

Abstract
The small GTPase Rho regulates cell morphogenesis through remodeling of the actin cytoskeleton. While Rho is overexpressed in many clinical cancers, the role of Rho signaling in oncogenesis remains unknown. mDia1 is a Rho effector producing straight actin filaments. Here we transduced mouse embryonic fibroblasts from mDia1-deficient mice with temperature-sensitive v-Src and examined the involvement and mechanism of the Rho-mDia1 pathway in Src-induced oncogenesis. We showed that in v-Src-transduced mDia1-deficient cells, formation of actin filaments is suppressed, and v-Src in the perinuclear region does not move to focal adhesions upon a temperature shift. Consequently, membrane translocation of v-Src, v-Src-induced morphological transformation, and podosome formation are all suppressed in mDia1-deficient cells with impaired tyrosine phosphorylation. mDia1-deficient cells show reduced transformation in vitro as examined by focus formation and colony formation in soft agar and exhibit suppressed tumorigenesis and invasion when implanted in nude mice in vivo. Given overexpression of c-Src in various cancers, these findings suggest that Rho-mDia1 signaling facilitates malignant transformation and invasion by manipulating the actin cytoskeleton and targeting Src to the cell periphery.
The small GTPase Rho functions as a molecular switch in cell morphogenesis through remodeling of the actin cytoskeleton (3, 14). Rho cycles between the inactive GDP-bound form and the active GTP-bound form. This process is controlled by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) specific to Rho; the former group catalyzes the exchange of GDP to GTP (50), and the latter accelerates the hydrolysis of bound GTP (24). When Rho is activated in fibroblasts, actin stress fibers are formed. Rho proteins are frequently overexpressed in human cancers, such as cancers of the colon and breast and lung and testicular germ cell tumors (34). A positive correlation between the expression level of RhoA and disease progression was also reported in breast cancer and testicular germ cell tumors. RhoC, on the other hand, has been repeatedly identified as a gene positively associated with metastasis (4, 21, 40). The clinical significance of Rho in cancer is further implicated by a discovery that a RhoA GAP named Dlc-1 (deleted liver cancer 1) functions as a tumor suppressor in humans (47). Thus, it was known for some time that heterozygous deletions on chromosome 8p22 are common in human tumors, such as cancers of the breast, prostate, lung (5, 22), and especially liver (15). Recent studies have revealed a strong association of deletion of DLC-1 in this region with clinical cancers, and complementary in vitro experiments showed that DLC-1 functions as a potent tumor suppressor, depletion of which causes RhoA hyperactivation and results in tumorigenesis in harmony with other oncogenes, such as Myc and Ras (47). Importantly, heterozygous deletions in chromosome 8p22 are found to be nearly as common as that of TP53 in clinical cancers, indicating the significant importance of DLC-1 and Rho signaling in clinical tumors (18). Consistent with these findings, there are several reports on the requirement of Rho activity in cell transformation in vitro. For example, coexpression of Raf and dominant active RhoA facilitates focus formation, and expression of dominant-negative RhoA suppresses oncogenic Ras-induced focus formation in NIH 3T3 cells (30). In addition, active forms of Rho GEFs, such as Dbl and Ect2, have potent transforming activities in cultured cells in vitro (31). Thus, there are ample in vitro and clinical data indicating the involvement of Rho signaling in oncogenesis.
Cell transformation often leads to a change in cell morphology. This morphological change associates with a change in the organization of actin filaments. Nontransformed cells often have thick bundled actin fibers known as stress fibers. When transformed by some oncogenes, such as Ras and v-Src, the actin stress fibers disappear and the cells dramatically alter their shape to the round refractile cell body (49). Alternatively, actin dot structures called podosomes are often formed. This remodeling of the cytoskeleton is believed to contribute to several aspects of the transformed phenotype, including adhesion-independent cell growth and increased migration abilities. Such actin remodeling associated with oncogenesis appears at odds with the requirement of Rho signaling in oncogenesis, because Rho activation leads to formation of actin fibers. Thus, there is a paradox of why transformed cells require Rho signaling yet show dissolution of actin cytoskeleton (27).
Among many Rho effectors, two effector molecules, named mDia (44) and ROCK (11), have important roles in actin cytoskeleton remodeling (27). mDia produces straight actin filaments by catalyzing actin nucleation and polymerization, and ROCK activates myosin to cross-link actin filaments for induction of actomyosin bundles and contractility. Further, mDia is potentially linked to Rac activation and membrane ruffle formation through c-Src-induced phosphorylation of focal adhesion proteins, and ROCK antagonizes this mDia action (42). Thus, actin remodeling inside the cell can be determined primarily by the balance between mDia and ROCK activities. Of the two, the involvement of ROCK in tumors has been widely examined by the use of its small molecule inhibitors, such as Y-27632 (26, 43), and the Rho-ROCK pathway has been strongly implicated in cancer migration and tumor metastasis and invasion. On the other hand, the role of ROCK in oncogenesis remains ambiguous. While its requirement in Ras-induced cell transformation was indicated by the use of Y-27632, examination in Ras-transformed cells revealed that the majority of ROCK is sequestered in an inactive pool by sustained extracellular signal regulated-kinase (ERK)-mitogen-activated protein (MAP) kinase activity under active Ras (33), which might be one of the mechanisms for dissolution of stress fibers found in Ras transformants. Thus, how Rho signaling contributes to oncogenesis remains an open question.
Study of Rho effectors other than ROCK has been hampered by the absence of available inhibitors. Recently we generated mDia1 knockout mice (36). Here, we used mouse embryonic fibroblast (MEF) cells derived from these mice and analyzed the involvement of mDia1 and its mechanism of action in v-Src-induced cell transformation and tumorigenesis. v-Src is the oldest widely studied oncogene, yet it remains unknown where in the cell it exerts its oncogenic potential. It was previously reported that temperature-sensitive (ts) v-Src accumulates in the perinuclear region at the restrictive temperature and migrates to the periphery upon a temperature shift in a manner dependent on the actin cytoskeleton and Rho (6, 37). However, the underlying mechanism of this v-Src targeting has not been fully elucidated, and whether this targeting is required for v-Src-induced oncogenesis remains to be shown. Using mDia1-deficient MEF cells, we have addressed these questions. Here we have shown that actin filaments produced by mDia1 are a prerequisite for v-Src targeting, and this v-Src targeting is critical for its role in cell transformation and tumorigenesis. Our results further show that the Rho-mDia1 pathway functions as a link between oncogenesis and invasion.
MATERIALS AND METHODS
Plasmids, antibodies, and reagents.
pEGFP-Src WT, pFL-mDia1-ΔN3, pEGFP-mDia1ΔN3, and pCX vectors (pCX4bsr SV40 and pCX4puro NY72) were described previously (1, 12, 37, 45). Primary antibodies used were monoclonal antibodies to mDia1 (clone 51; BD Transduction Laboratories), Src (EC10; Upstate), cortactin (4F11; Upstate), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (6C5; Ambion), and paxillin (Z035; Invitrogen); and rabbit polyclonal antibodies to pY416 Src (Cell Signaling Technology) and phosphotyrosine (Invitrogen); and rat monoclonal antibody to tubulin (YL1/2; Chemicon). Alexa Fluor 488-, 594-, and 633-conjugated goat antibodies against rabbit, rat, and mouse IgG(H+L), Oregon green-phalloidin, Alexa Fluor 633-phalloidin, and Texas Red X-phalloidin were purchased from Invitrogen. Horseradish peroxidase-conjugated sheep anti-mouse IgG, donkey anti-rabbit IgG, and goat anti-rat IgG antibodies were purchased from GE Healthcare.
Generation of MEF cells.
Mice lacking mDia1 were generated as described previously (36). All experiments involving animals were approved by the Animal Research Committee of Kyoto University Faculty of Medicine and carried out following guidelines in accordance with regulations and laws set by the Japanese government. Littermates of wild-type (WT) and mDia1−/− (KO) mouse embryonic fibroblasts (MEFs) were obtained from embryonic day 15.5 (E15.5) embryos from an mDia1 heterozygous mating. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). For retrovirus production, Plat-E cells (25), provided by T. Kitamura (Department of Hematopoietic Factors, Institute of Medical Science, University of Tokyo) were transfected with either pCX4bsr SV40 or pCX4puro NY72. After 48 h, the supernatant containing retrovirus was collected. To generate stable polyclonal cell populations (WT-SV40 and KO-SV40), MEFs (1 × 105 cells) were plated in 60-mm dishes. On the day of infection, the supernatant containing retrovirus encoding simian virus 40 (SV40) T antigens and a blasticidin S-resistant gene were added to 2 ml of growth medium in each well. After 24 h, the virus-containing medium was aspirated and replaced with fresh growth medium. MEF cells infected with viruses were expanded in medium for 2 days after infection and selected with blasticidin S at a concentration of 10 μg/ml for a week. To generate stable polyclonal cell lines (WT-NY72 and KO-NY72), WT-SV40 and KO-SV40 cells were infected with retrovirus encoding ts-v-Src (NY72) and a puromycin-resistant gene. The cells were selected with 2 mg/ml of puromycin for a week.
Activation of ts-v-src by temperature shift.
ts-v-Src-expressing cells were grown at 40°C (nonpermissive temperature) for 72 h prior to a temperature shift to 37°C (permissive temperature). For immunofluorescence, the cells were grown on fibronectin-coated coverslips (BD Biosciences) and then fixed at various time points with 3.7% formaldehyde in phosphate-buffered saline (PBS) prior to staining. For immunoblotting, the cells were grown on dishes and then lysed with Laemmli buffer at the appropriate time points.
Immunofluorescence.
All procedures were carried out at room temperature. Cells were fixed with 3.7% formaldehyde in PBS for 15 min and then permeabilized with 0.1% Triton X-100 in PBS for 5 min. The cells were then incubated with PBS containing 5% bovine serum albumin (BSA) for 1 h. Primary antibodies were diluted in PBS containing 5% BSA and included antibodies against Src (1:100), mDia1 (1:100), cortactin (1:200), and pY416-Src (1:100). The secondary antibodies used were Alexa Fluor 488- and/or 633- conjugated goat antibodies against rabbit and/or mouse IgG(H+L) (Invitrogen) diluted in PBS containing 5% BSA (1:200). F-actin was stained with Oregon-green phalloidin or Texas Red-X phalloidin (1:200). Immunofluorescent images were acquired using a Zeiss LSM510META confocal microscope with a 63× PlanAPO (1.4) oil immersion objective and a Leica SP5 confocal imaging system (Plan-Apo, 63/1.40 numerical aperture).
Fractionation.
For preparation of the P100 membrane fraction, the cells were lysed in a hypotonic buffer containing 25 mM Tris-HCl, pH 7.5, 5 mM EDTA, 5 mM β-mercaptoethanol, a phosphatase inhibitor cocktail (Roche), and a protease inhibitor mixture (Nacalai). For each sample, the lysate was centrifuged at 10,000 × g for 10 min at 4°C, and the supernatant was collected and centrifuged at 100,000 × g for 60 min at 4°C. The cell pellet (P100 fraction) was collected, dissolved in Laemmli buffer, and then analyzed by immunoblotting.
Proliferation assay.
Cells (1 × 104 cells per dish) were seeded onto 35-mm dishes and allowed to grow in DMEM supplemented with 10% FBS. The cells were trypsinized and counted daily by a hemocytometer.
Video microscopy.
Cell imaging using XTC cells was performed as described previously (10, 46). XTC cells were transfected with constitutively active pFL-mDia1-ΔN3 and pEGFP-Src WT, with either mCherry-lifeact or mPlum-paxillin. At 2 days after the transfection, cells were allowed to spread on a poly-l-lysine (PLL)-coated glass coverslip in 70% Leibovitz's L15 medium (Invitrogen) without serum. After spreading, the medium was changed to 70% Leibovitz's L15 medium supplemented with 10% FBS for 24 h. Cells were placed on the stage of a microscope (BX52 or IX71; Olympus) equipped with either 100-W mercury or 75-W Xenon illumination and with a cooled charge-coupled-device (CCD) camera (MMX1300-YHS, CoolSNAP HQ, or Cascade II:512 [Roper Scientific] or UIC-QE [Molecular Devices]). Time-lapse imaging was performed at 21 to 23°C using the MetaMorph software program (Universal Imaging Corp.). Fluorescent speckle microscopy was performed by observing cells expressing a small amount of enhanced green fluorescent protein (EGFP)-tagged proteins using a PlanApo 100× numerical-aperture 1.40 oil objective (Olympus). A restricted area near the cell edge was illuminated.
Soft agar colony formation assay.
Single-cell suspensions of 3 × 105 cells were plated per 60-mm dish in 3 ml of DMEM supplemented with 10% FBS and 0.36% agar on a layer of 5 ml of the same medium containing 0.7% agar. The plates were fed twice a week with 0.5 ml of DMEM supplemented with 10% FBS. Two weeks after plating, colonies were stained with 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide (MTT) (Kishida Chemicals), and the number of colonies was determined using the Image J software program (National Institutes of Health).
Focus formation assay.
Cells were plated on 60-mm dishes in a subconfluent fashion and cultured in DMEM supplemented with 5% FBS for a week. The appearance of colonies of proliferating cells was visualized by Giemsa staining, and visible colonies were counted.
In situ zymography.
Glass coverslips were coated with Oregon green 488-conjugated gelatin (0.2 mg/ml in PBS), cross-linked for 15 min in 0.5% glutaraldehyde in PBS, and incubated for 3 min with 5 mg/ml NaBH4 in PBS. After incubation in DMEM at 37°C, 7.5 × 104 cells maintained at 40°C were plated on coated coverslips in DMEM containing 10% FBS and then incubated at 37°C for 3 h before being processed for cell staining. Results were quantified by counting cells degrading matrix, as defined by the ability to form at least one degradation patch regardless of its size, and were represented as a percentage of the total (50 cells per treatment in at least three independent experiments) (2).
Matrigel invasion assay.
Transwell invasion chambers (BD BioCoat Matrigel Invasion Chamber) were used. Cells (1.5 × 104 cells per well) suspended in DMEM were added to the upper compartment, and DMEM supplemented with or without 10% FBS was applied to the lower compartment. The number of cells that migrated across the membrane was determined 24 h later.
In vivo tumorigenicity assay.
Cells (3 × 105 per site) in DMEM were injected subcutaneously into the bilateral flank of 6- to 8-week-old female nude athymic mice (BALB/c Slc-nu/nu). Widths and lengths of tumors were measured twice a week. The tumor volume (mm3) was calculated by using the following formula: (length × width2)/2. The mice were sacrificed at day 22, and subcutaneous tumors were removed and fixed with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4) for 2 to 3 days. These tumors were then embedded in paraffin, sectioned, and stained with hematoxylin and eosin.
Statistical analysis.
Data are presented as means ± standard errors (SEM). The comparison of two groups was analyzed using Student's unpaired two-tailed t test or Bonferroni's multiple-comparison test (see Fig. 2B). A P value of <0.05 was considered statistically significant.
RESULTS
v-Src-induced morphological transformation is impaired in mDia1-deficient MEFs.
We recently generated mice deficient in mDia1 (36). Here we prepared mouse embryonic fibroblasts (MEF cells) from wild-type (WT) and mDia1-deficient littermates produced by heterozygous matings and investigated the role of mDia1 in malignant cell transformation and tumorigenesis. WT and mDia1−/− (KO) MEFs were first immortalized with retroviral transfer of SV40 T antigen and the blasticidin S-resistant gene and drug selected to produce WT-SV40 and KO-SV40 cells. The two cell populations were then transduced with retrovirus encoding ts-v-Src, NY72, and the puromycin resistance gene. The tyrosine kinase of ts-v-Src is enzymatically active at 37°C (the permissive temperature) and inactive at 40°C (the restrictive temperature) (23). By puromycin selection, we obtained WT-NY72 and KO-NY72 cell populations (Fig. 1 A). WT-NY72 and KO-NY72 cells exhibited similar levels of expression of v-Src, and the activity of ts-v-Src at the permissive temperature was similar in that for WT-NY72 and KO-NY72 cell populations as examined by staining for pY416-Src (Fig. 1B). No statistically significant difference was observed in the growth of WT-NY72 and KO-NY72 cells and that of WT-SV40 and KO-SV40 cells at either 37°C or 40°C, although the growth of WT-NY72 cells always tended to be higher than that of KO-NY72 cells (Fig. 1C). Notably, when shifted to the permissive temperature for 24 h, WT-NY72 cells exhibited a morphological transformation typical of the Src-induced transformation, with a round, refractile cell body and elongated processes, whereas KO-NY72 cells were resistant to such morphological change and retained their extended shape even at the permissive temperature (Fig. 1D). Both WT-NY72 and KO-NY72 cells exhibited an array of actin bundles at 40°C, although KO-NY72 cells showed few transcytoplasmic actin bundles compared to WT-NY72 cells. This reduction in the actin bundles was also observed in KO-SV40 cells compared to results for WT-SV40 cells (data not shown). However, when maintained at 37°C, WT-NY72 cells lost actin bundles and instead formed dot-like actin accumulation, whereas actin bundles were retained in KO-NY72 cells (Fig. 1E).
FIG. 1.

mDia1 deficiency impairs v-Src-induced morphological transformation. (A) Generation of WT-NY72 and KO-NY72 cell populations. MEFs from WT and mDia1−/− (KO) littermates were immortalized with SV40 T antigen and then infected with retrovirus harboring ts-v-Src, NY72. Populations of cells stably expressing SV40 T antigen and NY72 were isolated by two-step drug selection. (B) Expression and activation of v-Src in WT-SV40, KO-SV40, WT-NY72, and KO-NY72 cells. The cells were maintained at 37°C. Cell lysates were prepared and probed for α-tubulin, mDia1, Src, and pY416-Src. Ratios of the densities of pSrc to Src bands are shown below the image. (C) Proliferation of WT-SV40, KO-SV40, WT-NY72, and KO-NY72 cells. Cells were seeded and cultured at 37°C in a 35-mm dish in DMEM containing 10% FBS and harvested at indicated times for cell counting. Means ± SEM of data from three independent experiments is shown. (D) v-Src-induced morphological changes in WT-NY72 and KO-NY72 cells. WT-NY72 and KO-NY72 cells were grown at either 40°C or 37°C and observed under phase-contrast microscopy. (E) Actin cytoskeleton in WT-NY72 and KO-NY72 cells. WT-NY72 and KO-NY72 cells were grown at either 40°C or 37°C, fixed, and stained with Oregon green-phalloidin. Bars in panels D and E, 50 μm.
Src translocation is impaired in mDia1-deficient MEFs.
The above results showed that the loss of mDia1 impaired Src-induced morphological transformation and cytoskeletal reorganization despite activation of its enzymatic activity at the permissive temperature. To explore the mechanism of this defect, we compared the localization of v-Src and the actin cytoskeleton in WT-NY72 and KO-NY72 cells at various time points following activation of ts-v-Src. ts-v-Src was reported to accumulate in vesicles in the perinuclear region at the restrictive temperature and move to the periphery upon the shift to the permissive temperature (6). During translocation, v-Src associates with actin stress fibers and gradually accumulates in focal adhesions. As reported, we observed the accumulation of v-Src in the perinuclear region at the restrictive temperature in both WT-NY72 and KO-NY72 cells (Fig. 2A). When shifted to 37°C, v-Src-containing vesicular structures began to disperse to the cytoplasm in WT-NY72 cells, and the v-Src signals accumulated in focal adhesions 4 h later (Fig. 2A; see also the figure in the supplemental material). At 24 h after the temperature shift, the v-Src signals distributed diffusely over the cell body. On the contrary, v-Src in KO-NY72 cells continued to stay in vesicles in the perinuclear region despite its activation even 24 h after the temperature shift. Coincidentally, upon v-Src activation, actin stress fibers were induced and v-Src vesicles were found associated with these stress fibers in WT-NY72 cells, whereas the induction of actin fibers was suppressed in KO-NY72 cells. At 24 h, WT-NY72 cells began to take a round shape with encircled actin bundles, while KO-NY72 cells remained extended. Given mDia1 as an actin-nucleating factor, these results indicated that v-Src activation somehow promotes mDia activation and mDia1-induced actin fibers facilitate v-Src translocation. Consistently, the actin content in the cell as measured by the fluorescence intensity of Texas Red phalloidin staining increased time dependently to 4 h and then decreased in WT-NY72 cells, whereas it remained almost at the same level during this period in KO-NY72 cells (Fig. 2B). The actin content decreased further as the Src-induced transformation progressed, as shown in Fig. 1E. We wondered whether this mDia1-mediated v-Src translocation is related to its membrane targeting. To this end, we prepared the 100,000 × g membrane fraction (P100) of WT-NY72 and KO-NY72 cells at various times after the temperature shift. The v-Src protein already was accumulating in the P100 fraction 1 h after temperature shift in WT-NY72 cells, whereas this accumulation was not found in the P100 fraction of KO-NY72 cells even at 24 h (Fig. 2C).
FIG. 2.

mDia1 deficiency impairs formation of actin fibers and peripheral targeting of v-Src upon temperature shift. (A) Time course of formation of actin fibers and v-Src translocation after temperature shift. WT-NY72 and KO-NY72 cells grown at the nonpermissive temperature were shifted to the permissive temperature and maintained for 0, 1, 4, and 24 h. The cells were fixed and stained with an antibody against Src and Texas Red X-phalloidin. A merged image of the two populations of cells 4 h after the temperature shift is shown in the figure in the supplemental material. Bar, 50 μm. (B) Quantification of F-actin intensity. WT-NY72 and KO-NY72 cells were cultured for 0, 1, 4, and 24 h after the temperature shift and stained for F-actin with Texas Red X-phalloidin. The fluorescence density, i.e., the intensity divided by cell area, of each cell was quantified and is shown (n = 20 each). (C) Accumulation of v-Src in the P100 fraction. WT-NY72 and KO-NY72 cells were shifted to the permissive temperature and harvested at indicated times. The P100 fraction was prepared from each population and subjected to Western blotting for Src. v-Src signal was detected 5 kDa below the molecular mass of c-Src. The c-Src signals were not visible under the present blotting conditions. The density of each band is shown below the blot.
To gain further insight into how mDia1 works in v-Src targeting to focal adhesions, we transfected Xenopus XTC fibroblasts with constitutively active mDia1 that lacks its N-terminal autoinhibitory domain (ΔN3) and Src-GFP, with either mCherry-lifeact or mPlum-paxillin. Forty-eight hours after transfection, live-cell imaging was carried out. Untransfected XTC fibroblasts were devoid of actin stress fibers, and there was no directed movement observed for Src-GFP. Introduction of mDia1-ΔN3 caused numerous actin bundles which run in parallel, as previously reported (12). Src-GFP was seen as a vesicular or tubular structure and was found to move along these parallel actin fibers (Fig. 3A; see also Movie S1 in the supplemental material). They sometimes moved from one site to another site by extending the tubular structure to another vesicle at a distant site and fusing with it (Fig. 3B; see also Movie S2). Speckle microscopy of GFP-mDia1ΔN3 revealed that they exhibit fast processive movement, as we reported previously (10). On the other hand, speckle microscopy of Src-GFP revealed no such processive movement within the time frame similar to that for GFP-mDia1ΔN3 (data not shown), suggesting that mDia1ΔN3 and Src do not move as a complex. Immunofluorescence of paxillin and live cell imaging with mPlum-paxillin showed that Src-GFP does not localize in large matured focal adhesions often seen near the cell periphery, but Src-GFP sometimes colocalizes with small paxillin signals neighboring the above matured adhesions (Fig. 3C).
FIG. 3.

Time-lapse video microscopy of Src-GFP in XTC cells transfected with mDia1ΔN3. (A) Coimaging of mCherry-lifeact and Src-GFP. XTC cells expressing mDia1ΔN3 were identified as cells showing parallel actin fibers, and Src-GFP and mCherry-lifeact signals were monitored. A still image is shown. Bar, 5 μm. (B) Tube extension and fusion of Src-GFP signals. Src signals boxed in panel A were video monitored at 400 ms per frame at the interval of 5 s. Arrows show Src-GFP moving along actin stress fibers. Arrowheads show Src in vesicular structures. (C) Colocalization of Src-GFP and signals for paxillin. XTC cells expressing Src-GFP and mDia1ΔN3 were fixed and stained for paxillin (red). Arrowheads indicate matured focal adhesions, and arrows indicate colocalization of paxillin and Src. Bar, 20 μm.
Impaired v-Src-mediated tyrosine phosphorylation in KO-NY72 cells.
The above results demonstrate that v-Src is not properly translocated in KO-NY72 cells. To examine the consequences of this impairment of v-Src in translocation, we analyzed tyrosine phosphorylation at different time points after the temperature shift (Fig. 4). Both WT-NY72 cells and KO-NY72 cells exhibited elevated tyrosine phosphorylation of various proteins, as demonstrated by immunoblotting of total cell lysates with an antiphosphotyrosine antibody (Fig. 4). Although mDia1 deficiency did not alter the overall pattern of tyrosine-phosphorylated proteins in v-Src-transformed cells, comparison of the signal intensity in WT-NY72 cells and KO-NY72 cells showed a visible decrease in the phosphotyrosine signal of several proteins, especially those in the relatively high molecular mass range (100 to 250 kDa), in KO-NY72 cells. These results suggest that impairment of v-Src translocation leads to downregulation of downstream signaling pathways.
FIG. 4.
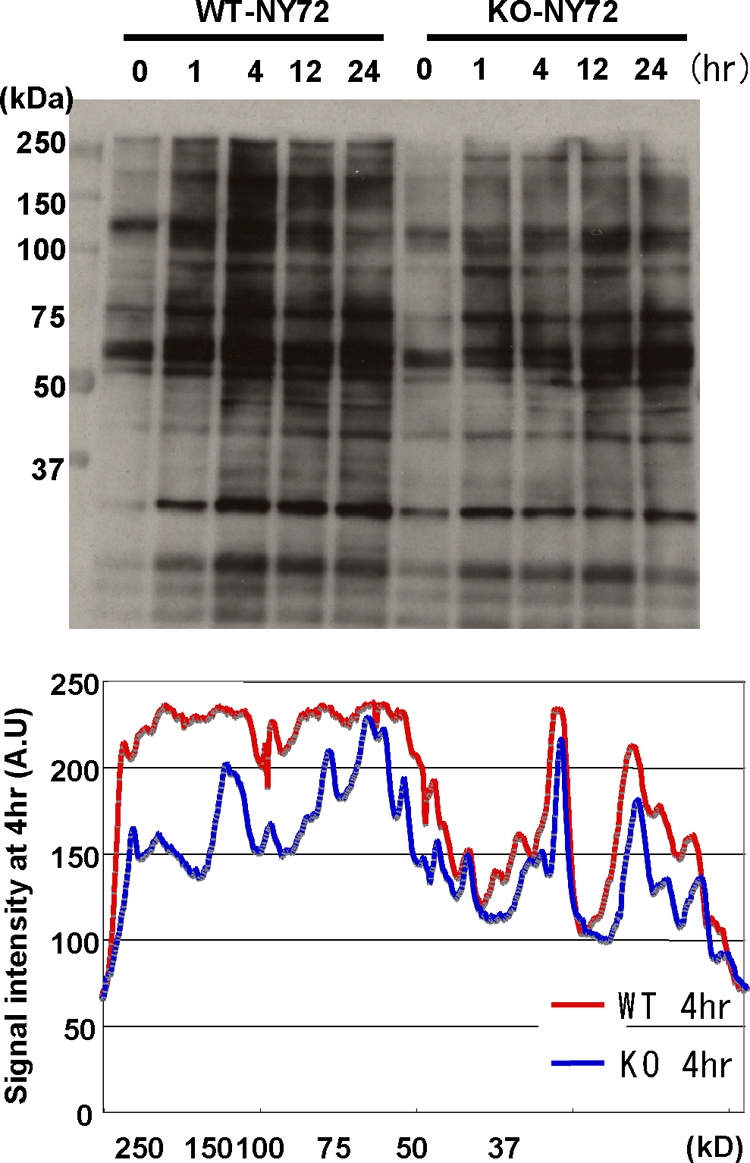
Impaired v-Src-mediated tyrosine phosphorylation in KO-NY72 cells. WT-NY72 and KO-NY72 cells were shifted to the permissive temperature and collected at the indicated times. Cell lysates were prepared and subjected to immunoblotting with an antiphosphotyrosine antibody. The intensity of the phosphotyrosine signal in the lysates of WT-NY72 and KO-NY72 cells at 4 h after the temperature shift was determined by densitometry and is shown below.
Impaired transformation and invasion in vitro of KO-NY72 cells.
The above findings demonstrate that in the absence of mDia1, v-Src does not translocate to the periphery and cannot mediate some of its signaling. We therefore next tested whether these failures result in a defect in cell transformation. We first subjected the two populations to colony formation in soft agar (Fig. 5A ). WT-NY72 cells formed a number of visible colonies in 2 weeks. On the other hand, colonies KO-NY72 cells formed were significantly less in number and much smaller in size than those of WT-NY72 cells. No colony formation was found with either WT-SV40 cells or KO-SV40 cells. We then examined focus formation of the two populations. Both WT-NY72 cells and KO-NY72 cells did not show focus formation at the restrictive temperature. While both showed focus formation at 37°C, the number of foci in KO-NY72 cells was significantly smaller than that in WT-NY72 cells (Fig. 5B).
FIG. 5.
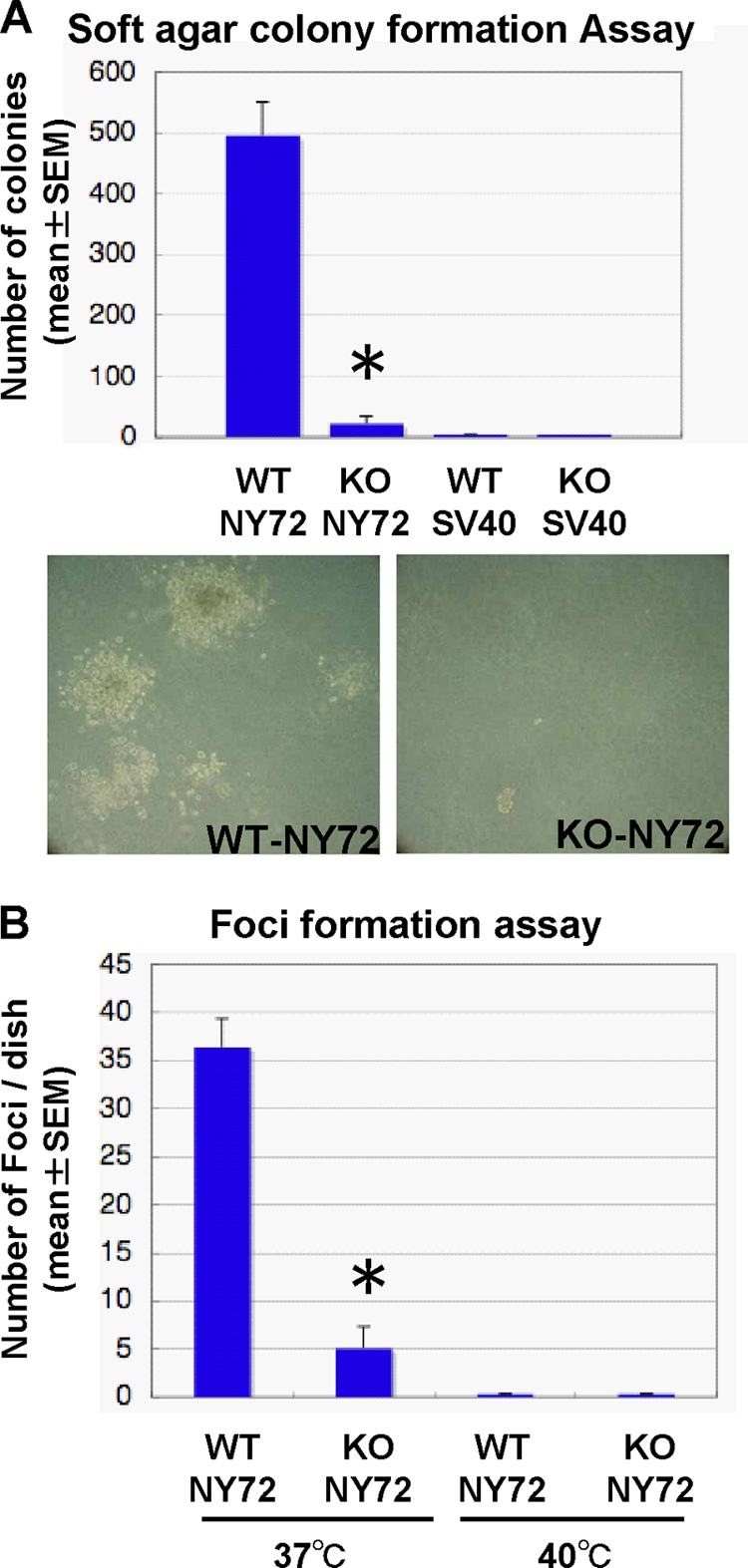
Impaired transformation in vitro of KO-NY72 cells. (A) Soft agar colony formation. WT-SV40, KO-SV40, WT-NY72, and KO-NY72 cells were cultured in 0.36% agarose for 2 weeks until colonies of WT-NY72 cells became visible. Colonies were stained with MTT, and the number of colonies in each dish was determined. The experiment was performed in triplicate independently three times. Shown are representative photomicrographs of WT-NY72 and KO-NY72 cells at the time of quantification. Results are means ± SEM of data from three experiments. (B) Focus formation. WT-NY72 and KO-NY72 cells were plated at subconfluence in 60-mm dishes and cultured for 1 week at 37°C and 40°C. Foci were stained with Giemsa solution, and the number was determined. Means ± SEM of three independent experiments are shown.
Expression of Src is also associated with an increase in the ability to invade through the extracellular matrix (ECM) (47). Podosomes are v-Src-induced actin-rich structures on the ventral surface, where proteolysis of matrix proteins occurs (20). We noted that dot-like actin-rich structures were formed in WT-NY72 cells, but few were formed in KO-NY72 cells maintained at the permissive temperature (Fig. 1E). These structures were costained with an antibody against cortactin, a marker of podosome, and an antibody against pSrc that is known to localize in podosomes (Fig. 6A ). These results verified that the actin-dot structures formed in WT-NY72 cells were indeed podosomes and the podosome formation was impaired in KO-NY72 cells. We examined a process in which podosomes are formed in WT-NY72 cells by monitoring mDia1 localization. At the restrictive temperature, most mDia1 localized in the cytoplasm, but a part was found in the extended edge of the cells, as previously reported (44), where it overlapped with a part of the active Src signal (data not shown). With a shift to the permissive temperature for 24 h, mDia1 accumulates in podosomes, where it colocalizes with active Src (Fig. 6A), suggesting that mDia1-mediated Src recruitment is important for podosome formation. To examine if these are functional podosomes, we placed WT-NY72 cells and KO-NY72 cells on Oregon green gelatin at 37°C for 3 h. This in situ zymography revealed extensive degradation of gelatin by WT-NY72 cells but not by KO-NY72 cells; the percentage of cells degrading the gelatin matrix was significantly higher in the WT-NY72 cell population than in the KO-NY72 cell population (Fig. 6B). Then, to examine whether the mDia1 deficiency affects cell invasion into ECM, we performed the Matrigel invasion assay. Cells were applied on the upper well, with or without serum in the lower well. WT-NY72 cells exhibited some extent of invasion without serum, but their invasion was significantly enhanced by the addition of serum. On the other hand, KO-NY72 cells showed little invasion without serum and significantly less invasion than WT-NY72 cells in the presence of serum (Fig. 6C). Independent culture confirmed that cell viability was not affected in either population by culturing on the Matrigel (data no shown).
FIG. 6.

Impaired invasion in vitro of KO-NY72 cells. (A) Colocalization of mDia1 and pSrc in podosomes. WT-NY72 cells were shifted to the permissive temperature for 24 h, fixed, and stained either for pSrc, F-actin, and mDia1 or for cortactin and F-actin. Bar, 50 μm on the left and 20 μm on the right. (B) In situ zymography of WT-NY72 and KO-NY72 cells. WT-NY72 and KO-NY72 cells maintained at 40°C were plated on Oregon green 488-conjugated gelatin cross-linked glass coverslips and cultured at 37°C for 3 h. Cells were fixed and stained for actin and cortactin, and cells degrading fluorescent gelatin were counted. (C) Invasion in Matrigel. WT-NY72 and KO-NY72 cells were applied to the upper well of the Matrigel invasion chamber with or without serum in the lower well. After 24 h, Matrigel was removed and cells that passed through Matrigel and penetrated the membrane were stained and their number determined. The numbers of invading cells are shown as means ± SEM from three independent assays. Typical photomicrographs are shown on the right.
mDia1-deficient cells are impaired in tumorigenesis and invasion in vivo.
We next addressed the question of whether mDia1 deficiency affects tumor formation in vivo. WT-NY72 cells and KO-NY72 cells were implanted subcutaneously into nude mice, and their growth was monitored. While palpable tumors developed in all sites injected with WT-NY72 cells (6 out of 6) within 2 weeks, KO-NY72 cells exhibited a reduced incidence of palpable tumor formation (2 out of 6) (Fig. 7A ). Determination of tumor size showed that tumors from WT-NY72 cells developed to about 700 mm3 in volume on average on day 18, while the size of tumors from KO-NY72 cells remained less than 10 mm3 on average (Fig. 7B). Three weeks after the cell injection, tumors were excised and their histology was analyzed (Fig. 7C). A tumor formed from WT-NY72 cells expanded to both sides of the muscle layer, suggesting that it invaded massively. On the other hand, KO-NY72 cells yielded only one tumor of significant size, which was small compared to tumors from the WT cells and showed no evidence of invasion.
FIG. 7.

mDia1-deficient cells are impaired in tumorigenesis and invasion in vivo. (A) Tumor formation in nude mice. Nude mice underwent subcutaneous injection of either WT-NY72 cells or KO-NY72 cells (3 × 105 per site) in the flank, three mice per group and two sites per mouse. Nineteen days after inoculation, tumor formation at the injected sites was photographed. Inoculation of KO-NY72 cells developed no or very small tumors compared to those from WT-NY72 cells (arrowheads). (B) Time course of tumor development. WT-NY72 and KO-NY72 cells were injected into nude mice as described above, and the tumor volumes were measured as described in Materials and Methods. Each point represents a mean ± SEM (n = 6). (C) Histological analysis. Tumors from WT-NY72 and KO-NY72 cells were taken at day 22 after inoculation, sectioned, and stained with hematoxylin and eosin. Photomicrographs of a representative tumor from WT-NY72 cells and a sole tumor formed from six injections of KO-NY72 cells are shown. Boxed areas are magnified below. Note that the tumor from WT-NY72 cells expands to both sides of the muscle layer (arrowheads). Bar, 5 mm.
DISCUSSION
In the present study, we have revealed that the Rho-mDia1 signaling facilitates translocation of v-Src to the periphery by way of producing specific actin cytoskeleton and that this translocation is essential in v-Src-induced oncogenesis. Our findings have addressed several long-standing questions on the oncogenic mechanism of Src, namely, the identity of a molecule responsible for Src translocation, the cellular location of Src mediating oncogenesis, a link between oncogenesis and invasion, and the clinical significance of Src activation. Our findings have also addressed questions on Rho and the actin cytoskeleton in oncogenesis, that is, the effector mechanism and the paradox of Rho signaling in oncogenesis.
mDia1-mediated Src translocation.
Here we have found that KO-NY72 cells are defective in actin stress fiber formation upon temperature shift and are impaired in v-Src translocation from the perinuclear region to the cell periphery. Previously it was reported that Src translocation to the periphery requires an intact actin cytoskeleton and Rho signaling (6, 38). However, the identity of an effector mediating Src translocation and its mechanism of action have been elusive. Our findings demonstrate that mDia1 is such an effector and mediates Src translocation by producing actin bundles upon temperature shift. So how does Src move to the periphery? The SH3 domain of c-Src is reported to be essential for this translocation, while the myristylation and kinase activity are dispensable (7, 8). This finding together with the finding of Tominaga et al. (41) that Src and mDia1 bind directly by their SH3 and FH1 domains, respectively, suggest a possibility that mDia1 and Src move together by processive actin polymerization by mDia1. Here we have tested this hypothesis by speckle imaging of GFP-mDia1ΔN3 in the presence of c-Src and of Src-GFP in the presence of mDia1ΔN3 in XTC cells. We have found that while GFP-mDia1ΔN3 exhibited fast processive movement as reported previously (10), signals of Src-GFP did not show such processive movement but associated with vesicular structures that move slower than mDia1 with frequent budding and fusion along actin filaments and overlap with some signals of paxillin. While further work is required to determine the precise interaction of F-actin fibers and Src vesicles, our results thus have identified mDia1 as a key molecule that is essential for Src vesicle targeting to the periphery. Recently Shi et al. used neutrophils from mDia1−/− mice and showed that mDia1 is also required for translocation of hematopoietic cell kinase (HCK), a Src family kinase in hematopoietic cells, to the leading-edge membrane and subsequent tyrosine phosphorylation of WASP (39). Thus, the regulation by mDia1 appears to be a general mechanism for membrane translocation of the Src family of kinases.
Paradox of Rho signaling and the actin cytoskeleton in oncogenesis.
There has been an issue for some time that can be called the Rho paradox in cell transformation, that is, while Rho is required for oncogenesis by v-Src and oncogenic Ras, the actin filaments that are induced by Rho activation usually disappear in transformed cells (27, 30). Sahai et al. reported that while pharmacological intervention suggested implication of ROCK in Ras-mediated transformation (32), the majority of ROCK is sequestered in an inactive pool, and they suggested that this sequestration might be one of the mechanisms for dissolution of stress fibers found in Ras transformants (33). However, the paradox of how the requirement for transformation and the downregulation of stress fibers could occur in the same context remains to be solved. Here we have revealed that mDia1 is required for actin filament formation, which is used for v-Src targeting to the periphery as discussed above, and that these actin fibers then undergo dissolution when targeted v-Src exerts its action on transformation. Thus, the Rho paradox could be explained by a phase-dependent use of the Rho-mediated actin cytoskeleton and its subsequent inhibition by a feedback mechanism. A remaining question is whether the above Rho-mediated mechanism is required only for initiation of transformation or functions in a more subtle way in transformed cells to maintain transformation. This should be clarified by future studies.
mDia1-mediated Src translocation is essential in oncogenesis.
Here we have found that KO-NY72 cells that are defective in Src translocation are impaired in both in vitro transformation, such as colony formation in soft agar and focus formation, and in vivo tumor formation in the xenograft transplantation model, suggesting that mDia1-mediated translocation of v-Src first to focal adhesions and then to the cell membrane is essential in oncogenic transformation. Src exhibits a variety of discrete subcellular distribution, including plasma membrane, adhesion plaques, cell-cell contact, and perinuclear membranes (16, 19, 28, 29). Our findings are consistent with the study by Hamaguchi and Hanafusa, who used various Src mutants and found correlation between cytoskeletal association and transforming activity (9), and the study by Liebl and Martin, who prepared chimera molecules of v-Src and motifs targeting to a specific subcellular site and found that v-Src targeted to adhesion plaques can induce a transformation phenotype (19). Indeed, v-Src-induced tyrosine phosphorylation of proteins was attenuated in KO-NY72 cells, particularly in proteins of molecular masses above 100 kDa. By analyzing tyrosine phosphorylation of individual proteins and comparing gene expression profiles of WT and KO cells, we have found impaired tyrosine phosphorylation of proteins such as STAT3, CAS, and p190 Rho GAP, as well as changes in gene expression in the KO cells (data not shown). We are in the process of analyzing which events are crucial for v-Src-induced cell transformation and tumorigenesis.
Rho-mDia1 functions as a link between oncogenesis and invasion.
Here we have found that KO-NY72 cells show impaired migration in the Matrigel invasion assay, suggesting that mDia1 is required for invasion. Consistently, WT-NY72 cells exhibited an invasive phenotype when transplanted into nude mice. The current model of tumor invasion proposes two interchangeable modes of movements: one is the proteolysis-guided mesenchymal movement, and the other is the actomyosin-driven amoeboid movement (35). While the Rho-ROCK pathway is critical in the latter, Rho signaling is also implicated in the former. In the former process, matrix metalloproteases (MMPs) are enriched in a specialized actin-rich structure called podosomes and actively degrade ECM fibers to make a path for tumor cells to invade. While the Cdc42-Arp2/3-N-WASP pathway regulates actin filament assembly of podosomes (48), Rho signaling has also been indicated to be involved in podosome induction. For example, Berdeaux et al. (2) reported that an active GTP-bound form of Rho accumulates in the podosome and that inactivation of Rho in v-Src-transformed cells suppresses podosome formation and inhibits Src-induced proteolytic degradation of ECM proteins. Notably, the podosome is resistant to treatment with Y-27632, which led those authors to conceive that a Rho effector(s) other than ROCK is involved in this process. Our results of impaired podosome formation in KO-NY72 cells and colocalization of mDia1 and Src in podosomes in wild-type cells suggest that mDia1 is such an effector and that the mDia1-mediated recruitment of Src to the membrane is linked to podosome induction. In addition to what we have found here, Kitzing et al. (17) found that mDia1 amplifies Rho-ROCK activation in a positive feedback loop via a Rho GEF, LARG, and also functions in amoeboid movement of MDA-MB-435 human cancer cells. These findings together have revealed that the same molecule, mDia1, functions as a link between transformation and invasion.
Clinical significance.
Rho is overexpressed in many clinical cancers (34), and its implication in human oncogenesis is strongly suggested by a discovery that DlC-1, one of the Rho GAPs, functions as a tumor suppressor in humans (47). Here we have demonstrated a critical role of the Rho-mDia1 pathway in v-Src-induced tumorigenesis. While v-Src itself is not a cause of human cancers, activation of Src is implicated in many human clinical cancers, including lung, skin, colon, breast, ovarian, endometrial, and head and neck malignancies, suggesting that c-Src facilitates oncogenesis by collaborating with other signaling pathways (13). Our results indicate that Rho signaling is one such pathway, functioning together with Src for oncogenesis. The next question we have to address is whether the Rho-mDia1 pathway has any role in oncogenesis induced by oncogenes other than the v-Src gene, particularly that of oncogenic Ras, because Ras-induced oncogenesis depends at least in part on Rho signaling. Using mDia1-deficient mice for skin oncogenesis and using MEF cells derived thereof for in vitro transformation, we have found that mDia1 is essential in Ras-mediated oncogenesis in vivo and in vitro (M. Tanji, T. Ishizaki, and S. Narumiya, unpublished results). Whether the mechanism we found in the present study operates in Ras-mediated oncogenesis or whether another novel mechanism operates there is a focus of our current study.
Supplementary Material
Acknowledgments
We thank the late H. Hanafusa for encouragement, N. Watanabe for XTC cell imaging, J. Monypenny, N. Yamana, S. Sakamoto, and S. Watanabe for advice on experiments, A. Mizutani and T. Fujiwara for animal care, and T. Arai and K. Nonomura for assistance.
This work was supported in part by a Grant-in-Aid for Specially-Promoted Research (no. 18002015) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Published ahead of print on 2 August 2010.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Akagi, T., K. Sasai, and H. Hanafusa. 2003. Refractory nature of normal human diploid fibroblasts with respect to oncogene-mediated transformation. Proc. Natl. Acad. Sci. U. S. A. 11:13567-13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berdeaux, R. L., B. Diaz, L. Kim, and G. S. Martin. 2004. Active Rho is localized to podosomes induced by oncogenic Src and is required for their assembly and function. J. Cell Biol. 166:317-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burridge, K., and K. Wennerberg. 2004. Rho and Rac take center stage. Cell 116:167-179. [DOI] [PubMed] [Google Scholar]
- 4.Clark, E. A., T. R. Golub, E. S. Lander, and R. O. Hynes. 2000. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 406:532-535. [DOI] [PubMed] [Google Scholar]
- 5.Durkin, M. E., B. Z. Yuan, X. Zhou, D. B. Zimonjic, D. R. Lowy, S. S. Thorgeirsson, and N. C. Popescu. 2007. DLC-1: a Rho GTPase-activating protein and tumour suppressor. J. Cell Mol. Med. 11:1185-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fincham, V. J., M. Unlu, V. G. Brunton, J. D. Pitts, J. A. Wyke, and M. C. Frame. 1996. Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J. Cell Biol. 135:1551-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fincham, V. J., and M. C. Frame. 1998. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 17:81-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fincham, V. J., V. G. Brunton, and M. C. Frame. 2000. The SH3 domain directs acto-myosin-dependent targeting of v-Src to focal adhesions via phosphatidylinositol 3-kinase. Mol. Cell. Biol. 20:6518-6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamaguchi, M., and H. Hanafusa. 1987. Association of p60src with Triton X-100-resistant cellular structure correlates with morphological transformation. Proc. Natl. Acad. Sci. U. S. A. 84:2312-2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higashida, C., T. Miyoshi, A. Fujita, F. Oceguera-Yanez, J. Monypenny, Y. Andou, S. Narumiya, and N. Watanabe. 2004. Actin polymerization-driven molecular movement of mDia1 in living cells. Science 303:2007-2010. [DOI] [PubMed] [Google Scholar]
- 11.Ishizaki, T., M. Maekawa, K. Fujisawa, K. Okawa, A. Iwamatsu, A. Fujita, N. Watanabe, Y. Saito, A. Kakizuka, N. Morii, and S. Narumiya. 1996. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 15:1885-1893. [PMC free article] [PubMed] [Google Scholar]
- 12.Ishizaki, T., Y. Morishima, M. Okamoto, T. Furuyashiki, T. Kato, and S. Narumiya. 2001. Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat. Cell Biol. 3:8-14. [DOI] [PubMed] [Google Scholar]
- 13.Ishizawar, R., and S. J. Parsons. 2004. c-Src and cooperating partners in human cancer. Cancer Cell 6:209-214. [DOI] [PubMed] [Google Scholar]
- 14.Jaffe, A. B., and A. Hall. 2005. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21:247-269. [DOI] [PubMed] [Google Scholar]
- 15.Jou, Y. S., C. S. Lee, Y. H. Chang, C. F. Hsiao, C. F. Chen, C. C. Chao, L. S. Wu, S. H. Yeh, D. S. Chen, and P. J. Chen. 2004. Clustering of minimal deleted regions reveals distinct genetic pathways of human hepatocellular carcinoma. Cancer Res. 64:3030-3036. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan, K. B., J. R. Swedlow, H. E. Varmus, and D. O. Morgan. 1992. Association of p60c-src with endosomal membranes in mammalian fibroblasts. J. Cell Biol. 118:321-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitzing, T. M., A. S. Sahadevan, D. T. Brandt, H. Knieling, S. Hannemann, O. T. Fackler, J. Grosshans, and R. Grosse. 2007. Positive feedback between Dia1, LARG, and RhoA regulates cell morphology and invasion. Genes Dev. 21:1478-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lahoz, A., and A. Hall. 2008. DLC1: a significant GAP in the cancer genome. Genes Dev. 22:1724-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liebl, E. C., and G. S. Martin. 1992. Intracellular targeting of pp60src expression: localization of v-src to adhesion plaques is sufficient to transform chicken embryo fibroblasts. Oncogene 7:2417-2428. [PubMed] [Google Scholar]
- 20.Linder, S., and M. Aepfelbacher. 2003. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 13:376-385. [DOI] [PubMed] [Google Scholar]
- 21.Ma, L., J. Teruya-Feldstein, and R. A. Weinberg. 2007. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 449:682-688. [DOI] [PubMed] [Google Scholar]
- 22.Matsuyama, H., Y. Pan, K. Oba, S. Yoshihiro, K. Matsuda, L. Hägarth, D. Kudren, K. Naito, U. S. Bergerheim, and P. Ekman. 2001. Deletions on chromosome 8p22 may predict disease progression as well as pathological staging in prostate cancer. Clin. Cancer Res. 7:3139-3143. [PubMed] [Google Scholar]
- 23.Mayer, B. J., R. Jove, J. F. Krane, F. Poirier, G. Calothy, and H. Hanafusa. 1986. Genetic lesions involved in temperature sensitivity of the src gene products of four Rous sarcoma virus mutants. J. Virol. 60:858-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon, S. Y., and Y. Zheng. 2003. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 13:13-22. [DOI] [PubMed] [Google Scholar]
- 25.Morita, S., T. Kojima, and T. Kitamura. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063-1066. [DOI] [PubMed] [Google Scholar]
- 26.Narumiya, S., T. Ishizaki, and M. Uehata. 2000. Use and properties of ROCK-specific inhibitor. Methods Enzymol. 325:273-284. [DOI] [PubMed] [Google Scholar]
- 27.Narumiya, S., M. Tanji, and T. Ishizaki. 2009. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28:65-76. [DOI] [PubMed] [Google Scholar]
- 28.Nigg, E. A., B. M. Sefton, T. Hunter, G. Walter, and S. J. Singer. 1982. Immunofluorescent localization of the transforming protein of Rous sarcoma virus with antibodies against a synthetic src peptide. Proc. Natl. Acad. Sci. U. S. A. 79:5322-5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parsons, J. T. 2003. Focal adhesion kinase: the first ten years. J. Cell Sci. 116:1409-1416. [DOI] [PubMed] [Google Scholar]
- 30.Qiu, R. G., J. Chen, F. McCormick, and M. Symons. 1995. A role for Rho in Ras transformation. Proc. Natl. Acad. Sci. U. S. A. 92:11781-11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rossman, K. L., C. J. Der, and J. Sondek. 2005. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6:167-180. [DOI] [PubMed] [Google Scholar]
- 32.Sahai, E., T. Ishizaki, S. Narumiya, and R. Treisman. 1999. Transformation mediated by RhoA requires activity of ROCK kinases. Curr. Biol. 9:136-145. [DOI] [PubMed] [Google Scholar]
- 33.Sahai, E., M. F. Olson, and C. J. Marshall. 2001. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 20:755-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahai, E., and C. J. Marshall. 2002. RHO-GTPases and cancer. Nat. Rev. Cancer 2:133-142. [DOI] [PubMed] [Google Scholar]
- 35.Sahai, E., and C. J. Marshall. 2003. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5:711-719. [DOI] [PubMed] [Google Scholar]
- 36.Sakata, D., H. Taniguchi, S. Yasuda, A. Adachi-Morishima, Y. Hamazaki, R. Nakayama, T. Miki, N. Minato, and S. Narumiya. 2007. Impaired T lymphocyte trafficking in mice deficient in an actin-nucleating protein, mDia1. J. Exp. Med. 204:2031-2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandilands, E., C. Cans, V. J. Fincham, V. G. Brunton, H. Mellor, G. C. Prendergast, J. C. Norman, G. Superti-Furga, and M. C. Frame. 2004. RhoB and actin polymerization coordinate Src activation with endosome-mediated delivery to the membrane. Dev. Cell 7:855-869. [DOI] [PubMed] [Google Scholar]
- 38.Sandilands, E., and M. C. Frame. 2008. Endosomal trafficking of Src tyrosine kinase. Trends Cell Biol. 18:322-329. [DOI] [PubMed] [Google Scholar]
- 39.Shi, Y., B. Dong, H. Miliotis, J. Liu, A. S. Alberts, J. Zhang, and K. A. Siminovitch. 2009. Src kinase Hck association with the WASp and mDia1 cytoskeletal regulators promotes chemoattractant-induced Hck membrane targeting and activation in neutrophils. Biochem. Cell Biol. 87:207-216. [DOI] [PubMed] [Google Scholar]
- 40.Suwa, H., G. Ohshio, T. Imamura, G. Watanabe, S. Arii, M. Imamura, S. Narumiya, H. Hiai, and M. Fukumoto. 1998. Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br. J. Cancer 77:147-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tominaga, T., E. Sahai, P. Chardin, F. McCormick, S. A. Courtneidge, and A. S. Alberts. 2000. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell 5:13-25. [DOI] [PubMed] [Google Scholar]
- 42.Tsuji, T., T. Ishizaki, M. Okamoto, C. Higashida, K. Kimura, T. Furuyashiki, Y. Arakawa, R. B. Birge, T. Nakamoto, H. Hirai, and S. Narumiya. 2002. ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. J. Cell Biol. 157:819-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uehata, M., T. Ishizaki, H. Satoh, T. Ono, T. Kawahara, T. Morishita, H. Tamakawa, K. Yamagami, J. Inui, M. Maekawa, and S. Narumiya. 1997. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389:990-994. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe, N., P. Madaule, T. Reid, T. Ishizaki, G. Watanabe, A. Kakizuka, Y. Saito, K. Nakao, B. M. Jockusch, and S. Narumiya. 1997. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 16:3044-3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe, N., T. Kato, A. Fujita, T. Ishizaki, and S. Narumiya. 1999. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1:136-143. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe, N., and T. J. Mitchison. 2002. Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science 295:1083-1086. [DOI] [PubMed] [Google Scholar]
- 47.Xue, W., A. Krasnitz, R. Lucito, R. Sordella, L. Vanaelst, C. Cordon-Cardo, S. Singer, F. Kuehnel, M. Wigler, S. Powers, L. Zender, and S. W. Lowe. 2008. DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 22:1439-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamaguchi, H., M. Lorenz, S. Kempiak, C. Sarmiento, S. Coniglio, M. Symons, J. Segall, R. Eddy, H. Miki, T. Takenawa, and J. Condeelis. 2005. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 168:441-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeatman, T. J. 2004. A renaissance for SRC. Nat. Rev. Cancer 4:470-480. [DOI] [PubMed] [Google Scholar]
- 50.Zheng, Y. 2001. Dbl family guanine nucleotide exchange factors. Trends Biochem. Sci. 26:724-732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.