

Abstract
Recent reports have begun unraveling the details of various roles of individual eukaryotic translation initiation factor 3 (eIF3) subunits in translation initiation. Here we describe functional characterization of two essential Saccharomyces cerevisiae eIF3 subunits, g/Tif35 and i/Tif34, previously suggested to be dispensable for formation of the 48S preinitiation complexes (PICs) in vitro. A triple-Ala substitution of conserved residues in the RRM of g/Tif35 (g/tif35-KLF) or a single-point mutation in the WD40 repeat 6 of i/Tif34 (i/tif34-Q258R) produces severe growth defects and decreases the rate of translation initiation in vivo without affecting the integrity of eIF3 and formation of the 43S PICs in vivo. Both mutations also diminish induction of GCN4 expression, which occurs upon starvation via reinitiation. Whereas g/tif35-KLF impedes resumption of scanning for downstream reinitiation by 40S ribosomes terminating at upstream open reading frame 1 (uORF1) in the GCN4 mRNA leader, i/tif34-Q258R prevents full GCN4 derepression by impairing the rate of scanning of posttermination 40S ribosomes moving downstream from uORF1. In addition, g/tif35-KLF reduces processivity of scanning through stable secondary structures, and g/Tif35 specifically interacts with Rps3 and Rps20 located near the ribosomal mRNA entry channel. Together these results implicate g/Tif35 and i/Tif34 in stimulation of linear scanning and, specifically in the case of g/Tif35, also in proper regulation of the GCN4 reinitiation mechanism.
The initiation phase of protein synthesis is promoted by numerous proteins or protein complexes called eukaryotic initiation factors (eIFs). The multiprotein eIF3 complex, together with eIFs 1, 1A, and 5, promotes recruitment of the Met-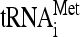 /eIF2/GTP ternary complex (TC) to the small ribosomal subunit (40S), producing the 43S preinitiation complex (PIC). At least in yeast, eIFs 1, 3, and 5 and the TC occur in a preformed unit called the multifactor complex (MFC), which enhances the efficiency of the 43S PIC assembly process (reviewed in reference 20). The eIF4F complex, containing the cap-binding eIF4E and the scaffold protein eIF4G, then mediates recruitment of an mRNA to the 43S PIC with the help of eIF3 and the poly(A)-binding protein. The resulting 48S PIC traverses the 5′ untranslated region (UTR) of mRNA, searching usually for the first AUG codon while unwinding secondary structures in an ATP-dependent reaction stimulated by helicases eIF4A and eIF4B (reviewed in reference 39). This intricate process is called scanning, and its precise molecular mechanism is still poorly understood. It is known that the presence of the TC and eIFs 1, 1A, and 3 in reconstituted mammalian 43S PICs is sufficient for scanning through the unstructured leaders of model mRNAs (38). eIFs 1 and 1A are thought to promote scanning by induction of a conformational change of the 40S head. This change, characterized by opening the latch formed by helices 18 (h18) and 34 (h34) of 18S rRNA and establishing a new interaction between RPS3 and h16, stabilizes the small subunit in an open/scanning-conducive state (36). When the start codon is recognized by the anticodon of Met-
/eIF2/GTP ternary complex (TC) to the small ribosomal subunit (40S), producing the 43S preinitiation complex (PIC). At least in yeast, eIFs 1, 3, and 5 and the TC occur in a preformed unit called the multifactor complex (MFC), which enhances the efficiency of the 43S PIC assembly process (reviewed in reference 20). The eIF4F complex, containing the cap-binding eIF4E and the scaffold protein eIF4G, then mediates recruitment of an mRNA to the 43S PIC with the help of eIF3 and the poly(A)-binding protein. The resulting 48S PIC traverses the 5′ untranslated region (UTR) of mRNA, searching usually for the first AUG codon while unwinding secondary structures in an ATP-dependent reaction stimulated by helicases eIF4A and eIF4B (reviewed in reference 39). This intricate process is called scanning, and its precise molecular mechanism is still poorly understood. It is known that the presence of the TC and eIFs 1, 1A, and 3 in reconstituted mammalian 43S PICs is sufficient for scanning through the unstructured leaders of model mRNAs (38). eIFs 1 and 1A are thought to promote scanning by induction of a conformational change of the 40S head. This change, characterized by opening the latch formed by helices 18 (h18) and 34 (h34) of 18S rRNA and establishing a new interaction between RPS3 and h16, stabilizes the small subunit in an open/scanning-conducive state (36). When the start codon is recognized by the anticodon of Met-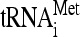 , the concerted action of eIFs 1, 1A, 2, and 5 stimulates a reverse conformational change of the 40S subunit that reforms the h18-h34 latch and arrests scanning (reviewed in reference 27). Upon subunit joining mediated by eIF5B, the 80S couple commences elongation.
, the concerted action of eIFs 1, 1A, 2, and 5 stimulates a reverse conformational change of the 40S subunit that reforms the h18-h34 latch and arrests scanning (reviewed in reference 27). Upon subunit joining mediated by eIF5B, the 80S couple commences elongation.
Over the last decade, functions of several subunits of the most complex initiation factor, eIF3, and its complete subunit composition have been investigated in yeasts, plants, and mammals (reviewed in reference 17). In Saccharomyces cerevisiae, eIF3 comprises five core essential subunits (a/Tif32, b/Prt1, c/Nip1, i/Tif34, and g/Tif35) and one noncore subunit (j/Hcr1). These all have corresponding orthologs in the more complex mammalian eIF3, which contains seven additional nonconserved subunits. Despite recent progress, the true composition of the core of mammalian eIF3 remains somewhat obscure. One study aimed at reconstitution of a human eIF3 in vitro suggested that the functional core contains three nonconserved subunits, e, f, and h, in place of eIF3i and -g (25), whereas other work based on tandem mass spectrometry and solution disruption assays identified three stable modules, one of which, composed of a, b, i, and g subunits, closely resembled the yeast eIF3 core (62).
A systematic effort was devoted to mapping the binding site of eIF3 on the 40S subunit. We found that the extreme N-terminal domain (NTD) of a/Tif32 forms a crucial intermolecular bridge between eIF3 and the 40S subunit (49) and that the RNA recognition motif (RRM) of b/Prt1 and the extreme C-terminal domain (CTD) of c/Nip1 also play direct roles in anchoring eIF3 to the ribosome (9, 33, 51). In addition, we observed that deleting the CTD of a/Tif32 reduced 40S association with the MFC when the connection between eIF3 and eIF5/Tif5 in the MFC was impaired by the tif5-7A mutation (51). Importantly, our findings that the a/Tif32 CTD interacts with helices 16 to 18 of 18S rRNA (51) and Rps2 and Rps3 (6), that the a/Tif32 NTD binds to ribosomal proteins Rps0A and Rps10A (51), and that the j/Hcr1 CTD interacts with Rps2 (9) suggested that yeast eIF3 associates with the solvent-exposed side of the 40S subunit, as others have proposed for mammalian eIF3 (45, 48).
Functional studies revealed that j/Hcr1, the only nonessential subunit of yeast eIF3, forms together with the a/Tif32 CTD and the RRM of b/Prt1 an eIF3 subassembly that ensures stringency of the AUG start codon selection by blocking leaky scanning (6, 9, 33). Likewise, the c/Nip1 subunit was implicated in regulation of the AUG decoding mechanism owing to the fact that its NTD associates directly or indirectly with the key actors in this process, such as eIF1, eIF5, and the TC (53). On the other hand, the prt1-1 point mutation in b/Prt1 and single point substitutions in the conserved KERR motif of a/TIF32-CTD were among other effects suggested to reduce the rate of ribosomal scanning (6, 31). Given the essentiality of all core subunits, a surprising result came from a biochemical study that suggested that the a/Tif32-b/Prt1-c/Nip1 subcomplex lacking g/Tif35 and i/Tif34 subunits is sufficient to stimulate the TC and mRNA recruitment to the 40S subunit and even to promote efficient translation in vitro (40). These findings were subsequently supported by pioneering work that used reconstituted mammalian eIF3 and suggested that eIF3i and -g are dispensable for active mammalian eIF3 formation in vitro (25).
Besides playing these canonical roles in general translation initiation, eIF3 was also implicated in the gene-specific translational control mechanism termed reinitiation (REI) in yeast, plant, and mammalian cells (35, 42, 49). REI is utilized to up- or downregulate translation of regulatory proteins, such as transcription factors and proto-oncogenes, in response to various environmental stimuli (22). In general, it relies on the ability of the small ribosomal subunit to remain attached to the mRNA following termination of translation on a short upstream ORF (uORF) in order to resume scanning on the same mRNA molecule. The next critical step of REI is de novo recruitment of the TC, which is required to recognize the next AUG codon; therefore, REI can be delicately regulated by manipulating the eIF2/GTP levels (8). The uORFs thus possess the exquisite potential to function as context-dependent cis regulators of translation.
The best-studied example of the REI mechanism is the translational control of yeast GCN4, a transcriptional activator of a large number of biosynthetic genes (reviewed in reference 18). The mRNA leader of GCN4 contains four short uORFs that differ dramatically in their capacity to promote efficient REI. Whereas uORFs 2 to 4 are very inefficient, uORF1 allows the majority of 40S ribosomes terminating at its stop codon to remain mRNA bound and resume scanning. This ability requires two segments of uORF1: enhancer sequences upstream of its start codon, and the last codon plus ∼12 nucleotides (nt) after the stop codon (12, 14). We have recently demonstrated that the 5′ enhancer of uORF1 functionally interacts with the extreme NTD of a/Tif32 at or near the mRNA exit channel of the posttermination 40S subunit. This interaction was proposed to be instrumental in stabilizing the 40S subunit on the mRNA to enable resumption of scanning for efficient REI on a downstream ORF (49). Under nutrient-replete conditions characterized by high levels of the TC, nearly all of the rescanning 40S ribosomes after uORF1 will rebind the TC before reaching uORFs 2 to 4, translate one of these uORFs, and dissociate from the mRNA. Amino acid starvation leads to phosphorylation of eIF2α by the kinase Gcn2, converting eIF2/GDP from a substrate to a competitive inhibitor of its GEF, eIF2B, thus reducing the concentration of TC. Low TC levels derepress GCN4 translation by allowing ∼50% of rescanning 40S ribosomes to rebind TC after bypassing the trap of uORFs 2 to 4 and to reinitiate at GCN4 instead. Failure to induce expression of GCN4 in response to a shortage of amino acids in various mutants confers increased sensitivity to inhibitors of amino acid biosynthetic enzymes, and this has been designated the Gcn− phenotype.
In this report, we performed functional analysis of two small essential eIF3 subunits, g/Tif35 and i/Tif34, whose contributions to general translation initiation were virtually unknown. Site-directed substitutions in the RRM of g/Tif35 in g/tif35-KLF produced no impact on its RNA-binding affinity, on the integrity of eIF3 in the MFC, or on formation of the 43S PICs. Nevertheless, the g/tif35-KLF mutation markedly reduced rates of translation initiation and decreased the processivity of scanning through a stable secondary structure inserted into the 5′-UTR of uORF-less GCN4 mRNA. In addition, g/tif35-KLF provoked a strong Gcn− phenotype owing to the inability of posttermination 40S subunits at GCN4's uORF1 to resume scanning for reinitiation downstream; this resembles the previously described phenotype of the a/tif32-Δ8 mutant (49). Detailed genetic analysis revealed, however, that the g/Tif35 RRM and the a/Tif32 NTD ensure efficient resumption of scanning by different mechanisms. Like g/tif35-KLF, the Q258R mutation of the WD40 repeat 6 of i/Tif34 also produced a severe Gcn− phenotype. However, in contrast to g/tif35-KLF, i/tif34-Q258R allowed resumption of scanning after uORF1 but significantly reduced the rate of scanning. Consistently, the Gcn− phenotype of i/tif34-Q258R was partially suppressible by cooverexpressing scanning-promoting factors eIF1 and eIF1A. Together these results provide the first insights into the functional contributions of the essential i/Tif34 and g/Tif35 subunits of yeast eIF3 to general translation initiation as well as to translational control of GCN4 expression.
MATERIALS AND METHODS
Construction of yeast strains and plasmids.
To create H464, H421 (Table 1) was first transformed with YEpTIF35-T (Table 2), and YEp-TIF35-U was evicted by growth on 5-fluoroorotic acid (5-FOA) medium. The resulting strain was transformed to Ura+ with the integrative GCN2 plasmid pHQ835 (kindly provided by Hongfang Qiu) digested with SnaBI. Ura− segregants were obtained by selecting for growth on medium containing 5-FOA, and the resulting H464 was tested for the presence of integrated GCN2 by growth on medium containing 3-aminotriazole (3-AT). YEp-TIF35-U was reintroduced into the verified strain, which was then grown on Trp+ medium to enable spontaneous loss of the TRP1 covering plasmid, YEpTIF35-T.
TABLE 1.
Yeast strains used in this study
| Straina | Genotype | Source or reference |
|---|---|---|
| H464§ | MATaleu2-3,112 ura3-52::GCN2 trp1Δ tif35Δ (YEp-TIF35-U) | This study |
| H421§ | MATaleu2-3,112 ura3-52 trp1 gcn2Δ tif35Δ (YEp-TIF35-U) | Klaus H. Nielsen |
| H111§ | MATaleu2-3,112 ura3-52::GCN2 trp1Δ tif35Δ (YCp22-g/TIF35-screen) | This study |
| H112§ | MATaleu2-3,112 ura3-52::GCN2 trp1Δ tif35Δ (YCp22-g/tif35-KLF) | This study |
| YBS47¶ | MATaleu2-3,112 ura3-52 trp1Δ gcn2Δ a/tif32Δ URA3::GCN2 ura3 (pRSeIF3a-HIS-L) | 49 |
| YBS53¶ | MATaleu2-3,112 ura3-52 trp1Δ gcn2Δ a/tif32Δ URA3::GCN2 ura3 (pRSeIF3a-Δ8-HIS-L) | 49 |
| H450‖ | MATaleu2-3,112 ura3-52::GCN2 trp1Δ tif34Δ (YEp-TIF34 (URA3)) | This study |
| H420‖ | MATaleu2-3,112 ura3-52 trp1 gcn2Δ tif34Δ (YEp-TIF34 (URA3)) | Klaus H. Nielsen |
| H120‖ | MATaleu2-3,112 ura3-52::GCN2 trp1Δ tif34Δ (YCp111-i/TIF34) | This study |
| H121‖ | MATaleu2-3,112 ura3-52::GCN2 trp1Δ tif34Δ (YCp111-i/tif34-Q258R) | This study |
Strains that share a footnote symbol (§, ¶, or ‖) are isogenic strains.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| p180 (YCp50-GCN4-lacZ) | Low-copy-number URA3 vector containing wild-type GCN4 leader | 29 |
| P227 | Low-copy-number URA3 vector containing GCN4 leader without uORFs | 26 |
| pM226 | Derivative of pM199; ORF of uORF1 extends into the GCN4-lacZ coding region | 13 |
| pA80z | Low-copy-number URA3 vector containing GCN4 leader with solitary uORF4 at the position of uORF1 | 1 |
| pG67 | Low-copy-number URA3 vector containing uORF1 only, placed 32 nt from GCN4-lacZ | 13 |
| pM199 | Low-copy-number URA3 vector containing uORF1 only, at the position of uORF4 (140 nt from GCN4-lacZ) | 13 |
| p209 | Low-copy-number URA3 vector containing uORF1 only, at its original position (350 nt from GCN4-lacZ) | 13 |
| P1014l | Low-copy-number URA3 vector containing uORF1 only, placed 640 nt from GCN4-lacZ | 13 |
| pBS64 | Derivative of pM128; low-copy-number URA3 vector containing GCN4 leader with uORF1 at its original position, with the CCCCGG substitution of the −21 AAAATT −16 region in its 5′-UTR | 49 |
| pBS62 | Derivative of pM128; low-copy-number URA3 vector containing GCN4 leader with uORF1 at its original position with 40-nt deletion in its 5′-UTR (from −61 to −21) | 49 |
| pVM11 | Derivative of pM128; low-copy-number URA3 vector containing GCN4 leader with uORF1 at its original position with 46-nt deletion in its 5′-UTR (from −61 to −15) | 49 |
| pBS63 | Derivative of pM128; low-copy-number URA3 vector containing GCN4 leader with uORF1 at its original position with 160-nt deletion in its 5′-UTR (from −181 to −21) | 49 |
| pWCB07 | GCN4-lacZ lacking all uORFs, with hairpin insertion between nt +481 and +482 in single-copy URA3 plasmid, from YCp50 | This study |
| pWCB06 | GCN4-lacZ lacking all uORFs, with hairpin insertion between nt +481 and +482 in single-copy URA3 plasmid, from YCp50 | This study |
| YCplac22 | Single-copy cloning vector, TRP1 | 11 |
| YEp-TIF35 | High-copy-number g/TIF35 URA3 plasmid from YEplac195 | 3 |
| YEp-TIF34 | High-copy-number i/TIF34 URA3 plasmid from YEplac195 | 3 |
| YCp22-g/TIF35-screen | Single-copy g/TIF35-His TRP1 plasmid from YCplac22 | This study |
| YCp22-g/tif35-KLF | Single-copy g/tif35-KLF-His TRP1 plasmid from YCplac22 | This study |
| pGEX-5X-3 | Cloning vector for GST fusions | 46 |
| pGEX-g/TIF35 | GST-g/Tif35 fusion plasmid from pGEX-5X-3 | 3 |
| pGEX-g/tif35-KLF | GST-g/tif35-KLF fusion plasmid from pGEX-5X-3 | This study |
| pGBK-T7-RPS3 | RPS3 ORF cloned into pGBKT7, TRP1 (Clontech) | 51 |
| pGBK-T7-RPS20 | RPS20 ORF cloned into pGBKT7, TRP1 (Clontech) | 51 |
| pGBK-T7-RPS2 | RPS2 ORF cloned into pGBKT7, TRP1 (Clontech) | 51 |
| pT7-Δ7-b/PRT1 | b/PRT1[641-724] ORF cloned under T7 promoter | 54 |
| pT7-i/TIF34 | i/TIF34 ORF cloned under T7 promoter | 3 |
| YEplac195 | High-copy-number cloning vector, URA3 | 11 |
| YEplac181 | High-copy-number cloning vector, LEU2 | 11 |
| YEplac112 | High-copy-number cloning vector, TRP1 | 11 |
| YEpSUI1-U | High-copy-number SUI1 URA3 plasmid from YEplac195 | 53 |
| pDSO22 | High-copy-number TIF11 URA3 plasmid from YEplac195 | 34 |
| YEp-SUI1+TIF11 | High-copy-number SUI1 TIF11 URA3 plasmid from YEplac195 | This study |
| YEp-SUI1+TIF11-W | High-copy-number SUI1 TIF11 TRP1 plasmid from YEplac112 | 10 |
| YEpTIF2(4A)-U | High-copy-number TIF2 URA3 plasmid from YEplac195 | 7 |
| YEpTIF2(4A)-L | High-copy-number TIF2 LEU2 plasmid from YEplac181 | This study |
| YEpTIF4631(4G)-U | High-copy-number TIF4631 URA3 plasmid from YEplac195 | 7 |
| pCF82 | High-copy-number SUI1 TRP1 plasmid from YEplac112 | 10 |
| YCpL-i/TIF34-HA | Single-copy i/TIF34-HA LEU2 plasmid from YCplac111 | 3 |
| YCpL-i/tif34-HA-3 (Q258R) | Single-copy i/tif34-HA-Q258R LEU2 plasmid from YCplac111 | 3 |
| YEpTIF35-T | High-copy-number g/TIF35 TRP1 plasmid from YEplac112 | 54 |
| YEpTIF34-T | High-copy-number i/TIF34 TRP1 plasmid from YEplac112 | 54 |
| pKA18 | β-Globin under SP6 promoter | 4 |
To produce H111 and H112, H461 was transformed with YCp22-g/TIF35-screen andYCp22-g/tif35-KLF, respectively, and the resident g/TIF35 URA3 plasmid was evicted on 5-FOA-containing medium.
To create H450, H420 (Table 1) was first transformed with YEpTIF34-T (Table 2), and YEp-TIF34-U was evicted by growth on 5-FOA medium. The resulting strain was transformed to Ura+ with the integrative GCN2 plasmid pHQ835 digested with SnaBI. Ura− segregants were obtained by selecting for growth on medium containing 5-FOA, and the resulting H450 was tested for the presence of integrated GCN2 by growing on medium containing 3-AT. YEp-TIF34-U was reintroduced into the verified strain, which was then grown on Trp+ medium to enable spontaneous loss of the TRP1 covering plasmid, YEpTIF34-T.
To produce H120 and H121, H450 was transformed with YCp111-i/TIF34 and YCp111-i/tif34-Q258R, respectively, and the resident i/TIF34 URA3 plasmid was evicted on medium containing 5-FOA.
YCp22-g/TIF35-screen was generated by fusion PCR. The following pairs of primers were used for separate PCR amplifications using Ycp22-g/TIF35-help (see below) as template: (i) 3gSalIr and 3gHistag and (ii) 3gNdeI and 3gHistagr. The PCR products thus obtained were used in a 1:1 ratio as templates for a third PCR amplification using primers 3gSalIr and 3gNdeI. The resulting PCR product was digested with SalI and NdeI and ligated with SalI-NdeI-cleaved Ycp22-g/TIF35-help (replacing wild-type [WT] g/TIF35 with 8×His-tagged g/TIF35-His). Ycp22-g/TIF35-help was constructed by inserting the 1,394-bp KpnI-SalI fragment from YEpTIF35-T into YCplac22 digested with KpnI-SalI.
YCp22-g/tif35-KLF was generated by fusion PCR. The following pairs of primers were used for separate PCR amplifications using YCp22-g/tif35-LF (see below) as template: (i) 3gKLF and 3gXhoIr and (ii) 3gNdeI and 3gKLFr. The PCR products thus obtained were used in a 1:1 ratio as templates for a third PCR amplification using primers 3gXhoIr and 3gNdeI. The resulting PCR product was digested with XhoI and NdeI and ligated with XhoI-NdeI-cleaved YCp22-g/tif35-LF (replacing g/tif35-LF with g/tif35-KLF-His).
YCp22-g/tif35-LF was also generated by fusion PCR. The following pairs of primers were used for separate PCR amplifications using Ycp22-g/TIF35-screen as template: (i) 3gLF and 3gXhoIr and (ii) 3gNdeI and 3gLFr. The PCR products thus obtained were used in a 1:1 ratio as templates for a third PCR amplification using primers 3gXhoIr and 3gNdeI. The resulting PCR product was digested with XhoI and NdeI and ligated with XhoI-NdeI-cleaved Ycp22-g/TIF35-screen (replacing WT g/TIF35-His with g/tif35-LF-His).
pGEX-g/tif35-KLF was made by inserting the XhoI-BamHI-digested PCR product, obtained with primers pGEX35NTD and pGEX35RRMr using the template YCp22-g/tif35-KLF, into XhoI-BamHI-digested pGEX-5X-3.
YEp-SUI1+TIF11was constructed by inserting the 1,134-kb SalI-SacI fragment from pDSO22 into YEpSUI1-U digested with SalI-SacI.
YEpTIF2(4A)-L was constructed by inserting the 2,026-kb SphI-BHI fragment from YEpTIF2(4A)-U into YEplac181 digested with SphI-BHI.
A list of all PCR primers named above can be found in Table 3.
TABLE 3.
Primers used in this study
| Primer name | Primer sequence (5′-3′) |
|---|---|
| 3gSalIr | CTGCAGGTCGACCTCTTCACGATCTGCAAAAGTCCCAA |
| 3gNdeI | CGACCATATGACCATGAACTGTCCATT |
| 3gHistagr | AAACAAGTGCAGAGCATATTCTGTGCATCTCGAGCTAATGATGATGATGATGATGATGATGTTCCTTAACCTTAGGTTTGGACCA |
| 3gHistag | ATGCACAGAATATGCTCTGCACTTGTT |
| 3gLF | CAAAGAAACAGGTAAATCAAGAGGTGCAGCCGCTGTTACCTTTTCGAGCGAAGAAGTTGCCGAACA |
| 3gLFr | TCTTGATTTACCTGTTTCTTTGTTTCT |
| 3gXhoIr | GTGCATCTCGAGCTAATGATGATGATGATG |
| 3gKLFr | AGTACACATATCATCACGTTCTCTAGA |
| 3gKLF | TGATGATATGTGTACTTTGGCAATTATGCAAGTTAATGAAAATGCCGATGAAAA |
| pGEX35NTD | AATAAGGATCCCCATGAGTGAAGTTGCACCAGAG |
| pGEX35RRMr | AATAACTCGAGCTATTCCTTAACCTTAGGTTTGGA |
Yeast biochemical methods.
Glutathione S-transferase (GST) pulldown experiments with GST fusions and in vitro-synthesized 35S-labeled polypeptides (see Table 2 for vector descriptions) were conducted as follows. Individual GST-fusion proteins were expressed in Escherichia coli, immobilized on glutathione-Sepharose beads, and incubated with 10 μl of 35S-labeled potential binding partners at 4°C for 2 h. The beads were washed three times with 1 ml of phosphate-buffered saline, and bound proteins were separated by SDS-PAGE. Gels were first stained with Gelcode Blue stain reagent (Pierce) and then subjected to autoradiography. Ni2+ chelation chromatography of eIF3 complexes containing 8×His-tagged g/Tif35 from yeast whole-cell extracts (WCEs) and Western blot analysis were conducted as described in detail previously (32). In short, WCEs were incubated at 4°C for 2 h with 4 μl of 50% Ni2+-nitrilotriacetic acid-silica resin (Qiagen) suspended in 200 μl of buffer A, followed by washing and elution. β-Galactosidase assays and polysome profile analysis were conducted as described previously (12, 55).
mRNA binding assay.
32P-labeled Xenopus laevis β-globin mRNA was prepared in vitro using the MAXIscript SP6 transcription kit (Ambion Inc.), [α-32P]UTP (10 mCi/ml), and the pKA18 vector (4) linearized with BamHI, according to the vendor's instructions. The transcript of the first 354 nucleotides of X. laevis β-globin mRNA was purified using a size exclusion column (NucAway spin column; Ambion, Inc.).
WT and mutant g/Tif35 proteins fused to the GST moiety and immobilized on glutathione-Sepharose beads were incubated with 100 ng of 32P-labeled X. laevis β-globin mRNA in 250 μl of the binding buffer (10 mM HEPES [pH 7.6], 3 mM MgCl2, 40 mM KCl, 5% glycerol, 1 mM dithiothreitol, 1.5% 2-β-mercaptoethanol) for 30 min at 26°C. (To increase specificity of binding, 200 ng of yeast total tRNA [Sigma] was added to each reaction mixture as a competitor RNA.) The beads were then washed three times with 1 ml of binding buffer, and bound β-globin mRNA was separated by electrophoresis on 5.5% denaturing (8 M urea) polyacrylamide gel, followed by autoradiography. For control experiments, the same procedure was carried out using beads containing only the GST moiety or beads preincubated with bacterial extracts derived from plain E. coli BL21 cells.
RESULTS
Multiple substitutions of the conserved residues of the RRM of g/Tif35 reduce efficiency of translation initiation.
The functionally important yet nonessential C-terminal domain of g/Tif35 is formed by the canonical RRM previously shown to possess nonspecific RNA-binding activity (15). A typical RRM contains two RNP sites in β-sheets 1 and 3, with four highly conserved positions always occupied by a set of either aromatic or basic residues that critically contribute to RNA binding, namely, RNP1 positions 1 (R or K), 3 (F/Y), and 5 (F/Y) and RNP2 position 2 (F/Y) (for a review, see reference 24). An unpublished nuclear magnetic resonance structure of the human heIF3g RRM (K. Tsuda, Y. Muto, M. Inoue, T. Kigawa, T. Terada, M. Shirouzu, and S. Yokoyama, unpublished data) (protein data bank accession code 2CQ0) (Fig. 1A) shows that RNP2 and RNP1 are typically aligned next to each other in a four-stranded anti-parallel β-sheet packed against two perpendicular α-helices. Interestingly, all of the eIF3g RRM homologs differ from the classical RRM at position 2 of RNP2, in that eIF3g has a basic as opposed to aromatic residue at that position (Fig. 1B). It is thus conceivable that the stacking interaction that the aromatic residue in position 2 normally establishes with mRNA is replaced by ionic attraction with a phosphate group in the RNA backbone. Surprisingly, the yeast protein also differs in position 3 of RNP1 (L versus F) (Fig. 1B).
FIG. 1.

The triple-Ala substitution of the highly conserved residues in RNPs of the g/Tif35 RRM in g/tif35-KLF impairs cell growth and the rate of general translation initiation. (A) The unpublished nuclear magnetic resonance solution structure of the human heIF3g RRM (K. Tsuda et al., unpublished; Protein Data Bank accession code 2CQ0) displays a canonical RRM fold with a four-stranded antiparallel β-sheet packed against two perpendicular α-helices. The highly conserved Arg242 in position 2 of RNP2 in β-sheet 1 and Phe282 and Phe284 in positions 3 and 5 of RNP1 in β-sheet 3 of human eIF3g RRM correspond to Lys194 and to Leu235 and Phe237, respectively, of yeast g/Tif35-RRM and are highlighted in red. (B) Amino acid sequence alignment of g/Tif35-RRM of Saccharomyces cerevisiae with that of other species. The amino acid sequence of S. cerevisiae g/Tif35 (accession number NP_010717) between residues 185 and 274 (the terminal residue) is aligned with its Schizosaccharomyces pombe (accession number CAA18400), Candida albicans (accession number Q59ZV5), Salmo salar (accession number ACI69727), Xenopus tropicalis (accession number Q28CY2), Bos taurus (accession number Q3ZC12), Mus musculus (accession number Q9Z1D1), and Homo sapiens (accession number O75821) homologs. The alignment was conducted with ClustalW (http://www.ch.embnet.org/software/ClustalW.html). Highly conserved sequences of RNP2 and RNP1 are shown in yellow and green, respectively, with the key residues that were subjected to site-directed mutagenesis highlighted in red. The sequence of g/tif35-KLF generated in this study is given at the bottom. (C) g/tif35-KLF severely impairs cell growth at elevated temperatures. The H464 (g/tif35Δ) strain was transformed with YCp22-g/Tif35-screen (top row) and YCp22-g/tif35-KLF (bottom row), and the resident YEp-TIF35-U (URA3) plasmid was evicted on medium containing 5-FOA. The resulting strains were then spotted in four serial 10-fold dilutions on SD medium and incubated at 30, 34, and 37°C for 2 days. The far right columns show results of Western analysis of WCEs from the very same strains grown at 34°C, using anti-g/Tif35 (g/Tif35 expression; lane 1) and anti-GCD11 (eIF2γ loading; lane 2) antibodies. (D) The g/tif35-KLF mutant reduces rates of translation initiation in vivo. Polysome profiles are shown for the strains in panel C cultured in YPD medium at 34°C and treated with cycloheximide just prior to harvesting. WCEs were separated by velocity sedimentation through a 5-to-45% sucrose gradient centrifugation at 39,000 rpm for 2.5 h. The gradients were collected and scanned at 254 nm to visualize the ribosomal species. Positions of 40S, 60S, and 80S species are indicated by arrows, and P/M ratios are given above the profiles.
In an effort to elucidate the role of g/Tif35 in translation, we first mutated conserved residues in either one or both RNPs (shown schematically in Fig. 1A and B) and tested the resulting mutants for slow growth (Slg−) and temperature-sensitive (Ts) phenotypes. Whereas neither individual RNP mutant showed any growth defects (data not shown), the combination of both RNP1 and RNP2 mutations in g/tif35-K194A-L235A-F237A (henceforth termed g/tif35-KLF) resulted in the Ts phenotype without having any effect on the steady-state levels of the g/Tif35 protein in cells (Fig. 1C). The g/tif35-KLF mutation was found to provoke substantial (∼3-fold) reductions in the polysome:monosome (P/M) ratio at 34°C (Fig. 1D), indicating a marked decrease in the rate of translation initiation in vivo.
g/Tif35 associates with eIF3 via two contacts with subunits i/Tif34 and b/Prt1; it was previously shown that its C-terminal RRM is not required for either of these interactions (3). Indeed, the g/tif35-KLF mutation did not affect binding of mutant g/Tif35 (fused to the GST moiety) to radiolabeled i/Tif34 and b/Prt1 in an in vitro pulldown experiment (Fig. 2A, second and third panels). It also had no impact on the integrity of the MFC as judged from affinity purification of the WT and mutant 8×His-tagged g/Tif35 proteins by nickel chelation chromatography followed by analysis of the yields of copurifying proteins by Western blot hybridization (Fig. 2B). Finally, no effect on the association of any of the MFC components with the 40S subunits in vivo was observed, as expected (data not shown).
FIG. 2.

The g/tif35-KLF mutant neither affects the integrity of eIF3 in the MFC nor impairs the RNA-binding activity of g/Tif35. (A) The g/tif35-KLF mutation does not reduce binding of g/TIF35 to b/Prt1, i/Tif34, or β-globin mRNA in vitro. Full-length WT g/Tif35 (lane 3) and mutant g/tif35-KLF (lane 4) fused to GST, and also GST alone (lane 2), were tested for binding to 35S-labeled b/Prt1 and i/Tif34 and to 32P-labeled β-globin mRNA. Lane 1 (In) contains 10% and 2.5% of input amounts of proteins and RNA, respectively, added to each reaction mixture. (B) The g/tif35-KLF mutation does not prevent g/Tif35 from associating with eIF3 in the MFC in vivo. WCEs were prepared from H421 (g/tif35Δ) bearing untagged g/Tif35 (lanes 1 to 4), H111 expressing 8×His-tagged g/Tif35 (lanes 5 to 8), and H112 expressing 8×His-tagged g/tif35-KLF (lanes 9 to 12). The WCEs were incubated with Ni2+-silica resin, and the bound proteins were eluted and subjected to Western blot analysis. Lanes 1, 5, and 9 contained 5% of the input WCEs (In); lanes 2, 6, and 10 contained 30% fractions eluted from the resin (E1); lanes 3, 7, and 11 contained 60% of the same fractions (E2); lanes 4, 8, and 12 contained 5% of the flowthrough (FT). The Western signals for a/Tif32, b/Prt1, eIF2, eIF5, and i/Tif34 in the E1 and E2 fractions for the WT g/TIF35 and mutant g/tif35-KLF strains were quantified, combined, normalized for the amounts of WT g/Tif35 in these fractions, and these data are plotted in the histogram on the right as percentages of the corresponding values calculated for the WT g/TIF35.
To examine whether mutating the RNPs of g/Tif35 affected its RNA-binding affinity, WT and mutant g/Tif35 fused to a GST moiety were incubated with in vitro-synthesized 32P-labeled β-globin mRNA, and the amount of bound mRNA species was quantified. As shown in Fig. 2A (fourth panel), the g/tif35-KLF mutation had no effect on the affinity of g/Tif35 for β-globin mRNA, indicating that none of the targeted residues critically contributes to g/Tif35's RNA-binding activity. Nevertheless, given the marked impact of the KLF mutation on the rate of translation initiation, we decided to explore the nature of its defect further.
The RRM of g/Tif35 is required for full derepression of GCN4 expression under starvation conditions.
According to previous reports, neither yeast nor mammalian eIF3g appears to be required for assembly of the 48S PIC, which includes sequential recruitment of the TC and mRNA to the 40S subunit (25, 40). Hence, we decided to examine whether g/Tif35 might contribute to processes following the assembly of the 48S PIC in living cells, such as scanning and AUG recognition. We tested the g/tif35-KLF mutant for specific phenotypes indicating impairment of translational control of GCN4 expression (18). This mechanism has been extensively used in the past as a valuable genetic tool for dissecting the contributions of individual eIFs to initiation (9, 10, 19, 31, 33, 49, 53, 61). Mutants defective in TC formation and/or its recruitment to the 40S subunit constitutively derepress GCN4 expression, imparting a so-called Gcd− phenotype, whereas mutants that fail to derepress GCN4 under starvation conditions provoke a Gcn− phenotype, which signals defects in the steps following assembly of 48S PICs such as processivity of scanning, AUG recognition, or subunit joining (9, 16, 23, 31, 53, 60).
In agreement with reports arguing against the involvement of g/Tif35 in stimulation of TC loading onto the 40S subunits, g/tif35-KLF did not display the Gcd− phenotype (data not shown). On the other hand, the g/tif35-KLF cells imparted the severe Gcn− phenotype, as they failed to grow in the presence of 3-AT (an inhibitor of histidine biosynthetic genes) at 34°C (Fig. 3A). The effect of 3-AT can only be overcome by sufficient upregulation of GCN4 expression, as illustrated by the fact that 30 mM 3-AT completely prevented growth of g/TIF35+ gcn2Δ cells, in which eIF2α cannot be phosphorylated and thus the TC levels remain high and GCN4 fully repressed (Fig. 3A). It should be noted that phosphorylation of eIF2α by Gcn2 upon starvation was not affected in g/tif35-KLF GCN2 cells, ruling out this possible mechanism for the Gcn− phenotype (data not shown). Using the GCN4-lacZ reporter containing all four uORFs, we measured derepression in g/tif35-KLF cells in response to 3-AT at 34°C and showed that it was reduced by a factor of ∼5 (Fig. 3B, construct i, row 2). The little to no reduction in expression from the uORF-less GCN4-lacZ construct clearly suggests that the observed derepression defect is not caused by changes in GCN4-lacZ mRNA levels (Fig. 3B, construct ii). (It should be noted that mRNAs produced from all GCN4-lacZ constructs used throughout the study are highly stable owing to the fact that they all contain an intact stabilizer element [STE] that prevents the natural GCN4 mRNA from undergoing nonsense-mediated decay [44, 49].) Together, these results imply that the RRM of g/Tif35 is required for proper upregulation of GCN4 expression upon starvation and indicate that its function in general translation initiation might be associated with the steps following formation of the 48S PIC.
FIG. 3.

g/tif35-KLF reduces processivity of scanning and interferes with the reinitiation process by preventing posttermination retention of the 40S ribosome on GCN4 mRNA. (A) g/tif35-KLF imparts a strong Gcn− phenotype, implicating g/Tif35 in regulation of translational control of GCN4 expression. Isogenic strain H464 (GCN2 g/TIF35; row 1) and H421 (gcn2Δ g/TIF35; row 2) transformed with an empty vector YCplac22 and strain H111 (GCN2 g/tif35Δ YCp22-g/TIF35-screen; row 3) and H112 (GCN2 g/tif35Δ YCp22-g/tif35-KLF; row 4) were spotted in four serial 10-fold dilutions on SD (left panel) or SD containing 30 mM 3-AT (right panel) and then incubated at 34°C for 3 or 6 days, respectively. (B, construct i) g/tif35-KLF reduces basal expression of GCN4-lacZ and prevents its full derepression upon starvation. H111 and H112 were transformed with p180 and grown in minimal medium for 6 h, and the β-galactosidase activities were measured in the WCEs and are expressed in units of nmol of o-nitrophenyl-β-d-galactopyranoside hydrolyzed per min per mg of protein. To induce GCN4-lacZ expression, strains grown in minimal medium for 2 h were treated with 10 mM 3-AT for 6 h. The mean values and standard deviations obtained from at least six independent measurements with three independent transformants, along with the activities the g/tif35-KLF mutant strain relative to the corresponding WT, are given in the table. White versus black squares indicate REI-permissive (uORF1) versus REI-nonpermissive (uORFs 2 to 4) uORFs. (Constructs ii to iv) The failure of g/tif35-KLF to derepress GCN4-lacZ is not caused by leaky scanning. H111 and H112 were transformed with p227 (ii), pM226 (iii), and pA80z (iv) and analyzed as for construct (i), except that they were not treated with 3-AT. Xs point to mutations eliminating the AUG start codons of uORFs 1 to 4. A paperclip symbol indicates an insertion of the nucleotide sequence. (Constructs v to viii) g/tif35-KLF blocks induction of GCN4 expression by reducing the amount of posttermination 40S ribosomes on uORF1, which resume scanning for reinitiation downstream. H111 and H112 were transformed with pG67 (v), pM199 (vi), p209 (vii), or p1014l (viii) and analyzed as described for constructs ii to iv. Scissors indicate deletions of the nucleotide sequence. (Constructs ix and x) g/tif35-KLF increases the translation-inhibitory effect of a stable stem-loop structure inserted in the 5′-UTR of uORF-less GCN4 mRNA. H111 and H112 were transformed with pWCB07 (ix) or pWCB06 (x) and analyzed as described for construct i. Both constructs contain the indicated sequences (with complementary bases underlined) inserted 21 nt 5′ of the GCN4 AUG codon. (C) β-Galactosidase activities obtained for constructs v to viii in panel B, with WT and g/tif35-KLF cells were plotted as a function of the intercistronic distance between uORF1 and the AUG start codon of GCN4-lacZ.
The RRM of g/Tif35 promotes resumption of scanning of posttermination ribosomes on uORF1 of GCN4.
To determine what postassembly step(s) might be perturbed by g/tif35-KLF, we analyzed a battery of GCN4-lacZ reporters that varied in their GCN4 mRNA leader sequences. Defects in AUG start codon recognition or subunit joining may lead to the Gcn− phenotype if the 48S PIC scans over the AUG start codon of uORF1 (leaky scanning); this can be monitored with a construct in which uORF1 is elongated and overlaps the beginning of GCN4. This elongated version of uORF1 blocks ∼99% of all scanning ribosomes from reaching the GCN4 start site, indicating that only ∼1% of ribosomes show leaky scanning of uORF1 in WT cells (13). As shown in Fig. 3B (construct iii), we observed a marginal ∼1.5-fold increase in GCN4-lacZ expression from this construct in g/tif35-KLF cells, which would by no means account for a strong derepression defect (Fig. 3B, construct i). Also, little to no increase in β-galactosidase activity was detected with a construct containing only uORF4 (Fig. 3B, construct iv), which allows a negligible level of REI and thus very effectively blocks translation of downstream ORFs (13). Together these results indicate that the KLF mutation does not significantly affect stringency of the start codon selection. It is worth noting that results in Fig. 3B (constructs iii and iv) additionally eliminate the possibility that induction of GCN4-lacZ is reduced in g/tif35-KLF cells due to a decrease in the reporter mRNA level, as this effect should apply equally to the mutant constructs, yet expression of these constructs was higher than in the WT cells.
The derepression defect in g/tif35-KLF cells can be also explained by a reduction in the rate of scanning of ribosomes progressing from uORF1 to uORF4 (i.e., slow scanning) or in their decreased stability on the mRNA during scanning (i.e., abortive scanning). To test this, we analyzed constructs carrying only uORF1 at four different positions relative to GCN4-lacZ (Fig. 3B, constructs v to viii). If the 40S subunits were more prone to dissociating from the mRNA during scanning, then a decrease in the GCN4-lacZ activity would be expected. This was indeed observed; however, one would also predict that the probability of the 40S falling off would increase with the increasing distance to scan through, thus producing a gradual decrease in activity, which is not the case (compare constructs v to viii). Therefore, it is highly unlikely that abortive scanning could explain the Gcn− phenotype. Slower scanning would, on the other hand, increase GCN4-lacZ expression from all four constructs (particularly those with shorter-than-normal spacers between uORF1 and GCN4-lacZ [constructs v and vi]) by providing the scanning 40S subunits more time to rebind TC before reaching the next AUG start codon. However, the fact that g/tif35-KLF decreased, rather than increased, expression from all four constructs to a similar extent (∼57 to 77%) strongly argues against both of these explanations. In fact, the observed phenotype closely resembles that of the a/tif32-Δ8 mutation only recently shown to interfere with the key first step of REI: resumption of scanning of posttermination ribosomes on the uORF1 stop codon (49).
As shown previously (13), GCN4-lacZ expression starts to plateau when the intercistronic distance between uORF1 and GCN4-lacZ exceeds the length of the natural 350-nt spacer, indicating saturated TC reacquisition (Fig. 3B, compare constructs viii and vii). The fact that g/tif35-KLF plateaus at an equal length but with expression of ∼40% of the WT level, as expected, further supports our conclusion that the REI-competent PICs in the mutant cells are reduced to 40% of normal (Fig. 3C). These results hence strongly suggest that besides the NTD of a/Tif32, the RRM of g/Tif35 represents yet another eIF3 domain that is critically required for retention of the posttermination 40S subunits on the mRNA, representing a crucial prerequisite for efficient REI.
The g/Tif35 RRM stimulates scanning through structured mRNA leaders.
Our finding that the g/tif35-KLF mutation impairs resumption of scanning from uORF1 implies that the utility of the GCN4 translational control as a tool to examine postassembly defects in general translation initiation is in this case limited. In particular, any effect of this mutation on the rate of scanning would be masked by its inability to start scanning downstream of uORF1.
To circumvent this obstacle, we compared WT and mutant g/tif35-KLF cells in terms of their abilites to scan through stem-loop structures inserted into the 5′-UTR of a GCN4-lacZ fusion lacking all four uORFs (Fig. 3B, constructs ix and x). Whereas a relatively weak stem-loop structure (ΔH, 0.3 kcal) reduced the GCN4-lacZ expression to the same degree in both WT and g/tif35-KLF cells (by ∼20%) (Fig. 3B, compare construct ix with ii), a stable stem-loop (ΔH, −8.7 kcal) had ∼4-fold stronger impact on translation in the mutant versus WT cells, where it diminished GCN4-lacZ expression by 85% (Fig. 3B, compare constructs x and ii). These results implicate g/Tif35 in stimulating processivity of scanning on mRNAs with structured 5′-UTRs—the first function in general translation initiation ever attributed to this small essential eIF3 subunit.
The a/Tif32 NTD and g/Tif35 RRM functional domains stimulate resumption of scanning by related but not identical mechanisms.
It is believed that linear scanning is promoted by several initiation factors, including eIFs 1, 1A, 4G, 4A, and 4B and a helicase, Ded1, and its yeast homolog Dbp1 (reviewed in reference 39). In addition, eIF4G and eIF4A were shown to be required for resumption of scanning after translation of a short uORF in mammalian cells (43). Thus, we next wished to investigate whether some of these proteins could suppress defects of g/tif35-KLF and a/tif32-Δ8 mutants in processivity of scanning and/or in resumption of scanning that produce the Gcn− phenotype. Toward this end, we overexpressed the proteins individually or in combinations in our mutant strains while scoring for growth effects on SD and 3-AT plates.
None of the factors suppressed the Gcn− phenotype of either of the mutants (Fig. 4A and C and data not shown); by contrast, an increased gene dosage of eIF1 (both alone and in combination with high-copy-number [hc] eIF1A) actually exacerbated the Gcn− phenotype of both of them (Fig. 4A and C, rows 5 and 6 versus 3). This effect of eIF1 was further verified in our reporter assay, which showed that g/tif35-KLF introduced with hc eIF1 conferred a severe derepression defect even at 30°C (Fig. 4B). In addition, the Gcn− phenotype of a/tif32-Δ8 cells was exacerbated by overexpression of eIF4A or its cooverexpression with eIF4G; however, no such effect was observed in g/tif35-KLF cells (Fig. 4A and C). Hence, even though we did not observe any suppression effect and have at present no solid explanation for the exacerbation defects, the latter findings at least indicate that the g/tif35-KLF and a/tif32-Δ8 mutations might differ in their effects on resumption of scanning.
FIG. 4.

Overexpression of eIF1 in g/tif35-KLF diminishes GCN4 upregulation upon starvation even at 30°C. (A) Overexpression of eIF1 in g/tif35-KLF produces the Gcn− phenotype at 30°C. Isogenic strains H111(g/TIF35) and H112 (g/tif35-KLF) were transformed with the following combinations of two vectors: empty vectors YEplac195 and YEplac181 (EV; row 3), pDSO22 and YEplac181 (hc eIF1A; row 4), YEpSUI1-U and YEplac181 (eIF1; row 5), YEp-SUI1 + TIF11 and YEplac181 (eIF1 + eIF1A; row 6), YEpTIF2(4A)-U and YEplac181 (hc eIF4A; row 7), YEpTIF4631(4G)-U and YEplac181 (eIF4G; row 8), YEpTIF2(4A)-L and YEpTIF4631(4G)-U (hc eIF4A + eIF4G; row 9). These strains, together with control strains H464 (GCN2; row 1) and H421 (gcn2Δ; row 2) transformed with YEplac195 and YEplac181, were spotted in four serial 10-fold dilutions on SD or SD medium containing 30 mM 3-AT and incubated at 30°C for 3 or 6 days, respectively. (B) High-copy-number expression of eIF1 in g/tif35-KLF severely blocks induction of GCN4-lacZ expression. Isogenic strains H111 (g/TIF35) and H112 (g/tif35-KLF) transformed either with empty vector YEplac112 (EV) or pCF82 (hc eIF1) were further transformed with p180 and analyzed as described for construct i in Fig. 3B. (C) Overexpression of eIF1 or eIF4A exacerbates the Gcn− phenotype of a/tif32-Δ8. Isogenic strains YBS47 (a/TIF32) and YBS53 (a/tif32-Δ8) were transformed with the same combinations of two vectors as described for panel A, spotted together with the control strains (as for panel A) in four serial 10-fold dilutions on SD or SD containing 30 mM 3-AT, and incubated at 30°C for 2 to 8 days, as indicated.
The high propensity of GCN4's uORF1 for REI depends on its short length (three codons) and enhancer sequences both 5′ and 3′ of uORF1 (reviewed in reference 18). The 5′ enhancer promotes retention of posttermination 40S subunits on the GCN4 mRNA by interacting with the NTD of a/Tif32 (49). To explore whether or not g/Tif35-RRM acts in a similar manner, we transformed the g/tif35-KLF strain with GCN4-lacZ constructs containing solitary uORF1 with progressive deletions of its 5′ enhancer sequence and examined effects of combining the KLF with the 5′ enhancer deletions on REI. Previously, epistatic interactions were observed between the 5′ enhancer deletion mutants and a/tif32-Δ8 (49). In agreement with this study, replacement of the nt −21-AAAATT-nt 16 stretch with CCCCGG or deletions of 40 (Δ40) or 46 (Δ46) nt from nt −21 upstream of the 5′ sequences of uORF1 progressively reduced the induction of GCN4-lacZ expression in WT cells (Fig. 5B, constructs i to iv). The g/tif35-KLF mutation reduced expression of GCN4-lacZ in the WT uORF1-only construct by ∼50% (Fig. 3B, construct vii, and 5B, construct i), and none of the other deletion constructs further decreased this activity. Essentially the same effect was previously observed in a/tif32-Δ8 cells. However, in striking contrast to a/tif32-Δ8, a complete deletion of the 5′ enhancer in Δ160, which diminishes GCN4-lacZ induction to ∼17% in both WT cells (Fig. 5B, construct v) and a/tif32-Δ8 cells (49), had a negligible additive effect in g/tif35-KLF cells (Fig. 5B, compare the ∼45% reduction with construct v versus the ∼51% reduction with construct iv). Together these findings further suggest that the g/Tif35 RRM and a/Tif32 NTD make synergistic yet independent contributions to the stabilization of posttermination 40S subunits on the GCN4 mRNA. The fact that combining the KLF and Δ8 mutations in the tif35Δ tif32Δ double deletion strain resulted in synthetic lethality corroborates this suggestion (data not shown). More importantly, however, our observations also indicate that even though the g/tif35-KLF mutation by itself substantially reduces the resumption-of-scanning capacity of 40S ribosomes after uORF1, it concurrently diminishes the requirement of the uORF1 5′ enhancer sequences for efficient REI (see Discussion).
FIG. 5.

The g/tif35-KLF mutation diminishes the requirement of uORF1 5′ enhancer sequences for efficient REI. (A) Schematic showing the predicted position of the 40S ribosome terminating at the stop codon of uORF1 from the GCN4 mRNA leader (based on data from reference 49). E, P, and A sites of the 40S ribosomes are aligned with the last two coding triplets and the TAA stop codon. The locations of the 5′ enhancer (labeled 5′-a/Tif32 enhancer to denote the interaction with the NTD of a/Tif32), linker, and buried parts of the sequences upstream of uORF1 are indicated at the top; the 3′ boundaries of the Δ40 deletion (identical to Δ160), Δ46 deletion, and the (C)4GG multiple substitution are shown below the line depicting mRNA. (B, constructs i to v) The same experiment as described for construct i in Fig. 3B, except that H111 and H112 were transformed with p209, pBS64, pBS62, pVM11, and pBS63 (constructs i to v, respectively) and analyzed without 3-AT treatment. Activities relative to WT are given as percentages in boldface in the table to the right of the schematics.
g/Tif35 interacts with the 40S beak proteins Rps20 and Rps3.
To gain more insight into the role of the g/Tif35 RRM in the REI mechanism, we wished to predict the g/Tif35 position on the 40S ribosome. Toward this end we tested GST-g/Tif35 for interactions against all 33 small ribosomal proteins (Rps), which were synthesized and 35S-labeled in rabbit reticulocyte lysates. Among all small ribosomal proteins, only Rps3 and Rps20 strongly interacted with GST-g/Tif35; these interactions were independent of the KLF mutation (Fig. 6A and data not shown). (Rps2 is shown for specificity, as it lies near Rps3 and 20 [Fig. 6B].)
FIG. 6.

g/Tif35 specifically interacts with Rps3 and Rps20 situated on the beak of the solvent-exposed side of the 40S subunit. Also shown in a revised model of the hypothetical location of eIF3 on the S. cerevisiae small ribosomal subunit. (A) g/Tif35 fused to GST (lane 3) or GST alone (lane 2) was tested for binding to 35S-labeled Rps3, -20, and -2 essentially as described for Fig. 2A. (B and C) Revised hypothetical location of the S. cerevisiae eIF3 on the back side of the 40S subunit, based on the data presented in this study and data from reference 51. The cryo-electron microscopy reconstruction of the 40S subunit is shown from the solvent side, with RNA segments in yellow and proteins in green. Positions of Rps3 and Rps20, i/Tif34, the RRM of g/Tif35, and the extreme CTD of b/Prt1 are highlighted in bold. The mRNA entry and exit channels are designated by an asterisk and an X, respectively. The blue lines represent mRNA. Positions of Rps2, -3, and -9 were modified according to findings described in reference 50.
Based on homology modeling with E. coli ribosomal proteins, both Rps3 and -20 were suggested to form the beak on the solvent-exposed side of the 40S subunit (47) (Fig. 6B). The fact that g/Tif35 specifically interacted only with two Rpss occurring next to each other is significant and suggests that this eIF3 subunit most likely occurs somewhere between the head-lobe and the beak regions (Fig. 6C). Based on these and other interactions identified between the a/Tif32 CTD and Rpss 2 and 3 (6) and between the c/Nip1 and the 40S head proteins (T. Kouba and L. Valášek, unpublished observations), we modified our original model for the position of eIF3 on the 40S subunit (51) as follows. The c/Nip1 CTD and the b/Prt1 CTD interacting with i/Tif34 and g/Tif35 were moved upwards to the head region, with the b/Prt1-CTD/i/Tif34/g/Tif35 module stretching toward the beak in proximity of the mRNA entry channel (Fig. 6C). The a/Tif32 NTD interacts with Rps0 and is thus thought to occur in the vicinity of the mRNA exit channel, where it could interact with the 5′ feature of uORF1 (49). Hence, since both a/Tif32-NTD and g/Tif35-RRM seem to occupy different positions on the back of the 40S, it is indeed conceivable that the nature of their involvement in the initial REI phase is mechanistically different, at least to a certain extent.
i/Tif34 stimulates the rate of scanning.
Since eIF3i was, alongside eIF3g, suggested to be dispensable for assembly of the functional 48S PIC in both yeast and mammals (25, 40), we next decided to examine the role of yeast i/Tif34 in scanning and other postassembly processes, in a similar manner as described above for g/Tif35. Yeast i/Tif34 is composed of seven WD40 repeats assembled into a propeller ring and has been shown to interact with the NTD of g/Tif35 and the extreme CTD of b/Prt1 (3, 52). Previously, three growth-defective i/Tif34 mutants were generated and characterized for binding defects (3). Of these, only the i/tif34-3-HA (henceforth i/tif34-Q258R) mutation, mapping into the WD repeat 6, had no effect on i/Tif34 interactions with g/Tif35 and b/Prt1, yet it showed a >2-fold reduction in the translational rates in vivo. These findings indicate that the Q258R mutations might have a functional defect rather than a simple assembly problem. Therefore, we selected the Q258R mutant for further analysis.
As expected, i/tif34-Q258R did not produce the Gcd− phenotype, which would indicate a defect in TC recruitment in vivo (data not shown). However, it did provoke a strong Gcn− phenotype in the presence of 3-AT (Fig. 7A ), resulting from an ∼3-fold reduction in the GCN4 derepression (Fig. 7B, construct i). (This defect was not caused by insufficient phosphorylation of eIF2α [data not shown].) Detailed examination revealed that i/tif34-Q258R modestly increases skipping of the AUG start codon of a short uORF preceding the GCN4-lacZ gene (by a factor of ∼2) (Fig. 7B, constructs iii and iv). More importantly, however, i/tif34-Q258R confers strong (∼3-fold and ∼2.5-fold) increases in expression of the GCN4-lacZ constructs containing solitary uORF1 with the 32-nt and 140-nt spacers, respectively (Fig. 7B, constructs v and vi), that are too large to be explained merely by the amount of leaky scanning (compare, for example, construct vi in Fig. 7B, with 1991 − 817, or 1,174 units, with construct iii, with 62 − 28, or 34 units).) Furthermore, increasing the spacing between uORF1 and the GCN4-lacZ start site to the natural 350 nt progressively increased GCN4-lacZ expression in both strains, as expected; however, the β-galactosidase activity was still ∼1.5-fold higher in i/tif34-Q258R versus WT cells (Fig. 7B, construct vii). These results are consistent with a defect in the rate of scanning, where the slower-scanning PICs are provided with more time to rebind TC before reaching the next start codon and the closer the solitary uORF1 is to GCN4-lacZ, the greater the increase in the frequency of REI. On the other hand, no effect on scanning through stem-loop structures inserted into the uORF-less leader of GCN4-lacZ was observed in i/tif34-Q258R cells (data not shown), in contrast to g/tif35-KLF (Fig. 3B, constructs ix and x). Together, these findings strongly suggest that i/tif34-Q258R provokes the Gcn− phenotype partially by modest leaky scanning over uORF1 but mainly by severe impairment of the rate of scanning between uORF1 and uORF4, which results in an increased number of 40S subunits that rebind TC before reaching inhibitory uORFs 2 to 4. Since i/tif34-Q258R has apparently no effect on resumption of scanning, we propose that both observed defects apply equally well to translation of any mRNA in the Q258R mutant cells. To our knowledge this is the strongest evidence obtained to date that implicates an eIF3 subunit in stimulating the rate of scanning in vivo.
FIG. 7.

Genetic evidence that i/tif34-Q258R prevents induction of GCN4 expression by a combination of modestly increased leaky scanning of uORF1 and a severe reduction in the rate of scanning from uORF1. (A) i/tif34-Q258R imparts a strong Gcn− phenotype, implicating i/Tif34 in regulation of translational control of GCN4. Isogenic strains H450 (GCN2 i/TIF34; row 1) and H420 (gcn2Δ i/TIF34; row 2) transformed with empty vector YCplac111 and strains H120 (GCN2 i/tif34Δ YCp111-i/TIF34; row 3) and H121 (GCN2 i/tif34Δ YCp111-i/tif34-Q258R; row 4) were spotted in four serial 10-fold dilutions on SD (left panel) or SD containing 30 mM 3-AT (right panel) and then incubated at 30°C for 6 or 8 days, respectively. (B, construct i) i/tif34-Q258R prevents full derepression of GCN4-lacZ expression upon starvation. Isogenic strains H120 (i/TIF34) and H121 (i/tif34-Q258R) were transformed with p180 and analyzed as described for construct i in Fig. 3B. (Constructs ii to iv) The i/tif34-Q258R mutation increases leaky scanning over the AUG start codon. H120 and H121 were transformed with p227 (ii), pM226 (iii), and pA80z (iv) and analyzed as described for constructs ii to iv in Fig. 3B. (Constructs v to vii) The i/tif34-Q258R mutation reduces the rate of scanning of posttermination 40S ribosomes from uORF1. H120 and H121 were transformed with pG67 (v), pM199 (vi), or p209 (vii) and analyzed as described for constructs ii to iv in Fig. 3B.
Finally, we examined whether some of the aforementioned scanning-promoting factors could suppress the Gcn− phenotype of i/tif34-Q258R. Strikingly, whereas an increased gene dosage of eIF1 exacerbated the Gcn− phenotype of the Q258R mutant, combined overexpression of eIF1 and eIF1A (i.e., two factors implicated in promotion of scanning [37, 38]) partially suppressed it (Fig. 8A, rows 6 versus 4). This suppression effect of eIF1 and eIF1A was further verified with the results of our reporter assay, where we showed that simultaneous overexpression of eIF1 and eIF1A increased the GCN4-lacZ activity in the i/tif34-Q258R cells treated with 3-AT by ∼3.5-fold but had no such an effect in the WT cells (Fig. 8B). Finally, we also observed that overexpression of the RNA helicase eIF4A, alone or in combination with eIF4G, greatly exacerbated the Slg− phenotype of i/tif34-Q258R, whereas overexpression of eIF4G alone partially suppressed it (Fig. 8A, rows 7 to 9). Although the molecular basis of these synthetic phenotypes remains to be elucidated, the observed genetic interactions of i/tif34-Q258R with the key scanning-promoting factors lend further support to our conclusion that i/Tif34 enhances the rate of the ribosomal scanning.
FIG. 8.

Simultaneous overexpression of eIFs 1 and 1A partially suppresses the Gcn− phenotype of i/tif34-Q258R. (A) Isogenic strains H120 (i/TIF34) and H121 (i/tif34-Q258R) were transformed with the same combinations of two vectors as for Fig. 4A, spotted together with H450 (GCN2; row 1) and H420 (gcn2Δ; row 2) transformed with YEplac195 and YEplac181 in four serial 10-fold dilutions on SD or SD containing 10 mM 3-AT, and incubated at 30°C for 5 or 8 days. (B) High-copy-number expression of eIFs 1 and 1A increases the GCN4-lacZ activity in the i/tif34-Q258R cells treated with 3-AT. Isogenic strains H120 (i/TIF34) and H121 (i/tif34-Q258R) transformed either with empty vector YEplac112 (EV) or YEp-SUI1+TIF11-W (hc eIF1 + eIF1A) were further transformed with p180 and analyzed as described for construct i in Fig. 3B.
DISCUSSION
In this paper we have focused on functional characterization of two small subunits of eIF3, g/Tif35 and i/Tif34, the cellular roles of which have remained highly elusive even though these subunits are essential for the viability of yeast cells (15, 21, 30). We found that substitutions of conserved residues of the RRM of g/Tif35 in g/tif35-KLF significantly reduced processivity of scanning through a stable stem-loop structure inserted into the uORF-less GCN4 mRNA leader; this prevented a large proportion of scanning PICs from reaching the coding region (Fig. 3). We also demonstrated that the single point mutation Q258R, mapping to the WD40 repeat 6 of i/Tif34, markedly impairs the rate of scanning of the 40S ribosomes that translate uORF1 and resume scanning downstream (Fig. 7B). The latter severe scanning defect, under starvation conditions with a limited supply of the TC, provides sufficient time for 40S ribosomes en route from uORF1 to GCN4 to rebind TC before reaching inhibitory uORFs 2 to 4 and thus blocks induction of GCN4 expression and produces the Gcn− phenotype (Fig. 7A).
It can be argued that the GCN4 model system used to explore the scanning properties of posttermination 40S subunits is not suitable for general studies of linear scanning by 48S PICs initiating de novo at the mRNA's 5′ end. However, as McCarthy and colleagues elegantly demonstrated, ribosomes scan with the same efficiency after termination on short uORFs as they do when starting from the 5′ cap; therefore, scanning in general is highly processive, with an extremely low off-rate (5). These observations thus validate the employed assay and make our findings applicable to the mechanism of canonical linear scanning. Hence, given that both i/Tif34 and g/Tif35 are dispensable for formation of the 48S PIC at the mRNA's 5′ end in both yeast and mammals (25, 40), and that neither the i/tif34-Q258R nor g/tif35-KLF mutation affects the integrity of the eIF3 complex (Fig. 2B and reference 3), our results provide the first evidence to date that directly implicates two eIF3 subunits in promoting the rate and processivity of scanning in living cells.
Ostensibly at odds with these conclusions, i/Tif34 and g/Tif35 were found to be dispensable for translation in in vitro cell-free systems. Specifically, using the ribosome-binding-toeprinting assay, the mammalian study measured formation of 48S PICs at the AUG codon of β-globin mRNA after a 5-min incubation of all components in a reaction buffer followed by a 10-min incubation with a reverse transcriptase mixture (25). The yeast study monitored the extent of a functional rescue of luciferase mRNA translation in a heat-inactivated b/prt1-1 extract 70 min after addition of various eIF3 subcomplexes purified by Ni chelation chromatography (40). In this way the a/Tif32-b/Prt1-c/Nip1 subcomplex was found to be nearly as active as the five-subunit complex, whereas the b/Prt1-i/Tif34-g/Tif35 subcomplex was practically inactive. It must be noted, however, that both approaches focused on the end points of the reactions that were monitored and did not follow the kinetics. As such, neither of them would likely be sensitive enough to detect qualitative defects in the rate and processivity of scanning.
At the same time, it is fair to note that the simple assumption that i/Tif34 and g/Tif35 are merely required to augment activities of other eIFs in scanning would not explain their essentiality for cellular viability in yeasts. To account for this fact, we propose the following. It has been established by numerous labs that the “strength” of a particular mRNA is determined by the presence of stable secondary structures in its 5′-UTR; in general, the closer these structures are to the 5′ cap, the more inhibitory they become (reviewed in reference 22). It is conceivable that a suboptimal rate of scanning might have dramatically diverse effects on translatability of mRNAs differing in both the length and the complexity of their 5′-UTRs. The essential character of both small eIF3 subunits might explained by suggesting that the loss of their stimulatory effects on scanning predominantly compromises translation of a subset of critical mRNAs encoding tightly regulated genes, such as those involved in cell cycle regulation, signal transduction, etc.—mRNAs which often have long 5′-UTRs rich in secondary structures (22, 56). This proposal is consistent with earlier observations showing that (i) specific mutations in i/Tif34 or overexpression of its fission yeast homolog Sum1 deregulates progression through the cell cycle and affects mating and the osmotic stress response (21, 57) and (ii) overexpression of human eIF3i, often observed in carcinomas, resulted in cell size increase, proliferation enhancement, cell cycle progression, and anchorage-independent growth (2). Indeed, we cannot rule out that there might be other essential functions for these two proteins in translation or even functions that are independent of eIF3. It will be important to subject those regions of both subunits that were not studied here to systematic mutagenesis and investigate their potential roles in translation and beyond.
In support of the i/TIF34 role in scanning, the Gcn− phenotype of its Q258R mutant was partially suppressed by simultaneous overexpression of eIFs 1 and 1A (Fig. 8), i.e., the two master regulators of scanning (38). Interestingly, overexpression of only eIF1 conversely worsened the effect of i/tif34-Q258R on derepression of GCN4, as did overexpression of eIF1 alone or in combination with eIF1A in g/tif35-KLF cells (Fig. 4). In addition, sole overexpression of the eIF4A helicase exacerbated the Slg− phenotype of i/tif34-Q258R but not that of g/tif35-KLF cells (Fig. 4 and 8). In both mutants, no effects were associated with other helicases implicated in translation initiation, such as Ded1, Dbp1, and eIF4B. Scanning consists of two linked processes: unwinding of secondary structures in the 5′-UTR and ribosomal movement along it. The 43S PICs can scan unstructured 5′-UTRs without factors associated with RNA unwinding and are thus intrinsically capable of movement along mRNA (38). Omission of eIF1A substantially reduces this ability and lack of eIF1 almost abrogates it, indicating that movement of the 43S complexes requires the open/scanning-conducive conformation induced by the synergistic actions of eIF1 and eIF1A (36). Furthermore, scanning along 5′-UTRs containing even weak secondary structures requires ATP and eIF4A, eIF4G, and eIF4B (38). However, the mechanism by which these factors assist scanning remains unknown. That said, our suppression and synthetic exacerbation genetic data might indicate that i/tif34-Q258R impedes full opening of the mRNA entry channel (i.e., the adoption of the scanning-conducive conformation), perhaps in a manner that could be corrected by the mass action of eIF1 and eIF1A. Alternatively, or in addition, both the Q258R and KLF mutations could predominantly interfere with the action of at least one of the initiation-specific helicases. Given the fact that g/Tif35 specifically interacts with Rps20 and mainly with Rps3 (Fig. 6), which is directly involved in transition between the closed/scanning-incompetent and open/scanning-conducive conformations of the 40S ribosome (36), it could be further proposed that this g/Tif35-Rps3 contact places i/Tif34 in a favorable position to influence the latter conformational transitions. (It should be noted that we did not detect any specific interactions between i/Tif34 and any of the 33 ribosomal proteins.) At the same time, the predicted position of g/Tif35 above the mRNA entry channel may enable its RRM to stimulate scanning by presenting mRNAs to the decoding center and/or by promoting the action of helicases. These options resonate with recent findings suggesting that mammalian eIF3 forms an extension of the mRNA-binding channel that might contribute to scanning (41). g/Tif35 could, for example, act in cooperation with eIF4B, a cofactor for the RNA helicase activity of eIF4F, since yeast g/Tif35 and eIF4B were found to interact (58). At odds with this scenario, however, we did not observe any binding between these two proteins in our GST pulldown assays (data not shown).
It is intriguing that the g/tif35-KLF mutation dramatically reduced induction of GCN4 expression under amino acid starvation by impairing resumption of scanning of posttermination ribosomes from uORF1. The strongest evidence supporting the nature of this defect was provided by demonstrating that irrespective of the intercistronic distance between uORF1 and the start codon of GCN4-lacZ, the efficiency of REI on GCN4 in the g/tif35-KLF cells remained at ∼30 to 40% of the WT (Fig. 3). These and other results thus implicate the RRM of g/Tif35, along with the a/Tif32 NTD, in stabilizing uORF1 posttermination 40S ribosomes on GCN4 mRNA and/or in promoting resumption of scanning for REI downstream. Intriguingly, eIF3g has also been shown to play a critical role in the REI mechanism in plants, where it interacted with the cauliflower mosaic virus transactivator, TAV, and promoted translation reinitiation of viral polycistronic mRNAs (35).
The key feature of GCN4's uORF1 is its ability to allow a high frequency of resumption of scanning after its translation; this ability depends on its short length (three codons) and its 5′ and 3′ enhancer sequences (18). Importantly, we have recently demonstrated that the postinitiation retention of eIF3 on 80S ribosomes translating uORF1 is likewise critical for uORF1's full REI capacity, owing to the fact that the 5′ enhancer sequences interact with the NTD of a/Tif32 and thus stabilize the posttermination 40S subunit on GCN4 mRNA (49).
Several lines of evidence suggest that even though both the a/Tif32 NTD and the g/Tif35 RRM are critical for this initial REI phase, their individual roles differ. First, combining g/tif35-KLF with the N-terminal deletion of a/Tif32 in a/tif32-Δ8 resulted in a synthetic lethal phenotype. Also, our suppression/exacerbation analysis with scanning-promoting eIFs at high copy numbers revealed different effects on the Gcn− phenotypes of g/tif35-KLF versus a/tif32-Δ8 mutants (Fig. 4). Third, the a/Tif32 NTD and the g/Tif35 RRM domains seem to occur on opposing pores of the mRNA-binding channel. Furthermore, our detailed genetic analysis strongly suggests that the g/Tif35 RRM does not promote resumption of scanning in cooperation with the 5′ enhancer of uORF1 (Fig. 5). Finally, the indisputable functional importance of uORF1's 5′ enhancer is practically eliminated in the background of the KLF mutation (Fig. 5B, construct v). Interestingly, somewhat similar results were observed when the GCN4 leader lacking the 5′ enhancer sequences of uORF1 was placed in front of the recombinant GAL1-lacZ gene (28). The resulting construct was unexpectedly fully capable of conferring the GCN4-like mode of translational control upon the GAL1-lacZ transcript. Besides the coding sequence, the only other region in which the GCN4-lacZ and GAL1-lacZ constructs differed were their 3′-UTRs, which corresponded to the genuine chromosomal sequence of each gene. These findings might imply that the GCN4 3′-UTR plays an inhibitory role in the GCN4 translational control mechanism, perhaps as a part of the 5′-cap/eIF4E/eIF4G/PAB1/3′-UTR closed-loop structure (59). It will be intriguing to examine how exactly the g/Tif35 RRM and the GCN4 3′-UTR contribute to reinitiation and whether there is any functional connection between them.
Acknowledgments
We are thankful to the members of the Valášek and Krásný laboratories for helpful comments and to Olga Krydová for technical and administrative assistance. We are also indebted to Monica Liu for critical reading of the manuscript and Olga Janoušková and Edit Rutkai for their help with site-directed mutagenesis of the g/Tif35-RRM and GST pulldown experiments with the small ribosomal proteins, respectively.
This research was supported by the Howard Hughes Medical Institute, The Wellcome Trust grant 076456/Z/05/Z, a Fellowship of Jan E. Purkyne from the Academy of Sciences of the Czech Republic, and Institutional Research Concept AV0Z50200510.
Footnotes
Published ahead of print on 2 August 2010.
REFERENCES
- 1.Abastado, J. P., P. F. Miller, B. M. Jackson, and A. G. Hinnebusch. 1991. Suppression of ribosomal reinitiation at upstream open reading frames in amino acid-starved cells forms the basis for GCN4 translational control. Mol. Cell. Biol. 11:486-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahlemann, M., R. Zeidler, S. Lang, B. Mack, M. Münz, and O. Gires. 2006. Carcinoma-associated eIF3i overexpression facilitates mTOR-dependent growth transformation. Mol. Carcinog. 45:957-967. [DOI] [PubMed] [Google Scholar]
- 3.Asano, K., L. Phan, J. Anderson, and A. G. Hinnebusch. 1998. Complex formation by all five homologues of mammalian translation initiation factor 3 subunits from yeast Saccharomyces cerevisiae. J. Biol. Chem. 273:18573-18585. [DOI] [PubMed] [Google Scholar]
- 4.Asano, K., H.-P. Vornlocher, N. J. Richter-Cook, W. C. Merrick, A. G. Hinnebusch, and J. W. B. Hershey. 1997. Structure of cDNAs encoding human eukaryotic initiation factor 3 subunits: possible roles in RNA binding and macromolecular assembly. J. Biol. Chem. 272:27042-27052. [DOI] [PubMed] [Google Scholar]
- 5.Berthelot, K., M. Muldoon, L. Rajkowitsch, J. Hughes, and J. E. G. McCarthy. 2004. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol. Microbiol. 51:987-1001. [DOI] [PubMed] [Google Scholar]
- 6.Chiu, W. L., S. Wagner, A. Herrmannova, L. Burela, F. Zhang, A. K. Saini, L. Valasek, and A. G. Hinnebusch. 28 June 2010. The C-terminal region of eIF3a promotes mRNA recruitment, scanning and, together with eIF3j and the eIF3b RRM, selection of AUG start codons. Mol. Cell Biol. doi: 10.1128/MCB.00280-10. [DOI] [PMC free article] [PubMed]
- 7.de la Cruz, J., I. Iost, D. Kressler, and P. Linder. 1997. The p20 and Ded1 proteins have antagonistic roles in eIF4E-dependent translation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 94:5201-5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dever, T. E., L. Feng, R. C. Wek, A. M. Cigan, T. D. Donahue, and A. G. Hinnebusch. 1992. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68:585-596. [DOI] [PubMed] [Google Scholar]
- 9.ElAntak, L., S. Wagner, A. Herrmannova, M. Karáskova, E. Rutkai, P. J. Lukavsky, and L. Valášek. 2010. The indispensable N-terminal half of eIF3j co-operates with its structurally conserved binding partner eIF3b-RRM and eIF1A in stringent AUG selection. J. Mol. Biol. 396:1097-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fekete, C. A., S. F. Mitchell, V. A. Cherkasova, D. Applefield, M. A. Algire, D. Maag, A. K. Saini, J. R. Lorsch, and A. G. Hinnebusch. 2007. N- and C-terminal residues of eIF1A have opposing effects on the fidelity of start codon selection. EMBO J. 26:1602-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gietz, R. D., and A. Sugino. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527-534. [DOI] [PubMed] [Google Scholar]
- 12.Grant, C. M., and A. G. Hinnebusch. 1994. Effect of sequence context at stop codons on efficiency of reinitiation in GCN4 translational control. Mol. Cell. Biol. 14:606-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grant, C. M., P. F. Miller, and A. G. Hinnebusch. 1994. Requirements for intercistronic distance and level of eIF-2 activity in reinitiation on GCN4 mRNA varies with the downstream cistron. Mol. Cell. Biol. 14:2616-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grant, C. M., P. F. Miller, and A. G. Hinnebusch. 1995. Sequences 5′ of the first upstream open reading frame in GCN4 mRNA are required for efficient translational reinitiation. Nucleic Acids Res. 23:3980-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanachi, P., J. W. B. Hershey, and H. P. Vornlocher. 1999. Characterization of the p33 subunit of eukaryotic translation initiation factor-3 from Saccharomyces cerevisiae. J. Biol. Chem. 274:8546-8553. [DOI] [PubMed] [Google Scholar]
- 16.Hannig, E. M., A. M. Cigan, B. A. Freeman, and T. G. Kinzy. 1992. GCD11, a negative regulator of GCN4 expression, encodes the γ subunit of eIF-2 in Saccharomyces cerevisiae. Mol. Cell. Biol. 13:506-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinnebusch, A. G. 2006. eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem. Sci. 31:553-562. [DOI] [PubMed] [Google Scholar]
- 18.Hinnebusch, A. G. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 59:407-450. [DOI] [PubMed] [Google Scholar]
- 19.Hinnebusch, A. G., K. Asano, D. S. Olsen, L. O. N. Phan, K. H. Nielsen, and L. Valasek. 2004. Study of translational control of eukaryotic gene expression using yeast. Ann. N. Y. Acad. Sci. 1038:60-74. [DOI] [PubMed] [Google Scholar]
- 20.Hinnebusch, A. G., T. E. Dever, and K. A. Asano. 2007. Mechanism of translation initiation in the yeast Saccharomyces cerevisiae, p. 225-268. In N. Sonenberg, M. Mathews, and J. W. B. Hershey (ed.), Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 21.Humphrey, T., and T. Enoch. 1998. Sum1, a highly conserved WD-repeat protein, suppresses S-M checkpoint mutants and inhibits the osmotic stress cell cycle response in fission yeast. Genetics 148:1731-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozak, M. 2005. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361:13-37. [DOI] [PubMed] [Google Scholar]
- 23.Lee, J. H., T. V. Pestova, B. S. Shin, C. Cao, S. K. Choi, and T. E. Dever. 2002. Initiation factor eIF5B catalyzes second GTP-dependent step in eukaryotic translation initiation. Proc. Natl. Acad. Sci. U. S. A. 99:16689-16694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maris, C., C. Dominguez, and F. H.-T. Allain. 2005. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 272:2118-2131. [DOI] [PubMed] [Google Scholar]
- 25.Masutani, M., N. Sonenberg, S. Yokoyama, and I. H. 2007. Reconstitution reveals the functional core of mammalian eIF3. EMBO J. 26:3373-3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller, P. F., and A. G. Hinnebusch. 1989. Sequences that surround the stop codons of upstream open reading frames in GCN4 mRNA determine their distinct functions in translational control. Genes Dev. 3:1217-1225. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell, S. F., and J. R. Lorsch. 2008. Should I stay or should I go? Eukaryotic translation initiation factors 1 and 1a control start codon recognition. J. Biol. Chem. 283:27345-27349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mueller, P. P., S. Harashima, and A. G. Hinnebusch. 1987. A segment of GCN4 mRNA containing the upstream AUG codons confers translational control upon a heterologous yeast transcript. Proc. Natl. Acad. Sci. U. S. A. 84:2863-2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller, P. P., and A. G. Hinnebusch. 1986. Multiple upstream AUG codons mediate translational control of GCN4. Cell 45:201-207. [DOI] [PubMed] [Google Scholar]
- 30.Naranda, T., M. Kainuma, S. E. McMillan, and J. W. B. Hershey. 1997. The 39-kilodalton subunit of eukaryotic translation initiation factor 3 is essential for the complex's integrity and for cell viability in Saccharomyces cerevisiae. Mol. Cell. Biol. 17:145-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nielsen, K. H., B. Szamecz, L. J. Valasek, A., B. S. Shin, and A. G. Hinnebusch. 2004. Functions of eIF3 downstream of 48S assembly impact AUG recognition and GCN4 translational control. EMBO J. 23:1166-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen, K. H., and L. Valášek. 2007. In vivo deletion analysis of the architecture of a multi-protein complex of translation initiation factors. Methods Enzymol. 431:15-32. [DOI] [PubMed] [Google Scholar]
- 33.Nielsen, K. H., L. Valášek, C. Sykes, A. Jivotovskaya, and A. G. Hinnebusch. 2006. Interaction of the RNP1 motif in PRT1 with HCR1 promotes 40S binding of eukaryotic initiation factor 3 in yeast. Mol. Cell. Biol. 26:2984-2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsen, D. S., E. M. Savner, A. Mathew, F. Zhang, T. Krishnamoorthy, L. Phan, and A. G. Hinnebusch. 2003. Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J. 22:193-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park, H. S., A. Himmelbach, K. S. Browning, T. Hohn, and L. A. Ryabova. 2001. A plant viral “reinitiation” factor interacts with the host translational machinery. Cell 106:723-733. [DOI] [PubMed] [Google Scholar]
- 36.Passmore, L. A., T. M. Schmeing, D. Maag, D. J. Applefield, M. G. Acker, M. A. Algire, J. R. Lorsch, and V. Ramakrishnan. 2007. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell 26:41-50. [DOI] [PubMed] [Google Scholar]
- 37.Pestova, T. V., S. I. Borukhov, and C. U. T. Hellen. 1998. Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394:854-859. [DOI] [PubMed] [Google Scholar]
- 38.Pestova, T. V., and V. G. Kolupaeva. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 16:2906-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pestova, T. V., J. R. Lorsch, and C. U. T. Hellen. 2007. The mechanism of translation initiation in eukaryotes, p. 87-128. In N. Sonenberg, M. Mathews, and J. W. B. Hershey (ed.), Translational control in biology and medicine. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 40.Phan, L., L. W. Schoenfeld, L. Valášek, K. H. Nielsen, and A. G. Hinnebusch. 2001. A subcomplex of three eIF3 subunits binds eIF1 and eIF5 and stimulates ribosome binding of mRNA and tRNAiMet. EMBO J. 20:2954-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pisarev, A. V., C. U. T. Hellen, and T. V. Pestova. 2007. Recycling of eukaryotic posttermination ribosomal complexes. Cell 131:286-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pöyry, T. A., A. Kaminski, E. J. Connell, C. S. Fraser, and R. J. Jackson. 2007. The mechanism of an exceptional case of reinitiation after translation of a long ORF reveals why such events do not generally occur in mammalian mRNA translation. Genes Dev. 21:3149-3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pöyry, T. A., A. Kaminski, and R. J. Jackson. 2004. What determines whether mammalian ribosomes resume scanning after translation of a short upstream open reading frame? Genes Dev. 18:62-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruiz-Echevarria, M. J., and S. W. Peltz. 2000. The RNA binding protein Pub1 modulates the stability of transcripts containing upstream open reading frames. Cell 101:741-751. [DOI] [PubMed] [Google Scholar]
- 45.Siridechadilok, B., C. S. Fraser, R. J. Hall, J. A. Doudna, and E. Nogales. 2005. Structural roles for human translation factor eIF3 in initiation of protein synthesis. Science 310:1513-1515. [DOI] [PubMed] [Google Scholar]
- 46.Smith, D. B., and K. S. Johnson. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31-40. [DOI] [PubMed] [Google Scholar]
- 47.Spahn, C. M., R. Beckmann, N. Eswar, P. A. Penczek, A. Sali, G. Blobel, and J. Frank. 2001. Structure of the 80S ribosome from Saccharomyces cerevisiae-tRNA ribosome and subunit-subunit interactions. Cell 107:373-386. [DOI] [PubMed] [Google Scholar]
- 48.Srivastava, S., A. Verschoor, and J. Frank. 1992. Eukaryotic initiation factor 3 does not prevent association through physical blockage of the ribosomal subunit-subunit interface. J. Mol. Biol. 220:301-304. [DOI] [PubMed] [Google Scholar]
- 49.Szamecz, B., E. Rutkai, L. Cuchalova, V. Munzarova, A. Herrmannova, K. H. Nielsen, L. Burela, A. G. Hinnebusch, and L. Valášek. 2008. eIF3a cooperates with sequences 5′ of uORF1 to promote resumption of scanning by post-termination ribosomes for reinitiation on GCN4 mRNA. Genes Dev. 22:2414-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor, D. J., B. Devkota, A. D. Huang, M. Topf, E. Narayanan, A. Sali, S. C. Harvey, and J. Frank. 2009. Comprehensive molecular structure of the eukaryotic ribosome. Structure 17:1591-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valášek, L., A. Mathew, B. S. Shin, K. H. Nielsen, B. Szamecz, and A. G. Hinnebusch. 2003. The yeast eIF3 subunits TIF32/a and NIP1/c and eIF5 make critical connections with the 40S ribosome in vivo. Genes Dev. 17:786-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valášek, L., K. H. Nielsen, and A. G. Hinnebusch. 2002. Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 21:5886-5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valášek, L., K. H. Nielsen, F. Zhang, C. A. Fekete, and A. G. Hinnebusch. 2004. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 24:9437-9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valášek, L., L. Phan, L. W. Schoenfeld, V. Valášková, and A. G. Hinnebusch. 2001. Related eIF3 subunits TIF32 and HCR1 interact with an RNA recoginition motif in PRT1 required for eIF3 integrity and ribosome binding. EMBO J. 20:891-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valášek, L., B. Szamecz, A. G. Hinnebusch, and K. H. Nielsen. 2007. In vivo stabilization of preinitiation complexes by formaldehyde cross-linking. Methods Enzymol. 429:163-183. [DOI] [PubMed] [Google Scholar]
- 56.van der Velden, A. W., and A. A. M. Thomas. 1999. The role of the 5′ untranslated region of an mRNA in translation regulation during development. Int. J. Biochem. Cell Biol. 31:87-106. [DOI] [PubMed] [Google Scholar]
- 57.Verlhac, M.-H., R.-H. Chen, P. Hanachi, J. W. B. Hershey, and R. Derynck. 1997. Identification of partners of TIF34, a component of the yeast eIF3 complex, required for cell proliferation and translation initiation. EMBO J. 16:6812-6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vornlocher, H. P., P. Hanachi, S. Ribeiro, and J. W. B. Hershey. 1999. A 110-kilodalton subunit of translation initiation factor eIF3 and an associated 135-kilodalton protein are encoded by the Saccharomyces cerevisiae TIF32 and TIF31 genes. J. Biol. Chem. 274:16802-16812. [DOI] [PubMed] [Google Scholar]
- 59.Wells, S. E., P. E. Hillner, R. D. Vale, and A. B. Sachs. 1998. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell 2:135-140. [DOI] [PubMed] [Google Scholar]
- 60.Williams, N. P., A. G. Hinnebusch, and T. F. Donahue. 1989. Mutations in the structural genes for eukaryotic initiation factors 2α and 2β of Saccharomyces cerevisiae disrupt translational control of GCN4 mRNA. Proc. Natl. Acad. Sci. U. S. A. 86:7515-7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamamoto, Y., C. R. Singh, A. Marintchev, N. S. Hall, E. M. Hannig, G. Wagner, and K. Asano. 2005. The eukaryotic initiation factor (eIF) 5 HEAT domain mediates multifactor assembly and scanning with distinct interfaces to eIF1, eIF2, eIF3, and eIF4G. Proc. Natl. Acad. Sci. U. S. A. 102:16164-16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou, M., A. M. Sandercock, C. S. Fraser, G. Ridlova, E. Stephens, M. R. Schenauer, T. Yokoi-Fong, D. Barsky, J. A. Leary, J. W. Hershey, J. A. Doudna, and C. V. Robinson. 2008. Mass spectrometry reveals modularity and a complete subunit interaction map of the eukaryotic translation factor eIF3. Proc. Natl. Acad. Sci. U. S. A. 105:18139-18144. [DOI] [PMC free article] [PubMed] [Google Scholar]