion (Fig. 3E). However, only low enzyme activity was observed in the presence of MgCl2 over a concentration range of 0.01 to 100 mM (data not shown). This suggests that Mn2+ is the metal ion necessary for LtaS enzyme function. Furthermore, as predicted for a metal-dependent enzyme, addition of the metal ion chelator EDTA inhibited enzyme function (Fig. 3B). Removal of EDTA using a NAP-10 desalting column restored Mn2+-dependent enzyme function (data not shown). The use of combinations of metal ions revealed that the addition of ZnSO4 inhibited Mn2+-dependent enzyme activity (Fig. 3C). This effect was specific for Zn2+; addition of MgCl2 or CaCl2 did not affect enzyme activity. Similar results were obtained by using ZnCl2 in place of ZnSO4, and enzyme inhibition was found to be Zn2+ concentration dependent (Fig. 3D). This result could indicate that Zn2+ can replace the Mn2+ ion in the active-site center of LtaS, but in a nonproductive manner. In summary, these data suggest that lipoteichoic acid synthase enzymes are Mn2+-dependent metal enzymes and that Zn2+ can act as a competitive inhibitor for enzyme function.
FIG. 3.

eLtaS requires Mn2+ for activity. (A) eLtaS in vitro reactions were set up in 10 mM sodium succinate (pH 6.0) buffer (ionic strength, 20 mM) in the absence or presence of the indicated metal ion at a final concentration of 10 mM. (B) EDTA inhibits eLtaS activity. eLtaS in vitro reactions were set up in 10 mM sodium succinate (pH 6.0) buffer (ionic strength, 50 mM) containing 10 mM MnCl2 with or without the addition of 16 mM EDTA. (C) Zn2+ inhibits eLtaS activity. eLtaS in vitro reactions were set up in 10 mM sodium succinate (pH 6.0) buffer (ionic strength, 20 mM) in the absence of ions or in the presence of a combination of metal ions at a final concentration of 10 mM each as indicated below each bar. (D) Zn2+ inhibits eLtaS activity in a dose-dependent manner. Enzyme reactions were set up in the presence of 10 mM MnCl2 and of ZnCl2 at concentrations in the range of 0.01 to 10 mM. (E) eLtaS activity is dependent on the MnCl2 concentration. Enzyme reactions were set up in the presence of MnCl2 at concentrations ranging from 1 to 250 mM. Two to four independent experiments were performed, and normalized average values and standard deviations were plotted as described in Materials and Methods. Student's t test was used to determine statistically significant differences between the enzyme activity represented by a dark grey bar in each graph and the activity with each of the other enzyme reaction conditions shown in the same plot. Double asterisks indicate statistically significant differences (P, <0.001).
eLtaS enzyme characteristics.
To determine additional enzyme parameters for the eLtaS enzyme, we performed in vitro assays at different pHs and ionic strengths, and we followed the reactions over time. To determine the pH profile, the NBD-PG substrate was brought into suspension by sonication in 10 mM sodium succinate (pH 4.5 to 6.5) and 20 mM HEPES (pH 7 to 8) buffers adjusted to an ionic strength of 20 mM with NaCl. To determine the optimal ionic strength, reactions were set up in 10 mM sodium succinate (pH 6.0) buffer, and the ionic strength was adjusted to 20, 50, 100, 200, or 300 mM with NaCl. As shown in Fig. 4A and B, the highest enzyme activity was seen at pH 6.5 and an ionic strength of 50 mM. Furthermore, a time course experiment revealed that in this assay, the level of product reached a plateau by 3 h (Fig. 4C). However, in contrast to that in B. cereus PLC control reactions, the hydrolysis of NBD-PG never proceeded to completion (see the example in Fig. 1B). At this time it is not clear whether the incomplete hydrolysis is due to suboptimal reaction conditions, the use of a shortened fragment of LtaS, or the absence from the reaction mixture of an additional component that is required for the transfer of the glycerolphosphate head group.
FIG. 4.
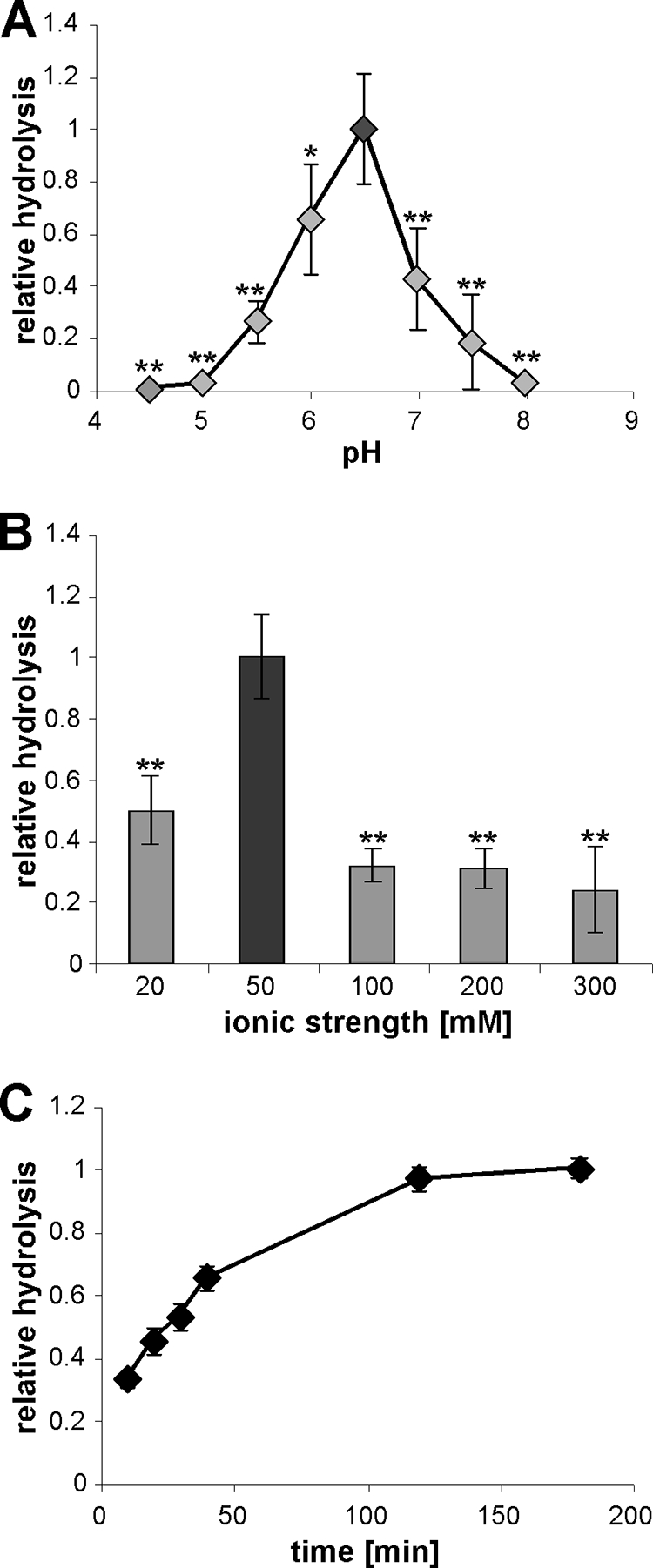
pH requirement, ionic strength profile, and reaction kinetics of eLtaS. (A) pH profile of eLtaS. NBD-PG vesicles were prepared in buffers ranging from pH 4.5 to pH 8 (for the exact buffer composition, see Materials and Methods). Reaction mixtures were incubated for 3 h at RT in the presence of 10 mM MnCl2, and lipids were extracted and analyzed by TLC. Data were plotted and analyzed as described for Fig. 3. (B) Ionic strength profile of eLtaS. NBD-PG vesicles were prepared in 10 mM sodium succinate (pH 6.0) buffer the ionic strength was adjusted to the indicated values with NaCl. Data were plotted and analyzed as described for Fig. 3. Asterisks indicate statistically significant differences (*, P values between 0.001 and 0.05; **, P values below 0.001). (C) Reaction kinetics of eLtaS. Enzyme reactions were set up in 10 mM sodium succinate (pH 6.0) buffer (ionic strength, 50 mM) containing 10 mM MnCl2, and reaction mixtures were incubated at 37°C for the indicated times. Three independent experiments were performed with duplicate or triplicate samples, and a representative graph is shown.
Glycerolphosphate cannot compete with PG lipid binding.
Structural studies have indicated that soluble glycerolphosphate can enter and bind to the active center of eLtaS (29). To test if glycerolphosphate could act as a competitive inhibitor of the enzyme, standard enzyme reactions were set up in the presence of a 10-fold excess of glycerolphosphate (GroP) over the NBD-PG lipid. No inhibition of enzyme activity was seen under these conditions (Fig. 5A). However, a 5-fold excess of unlabeled PG over fluorescently labeled PG could compete with the hydrolysis of the NBD-PG lipid (Fig. 5B). These results suggest that eLtaS binds structural features of the lipid substrate in addition to the head group.
FIG. 5.
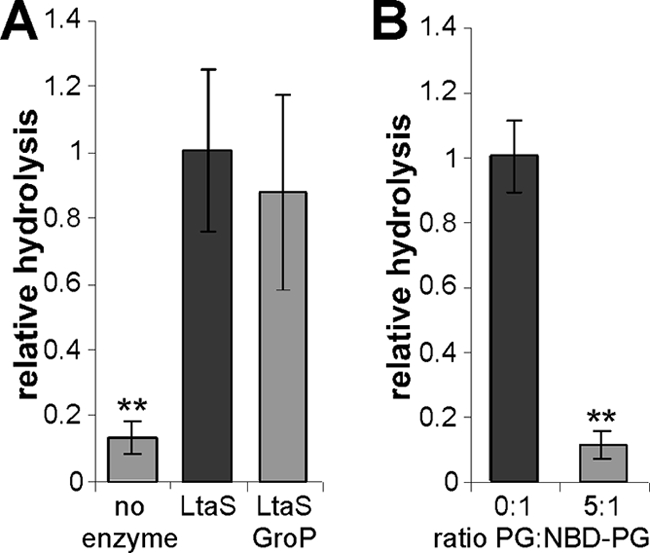
Unlabeled PG but not glycerolphosphate can compete with NBD-PG as a substrate. (A) Standard eLtaS reactions were set up with or without the addition of 175 μM glycerol-1-phosphate (GroP) (10-fold excess over the NBD-PG lipid substrate). (B) Unlabeled PG competes with NBD-PG. Vesicles containing only NBD-PG (0:1) or a mixture of PG and NBD-PG (5:1) were prepared, and eLtaS reactions were performed as described in Materials and Methods. Reaction products were separated by TLC, and the amount of the hydrolysis product was quantified. Data were plotted and analyzed as described for Fig. 3. Double asterisks indicate statistically significant differences (P, <0.001).
eLtaS has narrow lipid substrate specificity.
The eLtaS enzyme reaction observed in the in vitro assay is similar to that of the phospholipase C type of enzymes. Some phospholipases, such as the B. cereus PLC enzyme, have broad substrate specificity with the following order of substrate preference, from highest to lowest: phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (30). In addition, as shown here and as determined in a previous study, NBD-PG or PG can also serve as a substrate for B. cereus PLC (38). To investigate the substrate specificity of eLtaS, the ability of the enzyme to cleave NBD-PC, NBD-PE. and NBD-PS was tested. Hydrolysis of the phosphodiester bond in each of these lipid substrates would result in the production of an identical NBD-DAG-labeled lipid reaction product. As can be seen in Fig. 6, eLtaS can hydrolyze only the NBD-labeled PG lipid substrate; no hydrolysis of PC, PE, or PS was seen, indicating a narrow substrate specificity for this class of enzymes.
FIG. 6.

The substrate specificity of the S. aureus eLtaS enzyme was tested by setting up enzyme assays with NBD-PG, NBD-PS, NBD-PC, or NBD-PE, as indicated. As a control, no enzyme (no enz.) or the active-site variant eLtaS-T300A was added to reaction mixtures, as indicated above the panels. PLC reactions using NBD-PG as a substrate were run alongside to indicate the mobility of the hydrolysis product. Note that only the portions of the TLC plates that correspond to the location of the hydrolysis product are shown.
In vitro activities of other members of the lipoteichoic acid synthase enzyme family.
Next, we set out to test if the in vitro assay can also be used to analyze the activities of other members of the lipoteichoic acid synthase family of enzymes. In a previous study, we have shown that L. monocytogenes encodes two enzymes of the LtaS family (42). While both enzymes are involved in LTA synthesis, they have distinct enzymatic functions in vivo. LtaPLm (Lmo0644) serves as the LTA primase and initiates LTA synthesis by the initial transfer of a glycerolphosphate subunit to the glycolipid anchor, while LtaSLm (Lmo0927) is the LTA synthase, which polymerizes the glycerolphosphate backbone chain of LTA. However, it is assumed that both enzymes use PG lipid as a substrate. To test this experimentally, we purified recombinant N-terminally His-tagged versions of the extracellular domains of LtaPLm and LtaSLm (eLtaPLm and eLtaSLm) (see Fig. S1 in the supplemental material). These proteins were used in the standard in vitro assays with the NBD-PG lipid substrate and in the presence of 10 mM MnCl2. Both L. monocytogenes enzymes showed activity (Fig. 7). A total of six independent experiments were performed, and in all experiments, eLtaPLm showed activity similar to that of the S. aureus eLtaS enzyme, while the eLtaSLm enzyme showed higher activity, though to varying degrees. This is apparent from the relatively large error bar that resulted when the combined averages and standard deviations of the normalized values from all six independent experiments were plotted (Fig. 7). In sum, these results show that both L. monocytogenes enzymes, the LTA primase and the LTA synthase, can use NBD-PG lipid as a substrate, and they indicate that the in vitro assay can be used to study the activities of other members of the lipoteichoic acid synthase family.
FIG. 7.
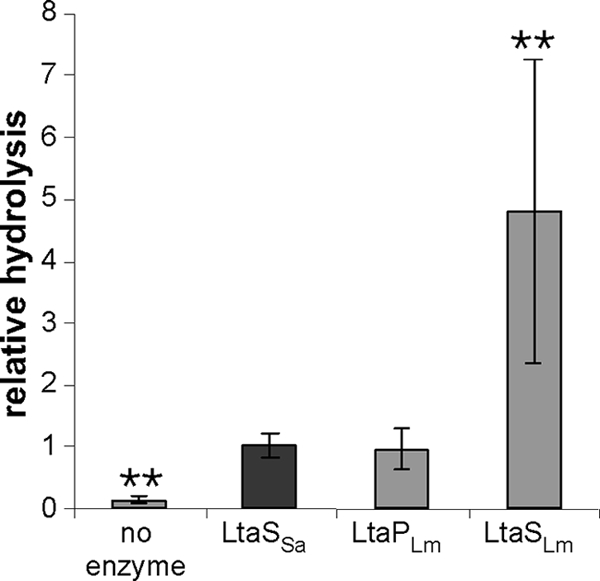
In vitro activities of L. monocytogenes LtaS-type enzymes. Enzyme assays were set up with eLtaPLm, eLtaSLm, and S. aureus eLtaS using NBD-PG as a substrate. Six independent experiments were performed with triplicate samples. Data were plotted and analyzed as described for Fig. 3. Double asterisks indicate statistically significant differences (P, <0.001).
DISCUSSION
In this study, we describe the first characterization of an enzyme belonging to the polyglycerolphosphate lipoteichoic acid synthesis protein family by using an in vitro assay system and defined purified components. We show that the extracellular domain of the S. aureus enzyme, eLtaS, is sufficient to cleave the phosphodiester bond within the substrate NBD-PG, resulting in the release of NBD-DAG (Fig. 1 and 2). This reaction requires the active-site threonine residue at position 300, as evidenced by the fact that incubation of NBD-PG with the active-site variant eLtaS-T300A does not lead to the production of NBD-DAG (Fig. 1 and 2). While all experiments in this study were performed with the soluble eLtaS lipoteichoic acid synthase domain, it should be noted that when full-length LtaS was overexpressed in E. coli, and membrane fractions were isolated and incubated with NBD-PG, we observed the production of a lipid with a mobility identical to that of the top band seen for PLC or eLtaS reactions in Fig. 1B. This lipid product was absent when membrane fractions from an E. coli strain expressing the active-site variant LtaS-T300A were used (unpublished results). This indicates that the full-length LtaS protein can also use the NBD-PG lipid as a substrate in a membrane environment.
eLtaS-dependent cleavage of NBD-PG is similar to that catalyzed by the phospholipase C type of enzymes. Detailed structural studies of the B. cereus PLC enzyme (used as a control in this study), in addition to an exhaustive biochemical analysis, established that PLC contains three Zn2+ ions in its active center and provided mechanistic details on the reaction mechanism (3, 15-17, 38, 40). Structural studies of LtaS enzymes revealed that these proteins also contain a metal ion within their active sites (29, 37). In the present study, we used the in vitro assay to provide experimental evidence that Mn2+ is the relevant cation necessary for enzyme function and that Zn2+ inhibits enzyme activity (Fig. 3). Hence, the enzymatic mechanism leading to PG hydrolysis will be quite different for PLC- and LtaS-type enzymes. The E. coli enzyme encoded by the mdoB gene is a Mn2+-dependent metal enzyme which we assume has a reaction mechanism similar to that of LtaS (18, 19). MdoB catalyzes the transfer of glycerolphosphate units from PG to periplasmic oligosaccharides, which are produced under low-osmolarity conditions (18, 19, 22). Interestingly, like LtaS, MdoB exists in two forms, a membrane-tethered form predicted to contain three N-terminal transmembrane helices and a second, soluble periplasmic form, the result of an internal protein cleavage event (27). The full-length membrane-embedded MdoB protein, also called phosphoglycerol transferase I, uses PG as a substrate and transfers the glycerolphosphate head group to membrane-linked oligosaccharides, while the soluble periplasmic protein domain, named phosphoglycerol transferase II, transfers these sugar-linked glycerolphosphate groups to soluble periplasmic oligosaccharides and/or exchanges glycerolphosphate groups between these soluble sugars (13, 18, 19, 27). It is interesting that the pathway is activated under low-osmolarity conditions (22). The accumulation of high levels of glycans, together with their glycerolphosphate and other modifications, will greatly modify the physical properties of the Gram-negative periplasm. It has been suggested that this will lead to an increase in the osmolarity within the periplasmic space, which helps bacteria to cope with low external osmolarity (21). Similarly, the product of the LtaS reaction, LTA, seems to be important for survival under low-osmolarity conditions. The growth of an S. aureus ltaS mutant strain, which is unable to produce LTA, can be rescued when bacteria are grown under high-osmolarity conditions, such as those established by the addition of salt or sucrose to the growth medium (35). Recent detailed cryoelectron microscopic analyses of the Gram-positive cell wall envelope have led to the description of a Gram-positive periplasm (the space between the membrane and the peptidoglycan layer) (31-34). Since LTA is an important constituent of this Gram-positive periplasm, it can be proposed that this polymer plays a role in regulating the local osmotic pressure on the outside of the cell membrane (33). However, additional studies are necessary to support such a model and to reveal the physiological function of LTA.
We never detected complete hydrolysis of the NBD-PG lipid substrate by eLtaS. Possibly, PG is not the preferred substrate for the soluble eLtaS domain. It should also be noted that the soluble periplasmic E. coli phosphoglycerol transferase II enzyme uses glycerolphosphate groups that are already linked to sugar moieties as substrates, and the enzyme does not act on PG (13). By analogy, glycerolphosphate groups already linked to the glycolipid anchor, an intermediate of LTA synthesis, could serve as preferred substrates for the processed S. aureus eLtaS domain. However, the role of LtaS cleavage in S. aureus remains unclear, and by extension, it remains unclear whether the cleaved eLtaS protein serves a function in the cell and, if so, which function. Currently, we know only that the eLtaS domain alone is not sufficient for LTA synthesis in S. aureus and that the full-length LtaS protein is required (M. E. Wörmann, unpublished results). Therefore, the full-length LtaS protein might be responsible for PG lipid cleavage in vivo and perhaps is needed for efficient lipid hydrolysis in an in vitro assay system. However, membrane proteins are notoriously difficult to purify and to study in in vitro systems. In the case of LtaS, this difficulty is combined with the fact that the full-length protein is inherently unstable and is cleaved (to release the eLtaS domain) even when expressed in E. coli (unpublished results). Therefore, it is particularly interesting that by broadening the analysis to other members of the lipoteichoic acid synthase enzyme family, we have identified an enzyme that is more active in an in vitro assay system (Fig. 7). The basis for the difference in activity between the S. aureus eLtaS enzyme and the L. monocytogenes eLtaSLm enzyme is not clear, and additional work is needed to improve our understanding. Nevertheless, the L. monocytogenes eLtaS enzyme might be better suited to the study of the enzyme mechanism of LTA synthases in more detail and to the investigation of the fate of the cleaved glycerolphosphate group and the formation of other predicted enzyme reaction products, such as the polyglycerolphosphate chain resulting from a glycerolphosphate transfer reaction. More active enzymes may also aid in the adaptation of the in vitro assay to a format that can be used to screen for and identify potential enzyme inhibitors. This study is a first step toward an enzymatic characterization of this important class of bacterial enzymes, and it opens the way to further structural and functional analysis.
Supplementary Material
Acknowledgments
This work was supported by funds made available by the Imperial College London Drug Discovery Center and by Medical Research Council grant G0701212 to A.G.
We thank Judit Nagy for technical assistance with the MALDI mass spectrometry instrument. We thank Paul Freemont, Duo Lu, Cathy Tralau-Stewart, Albert Jaxa-Chamiec, and Caroline Low for valuable discussions. We also acknowledge Christiaan van Ooij for valuable input and critical reading of the manuscript.
Footnotes
Published ahead of print on 13 August 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Abachin, E., C. Poyart, E. Pellegrini, E. Milohanic, F. Fiedler, P. Berche, and P. Trieu-Cuot. 2002. Formation of d-alanyl-lipoteichoic acid is required for adhesion and virulence of Listeria monocytogenes. Mol. Microbiol. 43:1-14. [DOI] [PubMed] [Google Scholar]
- 2.Archibald, A. R., J. J. Armstrong, J. Baddiley, and J. B. Hay. 1961. Teichoic acids and the structure of bacterial walls. Nature 191:570-572. [DOI] [PubMed] [Google Scholar]
- 3.Benfield, A. P., N. M. Goodey, L. T. Phillips, and S. F. Martin. 2007. Structural studies examining the substrate specificity profiles of PC-PLC(Bc) protein variants. Arch. Biochem. Biophys. 460:41-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cleveland, R. F., L. Daneo-Moore, A. J. Wicken, and G. D. Shockman. 1976. Effect of lipoteichoic acid and lipids on lysis of intact cells of Streptococcus faecalis. J. Bacteriol. 127:1582-1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins, L. V., S. A. Kristian, C. Weidenmaier, M. Faigle, K. P. Van Kessel, J. A. Van Strijp, F. Götz, B. Neumeister, and A. Peschel. 2002. Staphylococcus aureus strains lacking d-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J. Infect. Dis. 186:214-219. [DOI] [PubMed] [Google Scholar]
- 6.Duckworth, M., A. R. Archibald, and J. Baddiley. 1975. Lipoteichoic acid and lipoteichoic acid carrier in Staphylococcus aureus H. FEBS Lett. 53:176-179. [DOI] [PubMed] [Google Scholar]
- 7.Emdur, L., and T. Chiu. 1975. The role of phosphatidylglycerol in the in vitro biosynthesis of teichoic acid and lipoteichoic acid. FEBS Lett. 55:216-219. [DOI] [PubMed] [Google Scholar]
- 8.Emdur, L. I., and T. H. Chiu. 1974. Turnover of phosphatidylglycerol in Streptococcus sanguis. Biochem. Biophys. Res. Commun. 59:1137-1144. [DOI] [PubMed] [Google Scholar]
- 9.Fischer, W. 1994. Lipoteichoic acid and lipids in the membrane of Staphylococcus aureus. Med. Microbiol. Immunol. (Berl.) 183:61-76. [DOI] [PubMed] [Google Scholar]
- 10.Fisher, N., L. Shetron-Rama, A. Herring-Palmer, B. Heffernan, N. Bergman, and P. Hanna. 2006. The dltABCD operon of Bacillus anthracis Sterne is required for virulence and resistance to peptide, enzymatic, and cellular mediators of innate immunity. J. Bacteriol. 188:1301-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatlin, C. L., R. Pieper, S. T. Huang, E. Mongodin, E. Gebregeorgis, P. P. Parmar, D. J. Clark, H. Alami, L. Papazisi, R. D. Fleischmann, S. R. Gill, and S. N. Peterson. 2006. Proteomic profiling of cell envelope-associated proteins from Staphylococcus aureus. Proteomics 6:1530-1549. [DOI] [PubMed] [Google Scholar]
- 12.Glaser, L., and B. Lindsay. 1974. The synthesis of lipoteichoic acid carrier. Biochem. Biophys. Res. Commun. 59:1131-1136. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg, D. E., M. K. Rumley, and E. P. Kennedy. 1981. Biosynthesis of membrane-derived oligosaccharides: a periplasmic phosphoglyceroltransferase. Proc. Natl. Acad. Sci. U. S. A. 78:5513-5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gründling, A., and O. Schneewind. 2007. Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 104:8478-8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen, S., L. K. Hansen, and E. Hough. 1992. Crystal structures of phosphate, iodide and iodate-inhibited phospholipase C from Bacillus cereus and structural investigations of the binding of reaction products and a substrate analogue. J. Mol. Biol. 225:543-549. [DOI] [PubMed] [Google Scholar]
- 16.Hansen, S., E. Hough, L. A. Svensson, Y. L. Wong, and S. F. Martin. 1993. Crystal structure of phospholipase C from Bacillus cereus complexed with a substrate analog. J. Mol. Biol. 234:179-187. [DOI] [PubMed] [Google Scholar]
- 17.Hough, E., L. K. Hansen, B. Birknes, K. Jynge, S. Hansen, A. Hordvik, C. Little, E. Dodson, and Z. Derewenda. 1989. High-resolution (1.5 Å) crystal structure of phospholipase C from Bacillus cereus. Nature 338:357-360. [DOI] [PubMed] [Google Scholar]
- 18.Jackson, B. J., J. P. Bohin, and E. P. Kennedy. 1984. Biosynthesis of membrane-derived oligosaccharides: characterization of mdoB mutants defective in phosphoglycerol transferase I activity. J. Bacteriol. 160:976-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson, B. J., and E. P. Kennedy. 1983. The biosynthesis of membrane-derived oligosaccharides. A membrane-bound phosphoglycerol transferase. J. Biol. Chem. 258:2394-2398. [PubMed] [Google Scholar]
- 20.Jonquières, R., H. Bierne, F. Fiedler, P. Gounon, and P. Cossart. 1999. Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: a novel mechanism of protein association at the surface of gram-positive bacteria. Mol. Microbiol. 34:902-914. [DOI] [PubMed] [Google Scholar]
- 21.Kennedy, E. P. 1996. Membrane-derived oligosaccharides (periplasmic beta-d-glycans) of Escherichia coli, p. 1064-1071. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, Jr., B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella typhimurium: cellular and molecular biology, 2nd ed., vol. 1. ASM Press, Washington, DC. [Google Scholar]
- 22.Kennedy, E. P. 1982. Osmotic regulation and the biosynthesis of membrane-derived oligosaccharides in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 79:1092-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koch, H. U., R. Haas, and W. Fischer. 1984. The role of lipoteichoic acid biosynthesis in membrane lipid metabolism of growing Staphylococcus aureus. Eur. J. Biochem. 138:357-363. [DOI] [PubMed] [Google Scholar]
- 24.Koga, Y., M. Nishihara, and H. Morii. 1984. Products of phosphatidylglycerol turnover in two Bacillus strains with and without lipoteichoic acid in the cells. Biochim. Biophys. Acta 793:86-94. [PubMed] [Google Scholar]
- 25.Lai, Y., A. Di Nardo, T. Nakatsuji, A. Leichtle, Y. Yang, A. L. Cogen, Z. R. Wu, L. V. Hooper, R. R. Schmidt, S. von Aulock, K. A. Radek, C. M. Huang, A. F. Ryan, and R. L. Gallo. 2009. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat. Med. 15:1377-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambert, P. A., I. C. Hancock, and J. Baddiley. 1975. The interaction of magnesium ions with teichoic acid. Biochem. J. 149:519-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lequette, Y., E. Lanfroy, V. Cogez, J. P. Bohin, and J. M. Lacroix. 2008. Biosynthesis of osmoregulated periplasmic glucans in Escherichia coli: the membrane-bound and the soluble periplasmic phosphoglycerol transferases are encoded by the same gene. Microbiology 154:476-483. [DOI] [PubMed] [Google Scholar]
- 28.Loidl, A., R. Claus, H. P. Deigner, and A. Hermetter. 2002. High-precision fluorescence assay for sphingomyelinase activity of isolated enzymes and cell lysates. J. Lipid Res. 43:815-823. [PubMed] [Google Scholar]
- 29.Lu, D., M. E. Wörmann, X. Zhang, O. Schneewind, A. Gründling, and P. S. Freemont. 2009. Structure-based mechanism of lipoteichoic acid synthesis by Staphylococcus aureus LtaS. Proc. Natl. Acad. Sci. U. S. A. 106:1584-1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin, S. F., B. C. Follows, P. J. Hergenrother, and B. K. Trotter. 2000. The choline binding site of phospholipase C (Bacillus cereus): insights into substrate specificity. Biochemistry 39:3410-3415. [DOI] [PubMed] [Google Scholar]
- 31.Matias, V. R., and T. J. Beveridge. 2007. Cryo-electron microscopy of cell division in Staphylococcus aureus reveals a mid-zone between nascent cross walls. Mol. Microbiol. 64:195-206. [DOI] [PubMed] [Google Scholar]
- 32.Matias, V. R., and T. J. Beveridge. 2005. Cryo-electron microscopy reveals native polymeric cell wall structure in Bacillus subtilis 168 and the existence of a periplasmic space. Mol. Microbiol. 56:240-251. [DOI] [PubMed] [Google Scholar]
- 33.Matias, V. R., and T. J. Beveridge. 2008. Lipoteichoic acid is a major component of the Bacillus subtilis periplasm. J. Bacteriol. 190:7414-7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matias, V. R., and T. J. Beveridge. 2006. Native cell wall organization shown by cryo-electron microscopy confirms the existence of a periplasmic space in Staphylococcus aureus. J. Bacteriol. 188:1011-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oku, Y., K. Kurokawa, M. Matsuo, S. Yamada, B. L. Lee, and K. Sekimizu. 2009. Pleiotropic roles of polyglycerolphosphate synthase of lipoteichoic acid in growth of Staphylococcus aureus cells. J. Bacteriol. 191:141-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peschel, A., M. Otto, R. W. Jack, H. Kalbacher, G. Jung, and F. Götz. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 274:8405-8410. [DOI] [PubMed] [Google Scholar]
- 37.Schirner, K., J. Marles-Wright, R. J. Lewis, and J. Errington. 2009. Distinct and essential morphogenic functions for wall- and lipo-teichoic acids in Bacillus subtilis. EMBO J. 28:830-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shinitzky, M., P. Friedman, and R. Haimovitz. 1993. Formation of 1,3-cyclic glycerophosphate by the action of phospholipase C on phosphatidylglycerol. J. Biol. Chem. 268:14109-14115. [PubMed] [Google Scholar]
- 39.Simocková, M., R. Holic, D. Tahotna, J. Patton-Vogt, and P. Griac. 2008. Yeast Pgc1p (YPL206c) controls the amount of phosphatidylglycerol via a phospholipase C-type degradation mechanism. J. Biol. Chem. 283:17107-17115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sundell, S., S. Hansen, and E. Hough. 1994. A proposal for the catalytic mechanism in phospholipase C based on interaction energy and distance geometry calculations. Protein Eng. 7:571-577. [DOI] [PubMed] [Google Scholar]
- 41.Waters, C. M., H. Hirt, J. K. McCormick, P. M. Schlievert, C. L. Wells, and G. M. Dunny. 2004. An amino-terminal domain of Enterococcus faecalis aggregation substance is required for aggregation, bacterial internalization by epithelial cells and binding to lipoteichoic acid. Mol. Microbiol. 52:1159-1171. [DOI] [PubMed] [Google Scholar]
- 42.Webb, A. J., M. Karatsa-Dodgson, and A. Gründling. 2009. Two-enzyme systems for glycolipid and polyglycerolphosphate lipoteichoic acid synthesis in Listeria monocytogenes. Mol. Microbiol. 74:299-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weidenmaier, C., S. A. Kristian, and A. Peschel. 2003. Bacterial resistance to antimicrobial host defenses—an emerging target for novel antiinfective strategies? Curr. Drug Targets 4:643-649. [DOI] [PubMed] [Google Scholar]
- 44.Weidenmaier, C., and A. Peschel. 2008. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 6:276-287. [DOI] [PubMed] [Google Scholar]
- 45.Ziebandt, A. K., H. Weber, J. Rudolph, R. Schmid, D. Hoper, S. Engelmann, and M. Hecker. 2001. Extracellular proteins of Staphylococcus aureus and the role of SarA and sigma B. Proteomics 1:480-493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.