

Abstract
Sporadic mutations in the hMeCP2 gene, coding for a protein that preferentially binds symmetrically methylated CpGs, result in the severe neurological disorder Rett syndrome (RTT). In the present work, employing a wide range of experimental approaches, we shed new light on the many levels of MeCP2 interaction with DNA and chromatin. We show that strong methylation-independent as well as methylation-dependent binding by MeCP2 is influenced by DNA length. Although MeCP2 is strictly monomeric in solution, its binding to DNA is cooperative, with dimeric binding strongly correlated with methylation density, and strengthened by nearby A/T repeats. Dimeric binding is abolished in the F155S and R294X severe RTT mutants. MeCP2 also binds chromatin in vitro, resulting in compaction-related changes in nucleosome architecture that resemble the classical zigzag motif induced by histone H1 and considered important for 30-nm-fiber formation. In vivo chromatin binding kinetics and in vitro steady-state nucleosome binding of both MeCP2 and H1 provide strong evidence for competition between MeCP2 and H1 for common binding sites. This suggests that chromatin binding by MeCP2 and H1 in vivo should be viewed in the context of competitive multifactorial regulation.
DNA methylation constitutes an important epigenetic component in transcriptional regulation, with methylation generally leading to repression of nearby genes (6). However, the mechanism by which the epigenetic signal is passed to the regulatory machinery is not well understood. Research in this area has been focused on a small family of methyl-CpG binding proteins, best characterized by MeCP2 (19), mutations in which result in Rett syndrome (RTT), a debilitating neurodevelopmental disease in humans (2).
A mechanism of MeCP2-mediated gene silencing may involve recruitment of histone deacetylases upon methyl-specific binding (57). However, other mechanisms, which are not necessarily mutually exclusive, such as stabilization of large chromatin loops (29) and promotion of chromatin compaction (51), have also been suggested (14). Studies on in vivo distribution of MeCP2 in nuclei have revealed that, in addition to the expected occupancy of sites of CpG methylation, MeCP2 shows significant binding to unmethylated DNA (71). However, a recent analysis of MeCP2 occupancy has revealed that the genomic distribution of MeCP2 in mammalian neurons closely tracks methyl-CpG density (60). These results highlight our current lack of understanding of key questions pertinent to the binding of MeCP2 to DNA and chromatin. It is especially important, for example, to quantitate the modulation of binding by factors such as methylation density (8, 37, 48, 60) and the presence of adjacent A/T-rich sequences (31) that are reported to influence binding. In the present work, we have used a variety of quantitative approaches to show that, when bound to DNA, MeCP2 exhibits a cooperative monomer-dimer equilibrium, which is influenced by DNA length, methylation density, and the presence of nearby A/T repeats.
The interaction of MeCP2 with mononucleosomes is similar to its binding to naked DNA and strongly influenced by DNA methylation. The primary binding site in the linker DNA entry/exit region is reminiscent of histone H1 binding, which induces specific changes in chromatin architecture (5). In this report, using direct electron microscopy (EM) imaging of tetra-nucleosomes, we show that, like H1 binding, MeCP2 binding induces compaction by dramatically reducing the nucleosomal linker DNA entry/exit angle. Further, the steady-state fluorescence anisotropy studies reported here demonstrate competitive binding to methylated chromatosomes and reveal that MeCP2 is more potent at displacing H1 than vice versa.
In vivo, both linker histone H1 (36, 46) and MeCP2 (31, 32) have similar levels of intranuclear mobility and, in vitro, share binding sites on nucleosomes, suggesting that they may compete for chromatin binding. A competitive binding equilibrium between them would have important consequences for their nucleosome occupancy. Our fluorescence-recovery-after-photobleaching (FRAP) analysis reveals that in vivo chromatin binding by MeCP2 and the well-characterized histone H1 variant H10 follows a complex competitive equilibrium. Together, these data suggest that there is a dynamic interplay between MeCP2 and H10 that modulates local chromatin structure. Nevertheless, there are binding sites that are tightly bound either by MeCP2 or by H1 and cannot be exchanged by increasing the abundance of the other. These findings may explain the wide range of phenotypes associated with different levels of MeCP2 abundance and diversity of RTT mutations.
MATERIALS AND METHODS
MeCP2 and H10.
For synthesizing monocysteine (Cys413) MeCP2, site-directed-mutagenesis primers were designed to convert the two other cysteines (Cys339 and -429) to alanine. For cysteine-to-alanine conversion of Cys339, the pair of primers used were 5′ GGGAAAGGACTGAAGACCgcTAAGAGCCCTGGGCGGAAAAG 3′ and 5′ CTTTTCCGCCCAGGGCTCTTAgcGGTCTTCAGTCCTTTCCC 3′, and for cysteine-to-alanine conversion of Cys429, the pair of primers used were 5′ CACTGGAGAGCGACGGCgcCCCCAAGGAGCCAGCTAAG 3′ and 5′ CTTAGCTGGCTCCTTGGGGgcGCCGTCGCTCTCCAGTG 3′ (bases mutated for Cys-to-Ala conversion are lowercase). The construct was verified by sequencing.
Wild-type (WT) human MeCP2, the F155S RTT mutant, the R294X C-terminal-deletion mutant, and mono-Cys MeCP2 were expressed and purified as described previously (25, 51). The plasmid for expressing histone H10 with a C-terminal Ala-Cys-Ala extension (H10-ACA) was kindly provided by David T. Brown (University of Mississippi Medical Center, Jackson, MS). Wild-type H10 and H10-ACA were expressed and purified as described previously (38).
DNA and nucleosomal arrays.
Immunoelectrophoresis-high-performance liquid chromatography (IE-HPLC)-purified unlabeled (Table 1) or 5′-end-fluorescein-labeled duplex oligonucleotides were purchased from Integrated DNA Technologies (IDT; Coralville, IA). Except for 52bp 6-(CpG-A/Tn)met, all symmetrically methylated CpG-containing oligonucleotides were custom synthesized by IDT. 52bp 6-(CpG-A/Tn)met was prepared by methylating 52bp 6-(CpG-A/Tn) with SssI CpG methyltransferase (New England Biological, Ipswich, MA), and methylation efficiency was verified by testing resistance to HhaI digestion of an internal HhaI site which becomes refractory to digestion upon symmetric CpG methylation.
TABLE 1.
Synthetic oligonucleotides used in sedimentation equilibrium studies
| Primer name | Sequencea |
|---|---|
| 11bp-unmet | 5′ ATAACGCAAATG 3′ |
| 11bp-met | 5′ ATAA(Cmet)GCAAATG 3′ |
| 23bp-unmet | 5′ ATAAGCGCAATTCATAATGCCTT 3′ |
| 23bp CpG-less | 5′ ATAAGCACAATTCATAATGCCTT 3′ |
| 23bp-monomet | 5′ ATAAG(Cmet)GCAATTCATAATGCCTT 3′ |
| 45bp-unmet | 5′ TCAATGCCCTGGAACGGAATTCTTCTAATAAACGATGTATCATTT 3′ |
| 45bp-monomet | 5′ TCAATGCCCTGGAA(Cmet)GGAATTCTTCTAATAAACGATGTATCATTT 3′ |
| 45bp-tri-(CpG-A/T) | 5′ TGAATTCCGTTATAGTGAATTCCGTTATAGTGAATTCCGTTATAG 3′ |
| 45bp-trimet | 5′ TGAATTC(Cmet)GTTATAGTGAATTC(Cmet)GTTATAGTGAATTC(Cmet)GTTATAG 3′ |
| 52bp poly (CpG-ATn) | 5′ AATAACGGAATAACGGAATAACGGAATAGCGCAATAACGGAATAACGGAATA 3′ |
| 52bp poly (CpG-ATn)met | 5′ AATAA(Cmet)GGAATAA(Cmet)GGAATAA(Cmet)GGAATAG(Cmet)GCAATAA(Cmet)GGAATAA(Cmet)GGAATA 3′ |
The underlined sequences in 52bp poly (CpG-ATn) and 52bp poly (CpG-ATn)met were used for determining methylation efficiency by testing resistance to HhaI digestion. (Cmet)G is a symmetrically methylated CpG motif.
pUC19 plasmids harboring a tandem array of four 207-bp repeats, each containing the “601” nucleosomal positioning sequence (63), were kindly provided by S. A. Grigoryev (Pennsylvania State University School of Medicine, Hershey, PA). Maximum-efficiency Stbl2 competent cell transformants (Invitrogen, Carlsbad, CA) were grown and DNA was prepared as described previously (50). 601-4 DNA was excised using XbaI and SphI (New England Biological, Ipswich, MA) and resolved on 0.8% agarose gels. The corresponding gel band was excised, and DNA was electro eluted into TAE (Tris-acetate-EDTA) buffer using 6-8K Spectra/Por (Spectrum Labs, Rancho Dominguez, CA) cutoff membrane sleeves. DNA was purified using phenol-chloroform, followed by ethanol precipitation, and methylated as described previously (50). Tetranucleosomes were prepared by salt gradient dialysis from 2 M NaCl as described previously (50).
Nucleosome preparation for fluorescence anisotropy experiments.
A 172-bp (601) DNA construct was prepared by PCR amplification using 601-1 (207 bp inserted in the PUC19 plasmid) DNA as a template and HPLC-purified forward (5′-TCTAGATATCGGACCCTATACGCGGCCG-3′) and reverse (5′-CATGCACAGGATGTATATATCTGACACGTGCCTG-3′) primers purchased from IDT. The 180-bp amplicon produced by PCR was further digested with EcoRV to obtain the blunt-ended 172-bp amplicon containing the 146-bp core nucleosomal positioning sequence, a 14-bp-long entering nonnucleosomal (linker) DNA, and a 12-bp exiting linker DNA. DNA was purified as described previously (50).
Mass measurement by STEM.
Reconstituted mononucleosomes containing 207 bp of DNA with the “601” strong nucleosome positioning sequence and two approximately equal linker DNA segments (50) were incubated with 0 or 2 molecules of MeCP2 per nucleosome, fixed with 0.1% glutaraldehyde (see below), and prepared for scanning transmission electron microscopy (STEM) imaging as described previously (50). The rationale for using a molar input of 2 MeCP2 per nucleosome was based on electrophoretic mobility shift assays (EMSAs) showing that MeCP2 produced two distinct shifted bands with methylated mononucleosomes, indicative of two binding events (50). Low-dose images (61) were analyzed with the PCMASS 29 program using a custom model nucleosome based on the structure of the nucleosome core particle (39) to select image regions corresponding to individual mononucleosomes. The custom mask was manually aligned when automatic alignment failed. In cases where the linker could not be accurately masked by the model, a double-stranded-DNA model that could be adjusted for length and curvature was employed.
Electron microscopy.
Tetranucleosomes were fixed by dialysis against buffer at the desired monovalent ion concentration, containing 0.1% glutaraldehyde, for 4 h at 4°C, followed by two changes of buffer for a total of 16 h. Arrays at a DNA concentration of 50 μg/ml were incubated with MeCP2 at molar ratio of 2 MeCP2 molecules per nucleosome for 30 min at room temperature prior to fixation. For negative staining, fixed samples were diluted 10-fold with 50 mM NaCl, adsorbed to a glow-discharged carbon film, washed with double-distilled water, negatively stained with 0.5% uranyl acetate containing 0.1% glucose, and dried slowly in a 50% relative-humidity chamber. Some samples were rinsed briefly with water after uranyl acetate treatment to provide positive staining.
Samples were examined with a Tecnai 12 transmission electron microscope (TEM; FEI Co., Hillsboro, OR) operating at 120 kV, and images were recorded on a 2,048- by 2,048-pixel TVIPS slow-scan charge-coupled device (CCD; TVIPS GmbH, Gauting, Germany). Tilt pairs (25° separation) of negatively stained preparations were recorded with low-dose protocols, yielding <5 electrons/Å2 for each member of a tilt pair. Positively stained samples were recorded using tilted-beam dark-field optics. For tomographic reconstruction of negatively stained samples, tilt series were recorded with an image pixel size of 0.45 nm. The tilt data consisted of images at 2.5° intervals between −65° and +65°, with an average dose of 0.5 electrons/Å2. Alignment of tilt series by cross-correlation and reconstruction by weighted back-projection were performed using IMOD eTomo software (http://bio3d.colorado.edu/imod/).
For cryoelectron microscopy, samples at a tetramer concentration of ∼50 μg/ml were applied to holey carbon films and vitrified in liquid ethane in a Vitrobot (FEI, Hillsboro, OR). Samples were vitrified both fixed and unfixed; some fixed samples were positively stained on carbon films over holes prior to vitrification to enhance contrast in the vitrified sample. Grids were transferred under liquid nitrogen to the Tecnai 12 TEM in a Gatan 626 cryoholder (Gatan, Inc., Pleasanton, CA) and observed at −170°C. Tilt pairs were recorded as described above.
Image analysis.
Stereology was used to determine the three-dimensional (3-D) locations of features in the tilt pairs from which coordinates of nucleosome positions and linker trajectories were calculated and internucleosomal distances and angles derived (24). The effect of point-picking error on 3-D calculations was constrained to <3 pixels, which limits 3-D distance changes to <1 pixel and 3-D angle change to <1 degree (4). Solid models of selected tetramers were created by positioning objects of appropriate dimensions representing nucleosomes and linker DNA at the coordinates derived stereologically, or directly from tomographic reconstructions, and were examined interactively in three dimensions using Vertigo software (Vertigo, Inc., Vancouver, BC, Canada).
AFM.
DNA-MeCP2 complexes were deposited on the mica surface functionalized with 1-(3-aminopropyl)silatrane (APS-mica) by use of the protocol described in references 41 and 42. Briefly, 5 μl of the complex solution was placed on the APS-mica surface for 2 min, rinsed with deionized water (AquaMax Ultra; APS Water Services Corporation), and dried with a flow of argon. Atomic force microscopy (AFM) images were acquired in air using a MultiMode SPM NanoScope IV system (Veeco, Santa Barbara, CA) operating in tapping mode. Tapping mode silicon probes (Olympus; Asylum Research, Santa Barbara, CA) with a nominal spring constant of ∼42 N/m and a resonant frequency between 300 and 320 kHz were used. Image processing, cross-sectioning, and volume measurements were performed using Femtoscan (Advanced Technologies Center, Moscow, Russia). To obtain the molecular mass of the protein in kDa from the protein volume, the conversion coefficient from the linear plot of the protein molecular mass versus the protein volume (59) was used. The data for the volume measurements were analyzed using Origin 6.0 (Originlab, Northampton, MA). The histograms were approximated by Gaussians, and the maxima of the distributions were used as the most-probable values for the set.
By chance, some binding events will result in MeCP2 molecules being adjacent on DNA and indistinguishable from cooperative dimeric binding. We estimate the relative frequencies of these events as follows. Since MeCP2 occupies 11 bp of DNA, there are ∼76 binding sites per 601-4 (828 bp) DNA (i1 to i76). If the first MeCP2 molecule binds at site in, the probability of random binding of the second molecule to sites in+1 and in−1 is the addition of their individual probabilities, which sums to ∼2.6% of the frequency of monomer formation. However, due to the limited resolution of the AFM probe, MeCP2 molecules must be at least 11 nm apart to be resolved as separate particles. This length of DNA is sufficient to accommodate three MeCp2 molecules (one MeCP2 molecule per 11 bp, or ∼3.7 nm). Therefore, the effective probability of two MeCP2 molecules being identified as adjacent particles on the basis of chance is the sum of the probabilities of binding of a second MeCP2 molecule to sites in+1, in+2, in−1, and in−2, which is ∼5.2% of the frequency of monomer formation. The ratio of the areas under the Gaussian fits to the distribution (Fig. 1g) provide the actual relative frequencies of monomers and dimers.
FIG. 1.

DNA-MeCP2 interactions. (a to f) AFM images of MeCP2-DNA complexes. (a to c) DNA strands with circular (single arrows) and elliptical (double arrows) MeCP2 foci. (d and e) DNA loops with circular MeCP2 foci at the base. (f) Large complex containing multiple DNA strands and MeCP2 molecules. Scale bar = 50 nm. (g) Histogram of the calculated volumes of individual MeCP2 foci indicating two Gaussian distributions, with mean volumes of 88 nm3 and 157 nm3, close to those expected for the MeCP2 monomer and dimer, respectively. The elliptical foci center on the higher value. (h) Normalized lsg*(s)-versus-S20,w plot showing sedimentation coefficient distributions of 45 bp of DNA and complexes comprising 45 bp of DNA and MeCP2 at increasing (0-, 1-, 2-, 3-, 4-, and 5-fold) molar inputs of MeCP2. The fold input of MeCP2 increases from left to right. Saturation is reached at a 4-fold input. A small peak at 2.5 S represents free MeCP2 at the 5-fold protein input. (i) Normalized plot of change in fluorescence anisotropy of a fluorescein-labeled, unmethylated 23-bp DNA at various MeCP2/DNA ratios. Saturation is reached at a 2-fold input of MeCP2. Error bars represent standard errors of the means (SEM). (j) Multiwavelength, multispeed sedimentation equilibrium profile of the unmethylated 11-bp DNA substrate incubated with MeCP2 (3 μM DNA-3 μM protein) in 200 mM NaCl. Data were collected at 230 nm (circles), 260 nm (squares), and 280 nm (triangles) and speeds of 15,000 (green), 20,000 (red), and 25,000 (blue) rpm. Solid lines represent global fits obtained using the A+B heteroassociation model, where A represents DNA and B represents protein. The residuals of the fits are shown in the top panel.
Protein labeling and functional validation of the mono-Cys MeCP2 variant.
Mono-Cys MeCP2 and H10-ACA, each of which contains one modifiable thiol group, were incubated with tetramethyl rhodamine iodoacetamide (TMRIA) (Anaspec, Fremont, CA) for 2 h at 22°C and then overnight at 4°C in 50 mM NaCl, 50 mM Tris, pH 7.5, buffer in the dark. Excess label was removed by extensive serial dialysis in 10-kDa-molecular-mass-cutoff Microcon filter units (Millipore, Billerica, MA) in the same buffer, followed by 2 or 3 consecutive column purifications using PD MiniTrap (GE Healthcare, Waukesha, WI). Labeling efficiencies for MeCP2 and H1 were determined by measuring the absorbances at 280 nm (for the protein) and 546 nm (for rhodamine) using molar extinction coefficients (MECs) of 12,950 (MeCP2), 4,000 (H1), and 70,000 for rhodamine. An ∼40% labeling efficiency was routinely achieved for H1-ACA, while an ∼80% labeling efficiency was achieved for MeCP2.
Mono-Cys (Cys413) MeCP2, which lacks Cys339 and Cys429, is fully functional in our assays. It retains methyl-CpG specificity and DNA binding efficiency in in vitro EMSAs (not shown) (26, 51). Also, tetramethyl rhodamine (TMR)-labeled mono-Cys (Cys413) MeCP2 retains its capacity for nuclear localization signal (NLS)-mediated transport into the nuclei upon microinjection into the cytoplasm and shows the pericentromeric distribution typical of cells stably expressing MeCP2-GFP (see Fig. 6).
DNA saturation analysis using fluorescence anisotropy.
For DNA saturation analysis using fluorescence anisotropy, 0.1 μM IE-HPLC-purified fluorescein-labeled 23-bp DNA segment of mouse brain-derived neurotrophic factor (BDNF) promoter IV (23bp-unmet) (Table 1) was mixed with 0.4 μM unlabeled DNA of the same sequence and then incubated for 10 min prior to data acquisition with a range of molar inputs of MeCP2 (0, 0.25, 0.5, 0.75, 1, 2, 3, 4, and 5) in separate tubes in 10 mM Tris, pH 7.5, 100 mM NaCl, 0.5 mM EDTA buffer. The fraction of DNA bound to MeCP2 was determined by fluorescence anisotropy. For fluorescence anisotropy measurements, a PTI QM1 spectrofluorometer (Photon Technology International, Birmingham, NJ) equipped with an excitation and emission polarizer was used. The excitation wavelength (λexcitation) and λemission used were 480 nm and 515 to 525 nm, respectively. Fluorescence anisotropy was calculated using methods described earlier (26).
Salt titration of DNA-MeCP2 complexes.
To study the role of electrostatics in MeCP2-DNA interaction, 0.1 μM fluorescein-labeled monomethylated or trimethylated DNA (45 bp) was incubated with a 2-fold molar excess of MeCP2 in 10 mM Tris, pH 7.5, 0.5 mM EDTA buffer containing NaCl concentrations ranging from 100 mM to 500 mM in separate increments of 25 mM. The fraction of DNA bound to MeCP2 was determined by measuring fluorescence anisotropy using a PTI QM1 spectrofluorometer equipped with an excitation and emission polarizer (26).
DNA saturation analysis using boundary velocity sedimentation.
For DNA saturation analysis using sedimentation velocity, a 45-bp segment of BDNF promoter IV DNA was incubated at 2 μM with a 0-, 1-, 2-, 3-, 4-, or 5-fold molar excess of MeCP2 in 10 mM Tris, pH 7.5, 100 mM NaCl, 0.5 mM EDTA buffer and examined using a Beckman Optima XL-I analytical ultracentrifuge (Beckman Coulter, Brea, CA) with absorbance optics at a velocity of 40,000 rpm and a temperature of 20 ± 0.1°C. Data were analyzed using SEDFIT (34, 54) with the lsg*(s) method. For Fig. 1h, the lsg*(s) maximum value for each profile has been normalized to 1.
Sedimentation equilibrium.
Interactions between MeCP2 and defined oligonucleotides were studied using multiwavelength (230 nm, 260 nm, 280 nm) sedimentation equilibrium data acquired at multiple rotor speeds and multiple concentrations using absorbance optics (Beckman Optima XL-I analytical ultracentrifuge). For DNA, the molar extinction coefficient (MEC) at 260 nm was determined for each oligonucleotide by use of an IDT biophysics UV spectrum tool after application of Cavaluzzi Borer correction (13). Buffer density, buffer viscosity, the MEC of MeCP2 variants at 280 nm as well as 260 nm, and their partial specific volumes (Vbar) were calculated using SEDNTERP (33). For obtaining apparent molar masses of each individual component, multispeed, multiwavelength, multiconcentration equilibrium data of each component were globally fit to a monomer model with mass conservation constraints using SEDPHAT (65). In the fits to the DNA equilibrium data, the MEC at 260 nm was fixed for DNA and floated for 230 nm and 280 nm to obtain relative extinction coefficients at these wavelengths. The relative molar extinction coefficients of MeCP2 at 230 nm and 260 nm were determined from the ratio of absorbances at 230 nm and 280 nm and at 260 nm and 280 nm, respectively, multiplied by the known molar extinction coefficient at 280 nm. The MEC of MeCP2 at 260 nm obtained by this procedure agreed closely with that calculated using SEDNTERP (33). Also, fixing the MEC of MeCP2 at 280 nm and floating it at 230 nm and 260 nm in the fits to the MeCP2 equilibrium data yielded similar values for relative MEC of MeCP2 at these wavelengths. All molar masses of DNA substrates were calculated using the Vbar value of MeCP2, as this allows the calculation of the molar mass of the DNA-MeCP2 complex by simple addition of the apparent molar masses of the individual components, as is the case for their buoyant molar masses. Global fits to MeCP2-DNA complex equilibrium data were performed for each experiment by use of the A+B, A+B+B, and A+B+B+B heteroassociation models, where A is the DNA and B is the protein (SEDPHAT) (65). The MECs and apparent molar masses of protein and DNA obtained by the methods mentioned above were fixed in global fits to MeCP2-DNA complex equilibrium data. Models giving the best fit for each experiment on the basis of goodness-of-fit statistics and the corresponding root mean square deviation (RMSD) of fit are shown in Table 2. To compare only the first and second bindings, the molar ratios of MeCP2/DNA were kept at or below 1. The DNA-MeCP2 inputs used in these experiments were (in μM) 1.3:1.3, 1.3:0.65, 2:1.5, 0.65:0.65, 3:3, 6:6, 6:4, and 1.5:1.5.
TABLE 2.
Equilibrium dissociation constants for the first binding and enhancement factors for the second binding as determined by sedimentation equilibrium experiments and analyzed using the SEDPHAT global-fit optiona
| DNA | MeCP2 type | Exptl conditionsb | Heteroassociation model providing best global fit | Kd A+B global fit (μM) | Enhancement factor (Kd A+B/Kd A+B+B) obtained by global fit | Mean RMSD of all local fits |
|---|---|---|---|---|---|---|
| 11bp-unmet | WT | 3, 3, 3 | A+B | 15.0 | NA | 0.007 ± 0.0010 |
| 11bp-met | WT | 4, 3, 3 | A+B | 1.1 | NA | 0.004 ± 0.0007 |
| 23bp-unmet | WT | 2, 3, 3 | A+B+B | 0.25 | 0.13 | 0.007 ± 0.001 |
| 23bp CpG-less | WT | 3, 3, 3 | A+B+B | 0.31 | 0.10 | 0.005 ± 0.001 |
| 23bp-monomet | WT | 2, 3, 3 | A+B+B | 0.13 | 0.35 | 0.005 ± 0.0008 |
| 45bp-unmet | WT | 3, 3, 3 | A+B+B | 0.16 | 0.4 | 0.002 ± 0.0001 |
| 45bp-monomet | WT | 3, 3, 3 | A+B+B | 0.10 | 1.4 | 0.003 ± 0.0007 |
| 45bp tri-(CpG-A/T) | WT | 2, 3, 3 | A+B+B | 0.13 | 0.7 | 0.005 ± 0.0006 |
| 45bp-trimet | WT | 3, 3, 3 | A+B+B | 0.05 | 5.0 | 0.003 ± 0.0006 |
| F155S | 2, 3, 3 | A+B+B | 0.34 | 0.7 | 0.004 ± 0.0004 | |
| R294X | 3, 3, 3 | A+B+B | 0.08 | 0.8 | 0.003 ± 0.0008 | |
| 52bp poly (CpG-ATn) | WT | 3, 3, 3 | A+B+B | 0.11 | 1.4 | 0.004 ± 0.0009 |
| 52bp poly (CpG-ATn)met | WT | 3, 3, 3 | A+B+B | 0.05 | 30 | 0.003 ± 0.0004 |
A, DNA; B, MeCP2; WT, wild type; NA, not applicable.
The first, second, and third numerals represent the number of molar concentrations, the number of wavelengths (230 nm, 260 nm, and 280 nm), and the number of speeds, respectively.
Fluorescence anisotropy assay of MeCP2-H1 competition.
Nucleosomes (172 bp) were incubated for 10 min with equimolar amounts of MeCP2-TMR and H10-TMR and excited with vertical or horizontal polarized 552-nm light, and emission over a 5-nm window between 578 nm and 582 nm was collected in the horizontal and vertical directions. Data were also collected after incubation of the binary complex with increasing amounts of the alternate unlabeled protein. For each input of the unlabeled protein, the binary complex was incubated for 10 min, after which the unlabeled protein was added and incubated for another 5 min, after which anisotropy was measured. Fluorescence anisotropy values were calculated as described previously (26).
Tissue culture, transfection, and microinjection.
An H10-GFP BALB/c 3T3 cell line expressing H10-GFP was kindly provided by David T. Brown. Cells were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) lacking phenol red, supplemented with 15% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA), 1% antibiotic-antimycotic solution (Sigma-Aldrich, St. Louis, MO), 4 mM glutamine, and 400 μg/ml G418 at 37°C under 5% CO2. BALB/c 3T3 cells expressing GFP fusions of MeCP2 isoform I or II were kindly provided by Carolyn C. Schanen (Department of Biological Sciences, University of Delaware, Newark, DE) and grown as described for H10-GFP-expressing cells. Chinese hamster ovary (CHO) cells stably expressing HP1α-GFP were kindly provided by Tom Misteli (National Cancer Institute, NIH, Bethesda, MD). A MeCP2-monomeric red fluorescent protein (mRFP) overexpression plasmid containing a strong cytomegalovirus (CMV) promoter was kindly provided by Christina Cardoso (Max Delbruck Center for Molecular Medicine, Germany). Transient transfection of H10-GFP cells with MeCP2-mRFP was carried out with Lipofectamine 2000 reagent (Invitrogen) in accordance with the manufacturer's instructions.
Cells were grown on Mattek 50-mm glass bottom dishes (Ashland, MA) for 48 h. Prior to microinjection, the medium was replaced with DMEM supplemented with 5 mM HEPES, pH 7.5. The concentrations of labeled protein used for microinjection were 0.1 mM for H10-ACA and MeCP2 mono-Cys, 0.25 mM and 0.5 mM for unlabeled H10, and 0.25 mM for MeCP2. Microinjected cells were detected by the presence of high-molecular-mass (70-kDa) Texas Red-dextran (Invitrogen) in the injectate. Cells were injected using an Eppendorf model 5242 microinjector (Eppendorf, Hauppauge, NY) and a Zeiss IM35 inverted microscope (Thornwood, NY). Micropipettes for injection were made by pulling 1.2-mm by 0.9-mm by 100-mm borofilament capillary tubes (Friedrich and Dimmock, Millville, NJ) using a model P80 Brown Flaming micropipette puller (Sutter Instruments, Novato, CA) using a velocity index of 117, a pull index of 160, and a heat index of 380. Microscopy was performed 30 min, 1 h and 2 h after injection.
5-Azadeoxycytidine treatment of cells.
MeCP2-GFP-expressing BALB/c 3T3 cells were grown on Mattek 50-mm glass bottom dishes for 24 h and treated with medium supplemented with 1 μM 5-deoxy-azacytidine (5-Aza-dC) (Sigma-Aldrich) for 4 to 5 days, with replenishment every 24 h.
Fluorescence microscopy.
H10-GFP-expressing cells microinjected with TMR-MeCp2 and MeCP2-GFP-expressing cells microinjected with TMR-H10 were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min, permeabilized with 0.2% Triton X-100 in PBS for 10 min, blocked with 2% (wt/vol) bovine serum albumin (BSA), 15% milk, 0.05% Triton X-100 in PBS for 30 min, and stained with Hoechst 33258 (0.1 μg/ml) in PBS. Fluorescence microscopy was performed using a Nikon Eclipse microscope with a 40× PlanApo lens.
For quantification of DNA demethylation upon 5-Aza-dC treatment, treated and untreated MeCP2-GFP-expressing cells were fixed with 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.2% Triton X-100 in PBS for 1 h, treated with 4 N HCl containing 0.1% Triton X-100 for 30 min, neutralized with 0.1 M borate buffer (pH 8.5) for 30 min, and incubated in blocking buffer (0.2% [wt/vol] BSA, 0.05% Tween in PBS) overnight. For immunodetection of methyl cytosine, cells were then incubated with 2 μg/ml primary antibody (anti-5-methyl cytosine mouse monoclonal antibody [MAb]; EMD Chemicals, Gibbstown, NJ) in blocking buffer for 1.5 h and then in 10 μg/ml secondary antibody (Alexa Fluor 568 goat anti-mouse IgG; Invitrogen) in blocking buffer for 1 h. To measure the effect of 5-Aza-dC on MeCP2 distribution in pericentromeric heterochromatin (PHC), treated and untreated MeCP2-GFP-expressing cells were fixed with 4% paraformaldehyde in PBS for 15 min and then permeabilized with 0.2% Triton X-100 in PBS for 10 min, blocked (2% [wt/vol] BSA, 15% milk 0.05% Triton X-100 in PBS) for 30 min, and stained with Hoechst 33258 (0.1 μg/ml) in PBS. Fluorescence microscopy was performed using a Nikon Eclipse microscope with a 20× PlanApo lens.
FRAP.
Fluorescence recovery after photobleaching was performed as described previously (52), using a Zeiss (Thornwood, NY) LSM Meta 510 confocal microscope. The 488-nm argon line was used for imaging H10/HP1α/MeCP2-GFP and the 543 nm HeNe line used for rhodamine-labeled H10/MeCP2. For FRAP, three prebleaching images were typically acquired, followed by bleaching of a spot with a 1-μm radius encompassing either the heterochromatic or the euchromatic domains, and subsequent acquisition of images every 1 s and 3 s for MeCp2-GFP and H10-GFP, respectively. For HP1α-GFP, images were acquired every 0.5 s. For imaging prebleaching and postbleaching images, the 488-nm argon laser was used at 3 to 4% efficiency for uninjected cells and 5 to 7% efficiency for microinjected cells. Detector gain and amplifier offset were optimized to avoid saturation. For bleaching, the 488-nm laser power was set to 100%. To compensate for the low MeCP2-GFP fluorescence intensity in PHC resulting from 5-Aza-dC treatment, data were recorded with higher laser intensity and gain than were used for untreated cells. Since FRAP data were collected only from nuclei in which the fluorescence intensities were at least half of the mean normal fluorescence intensity, the effect of demethylation on binding kinetics is underestimated. FRAP recovery plots were generated from the fluorescence intensities in the bleached region of interest (ROI) and reference ROIs, and background fluorescence was measured at each time step. The fluorescence recovery in the ROI was double normalized using the equation Ft = [(It − Bt)/(I0 − B0)]/[(Rt − Bt)/(R0 − B0)], where Ft is the normalized fluorescence recovery at each time point t, It is the mean intensity of the bleached ROI at each time point t, I0 is the prebleaching fluorescence intensity of the ROI, Rt is the reference intensity at each time point t, R0 is the initial reference intensity, and B0 and Bt are the mean background intensities at the original and each subsequent time point t.
The times for 50% recovery (T50 values) and the percents mobile and immobile fractions were obtained by fitting individual recovery curves to the equation 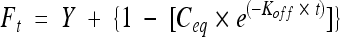 (62), where Ft is the normalized fluorescence recovery at time t, Y is the value of the fluorescence recovery plateau, Ceq is the equilibrium concentration of protein bound to chromatin, and Koff is the dissociation rate constant which has been shown to be identical to the rate constant for FRAP recovery (9). The T50 values were calculated using the equation T50 = ln (0.5)/(−Koff), and the mobile fraction was calculated using the equation Mf = Ceq/(Ceq − Y).
(62), where Ft is the normalized fluorescence recovery at time t, Y is the value of the fluorescence recovery plateau, Ceq is the equilibrium concentration of protein bound to chromatin, and Koff is the dissociation rate constant which has been shown to be identical to the rate constant for FRAP recovery (9). The T50 values were calculated using the equation T50 = ln (0.5)/(−Koff), and the mobile fraction was calculated using the equation Mf = Ceq/(Ceq − Y).
RESULTS
MeCP2 binding produces monomeric and dimeric foci on DNA.
Although MeCP2 exists strictly as a monomer in solution (1), direct EM imaging suggests that clustering may occur upon binding to DNA (51). This is seen with atomic force microscopy (AFM) (Fig. 1a to f), which shows MeCP2 binding to a methylated DNA consisting of 4 repeats of 207 bp containing a “601” nucleosome positioning sequence (63). Sites of bound protein appear predominantly as circular or elliptical foci (Fig. 1a to c). For AFM images, it is possible to estimate the volume of a particle and derive the approximate mass (41, 40, 42, 58, 59). The volumes of these foci show a bimodal distribution (Fig. 1g), which when fitted to a Gaussian function indicates maxima corresponding to 88 and 157 nm3, close to the expected volumes of a MeCP2 monomer and dimer. The ratio of the areas under the Gaussian fits to the monomer and dimer is 1.5:1, suggesting that dimers form at a frequency of ∼67% of monomers, much higher than the ∼5% expected for chance (see Materials and Methods).
A common conformation seen in ∼27% of MeCP2-bound molecules involves a DNA loop resulting from intra-DNA bridging by MeCP2 (Fig. 1d and e). The volume distribution of particles involved in looped complexes shows a single peak corresponding to monomeric MeCP2. This visual demonstration of multistrand capture by a single MeCP2 molecule is likely due to multiple independent DNA binding domains in MeCP2 (26, 51). Occasionally, complexes containing multiple DNA strands emanating from large MeCP2 foci were also seen (Fig. 1f). The frequencies of these complexes increase dramatically as the ratio of MeCP2 to DNA is increased. The ability of MeCP2 to induce DNA looping and complex macromolecular assemblies is consistent with its suggested role in the formation and maintenance of large-scale chromatin loops in vivo (29).
Systematic analysis of cooperative binding of MeCP2 to DNA.
The apparent dimeric binding mode of MeCP2 evident from the AFM images of MeCP2-DNA complexes suggested a possible cooperative binding and was surprising in view of the failure of MeCP2 to self-associate in solution (1). To investigate this further, we embarked on a systematic analysis of the effects of DNA length, CpG motif, CpG methylation, and methylation density on MeCP2 binding and the role of A/T-rich stretches adjacent to methylated CpGs, which have been reported to increase binding affinity (31). For these studies, a set of defined DNA sequences was prepared (Table 1), including a 45-bp section of the BDNF promoter (15, 44), containing single methylatable CpG unit flanked on each side by A/T stretches (45bp-unmet and 45bp-monomet) (Table 1). Smaller constructs that were approximately one-half and one-fourth as long (23bp-unmet, 23bp-monomet, 23bp CpG-less, 11bp-unmet, and 11bp-monomet) were also prepared (Table 1). To examine the effect of tandemly repeated CpG-A/T≥4 clusters, a modification of the 45-bp sequence containing 3 such clusters spaced 15 bp apart [45bp-trimet and 45bp tri(CpG-A/Tn)met] (Table 1) and a 52-bp sequence with 6 such clusters [52bp poly (CpG-A/Tn) and 52bp poly (CpG-A/Tn)met] were also prepared (Table 1). All sedimentation equilibrium experiments were carried out with 200 mM NaCl buffer, as salt titration experiments of MeCP2 dissociation from DNA with various methylation densities (see Materials and Methods) revealed substrate-specific differences only above 150 mM NaCl (not shown).
One MeCP2 molecule binds to 11 bp of DNA.
Salt titration experiments reveal that at monovalent ion concentrations below ∼150 mM, there is no discrimination between MeCP2 binding to methylated DNA and MeCP2 binding to unmethylated DNA. Thus, at the low salt concentration, the DNA binding stoichiometry of MeCP2 is independent of the methylation status of DNA. Also, any preferential positioning of MeCP2 on a DNA substrate mediated by methyl-CpG can complicate the estimation of binding stoichiometry. Therefore, to focus on binding stoichiometry, unmethylated DNA in 100 mM salt buffer was used in these experiments. To ensure that all saturable sites were occupied, the concentrations of DNA and protein (≥0.5 μM) in saturation experiments were kept well above the dissociation constant (Kd) of MeCP2 binding to DNA at a 100 mM monovalent ion concentration (<1 nM) (26).
We first determined the stoichiometry of MeCP2 binding to the 45-bp unmethylated DNA by use of sedimentation velocity monitored at 250 nm, where proteins have negligible absorbance (30, 34, 54). The lsg*(s) values at different molar inputs of MeCp2 show an increase in sedimentation coefficient for the complexes at molar excesses of up to 4-fold (Fig. 1h). At 5-fold excess, the lsg*(s) peak showed no further change. The small peak at ∼2.5 S, seen only at 5-fold input, represents unbound MeCP2 (1). The results show that a maximum of 4 MeCP2 molecules can bind to 45 bp of DNA and therefore that each molecule occupies 10 or 11 bp. To refine this estimate, we examined MeCP2 binding to a 5′ fluorescein-labeled, unmethylated 23-bp DNA by following fluorescence anisotropy. There was a linear increase in anisotropy at MeCP2/DNA ratios of up to 2, followed by a plateau at higher inputs (Fig. 1i), confirming that a single MeCP2 molecule occupies 10 or 11 bp of DNA. Previous work has established that the MBD domain alone occupies ∼6 bp (28), suggesting that the remainder of the molecule either occupies or sterically limits access to an additional 4 or 5 bp of DNA.
Neither anisotropy nor sedimentation velocity approaches can exclude the possibility that, upon initial binding to DNA, subsequent binding occurs solely through protein-protein interactions. In that case, the DNA binding stoichiometry of MeCP2 would be 1 per 23 bp. To clarify this issue, different concentrations of unmethylated or methylated 11-bp DNA were incubated with MeCP2 at different molar ratios and multispeed, multiwavelength sedimentation equilibrium data were obtained. Applying a global fit to the data using mass conservation constraints allows discrimination between various modes of binding and also yields Kd values (55, 64, 65). The data gave the best fit to a 1:1 binding model (the A+B heteroassociation model, where A is DNA and B is MeCP2) for both the unmethylated and the methylated 11-bp substrates (Table 2 and Fig. 1j). There was, however, a striking ∼14-fold increase in binding affinity when the single CpG unit was symmetrically methylated. This clearly indicates that one MeCP2 molecule occupies 11 bp of DNA and that a second binding would require additional DNA. It is interesting to note that in an earlier study (50), we found that MeCP2 confers ∼11-bp protection from micrococcal nuclease (MNase) digestion on the nucleosomal linker DNA proximal to the nucleosomal dyad. However, it was not possible to determine the number of MeCP2 molecules required to confer this protection. Our new data suggest that a single MeCP2 molecule would be sufficient to confer the 11-bp protection on nucleosomal linker DNA.
MeCP2 binding affinity is DNA length dependent.
Importantly, when sedimentation equilibrium was used to study MeCP2 binding to a 45-bp substrate, the A+B heteroassociation model gave a poor fit (Fig. 2a), whereas the A+B+B model gave an excellent fit to the data (Fig. 2b). At molar ratios of MeCP2/DNA of 1 or below, the A+B+B heteroassociation model gave the best fit with all sequences longer than 11 bp (Table 2), irrespective of sequence and methylation status. At higher molar ratios of MeCP2/DNA, such as a 3:1 ratio of MeCP2 to 45-bp unmethylated DNA, the A+B+B+B heteroassociation model provided the best fit to the sedimentation equilibrium data (Fig. 2d and e). On the basis of AFM imaging, which shows that MeCp2 binding follows a monomer-dimer equilibrium, on DNA long enough to accommodate more than two MeCP2, the third binding is likely to be distributive. It is, however, possible that on DNA with high methylation density, cooperative binding would lead to MeCP2 oligomerization.
FIG. 2.

Sedimentation equilibrium reveals mechanistic details of DNA binding by MeCP2. (a and b) Multiwavelength, multispeed sedimentation equilibrium of MeCP2 with an equimolar amount of trimethylated 45-bp DNA in 200 mM NaCl (1.3 μM DNA-1.3 μM protein). Data were collected at 230 nm (circles), 260 nm (squares), and 280 nm (triangles) and speeds of 10,000 (green), 14,000 (red), and 18,000 (blue) rpm. Solid lines represent global fits obtained using an A+B heteroassociation model (a) or an A+B+B model (b). Note the very small residuals in panel b, indicating that MeCP2 binding to longer DNA follows an A+B+B heteroassociation model at protein/DNA ratios of ≤1. (c) Summary of the sedimentation equilibrium data showing for each DNA substrate the fraction existing as A, AB, and ABB, as determined from the binding parameters given in Table 2, using the mass action law option in SEDPHAT and input concentrations of 0.5 μM for MeCP2 and 0.5 μM for DNA. The error in the calculations is the same as that presented in Table 2 in the “Mean RMSD of all local fits” column. (d and e) Multiwavelength, multispeed sedimentation equilibrium of the interaction of MeCP2 with 45 bp of monomethylated DNA in 200 mM NaCl at an input ratio of 1:3 (DNA/MeCP2) and data collected at 230 nm (circles), 260 nm (squares), and 280 nm (triangles) and speeds of 10,000 (green), 14,000 (red), and 18,000 (blue) rpm. Solid lines represent global fits obtained using the A+B+B+B heteroassociation model (d) and the A+B+B heteroassociation model (e). Residuals show that at this input ratio, the A+B+B+B model provides a better fit.
To limit our analysis to the two-binding-event scenario, which is particularly useful in dissecting the relationship between the first and second binding events, we carried out sedimentation equilibrium analysis at molar ratios of MeCP2/DNA of ≤1. Under these conditions, there were important differences in the binding affinities for the different substrates. A comparison of the first binding (Kd A+B) to the different substrates demonstrates a strong DNA length dependence for MeCP2-DNA binding (Table 2). Even though 10 or 11 bp is the stoichiometric binding length of DNA for MeCP2, when presented with the unmethylated 11-bp substrate, MeCP2 binds with low affinity (∼15 μM). With the corresponding methylated substrate (11bp-met), binding affinity increased ∼14-fold (Table 2). Longer methylated DNA gave a further substantial increase in affinity (Table 2). For example, when the DNA length was increased to 23 bp (23bp-unmet) or 45 bp (45bp-unmet), the binding affinity (Kd A+B) increased to ∼0.25 μM or ∼0.16 μM, respectively. Furthermore, with the 23-bp substrate (23bp-unmet), the second MeCP2 molecule, for which a maximum of 11 bp is available, bound ∼8-fold more strongly than was seen with single MeCP2 binding to the 11-bp oligonucleotide. This indicates that the presence of the first MeCP2 molecule strongly facilitates the second binding (Table 2). In the case of 23-bp DNA, the DNA was designed with an end-located CpG motif (Table 1). This was done in order to avoid steric exclusion of a second molecule by a methyl-CpG-bound MeCP2 molecule, in case methyl-CpG-specific positioning in 23bp-monomet with a centrally located methyl-CpG restricted the translational dynamics of MeCP2.
A/T-rich sequence motifs markedly improve MeCP2 binding to DNA.
In order to determine whether the [CpG-(A/T)≥4] sequence motif influences cooperative MeCP2 binding to DNA in the presence or absence of methylation, we carried out sedimentation equilibrium analysis of MeCP2 binding to 45bp-unmet DNA [single CpG-(A/T)≥4], 45bp tri-CpG-(A/T) DNA [three CpG-(A/T)≥4], and 52bp poly (CpG-A/Tn) DNA [six CpG-(A/T)≥4] (Table 1). Repeats of the CpG-(A/T)≥4 motif clearly improve MeCP2 binding even on unmethylated sequences. The consistent increase in the affinity of the second binding obtained with increasing CpG-(A/T)≥4 motif density is a further clear indication of cooperative binding (Table 2 and Fig. 2c).
Cooperative binding increases with methylation density.
With 23-bp monomethylated DNA, the first and the second bindings occurred with affinities ∼2.0-fold- and ∼3-fold-higher, respectively, than those observed for 23-bp unmethylated DNA (Table 2 and Fig. 2c). Binding to 23-bp unmethylated DNA did not differ significantly from binding to 23-bp CpG-less DNA, suggesting that a single CpG motif by itself has little effect on MeCP2 binding (Table 2 and Fig. 2c). Moreover, comparison of binding to 45bp-Tri-(CpG-A/T) DNA versus binding to 45bp-trimet DNA shows that increasing the methylation density to the trimethylated state results in a further marked increase in binding affinity, especially for the second binding (Table 2 and Fig. 2b and c). Increasing the methylation density still further to 6 methylated CpG-(A/T)≥4 motifs in the 52-bp substrate produced the most dramatic increase in second-binding affinity compared to the level for the first binding (Table 2 and Fig. 2c). It is important to note that this 30-fold enhancement of the affinity of the second binding could be an underestimation, as the second-binding affinity is near the lower limit of sensitivity of the fitting program (65). The relative abundances of A, AB, and ABB, as determined using the mass law calculator with the binding parameters given in Table 2, clearly demonstrate the striking increase in the ABB component with methylated 52bp-met DNA (Fig. 2c). While the correlated increases in first- and second-binding affinities obtained with methylation density could be explained by a higher probability of methylation-specific binding on densely methylated DNA, the increase in second-binding affinity compared to the level for the first binding indicates a strong cooperative association. We also examined the interaction of the RTT-causing MeCP2 F155S mutant, which is severely misfolded (25), and the R294X C-terminal-deletion mutant with DNA. The F155S mutant, which was selected since its effect on methylation specificity and overall binding affinity shows an intermediate level of deficiency (51), exhibited an overall diminution in binding affinity when presented with the trimethylated 45-bp substrate (Table 2 and Fig. 2c). Interestingly, the R294X mutant, which lacks the C terminus of the protein, showed no sign of cooperative binding to the 45bp-trimet substrate (see Fig. 5E; also Table 2). This indicates a role for the MeCP2 C terminus in the cooperative dimerization of MeCP2 that occurs upon DNA binding.
In terms of the in vitro relevance of these results, it is important to note that the cooperative binding of MeCP2 occurs over a wide range of concentrations and with MeCP2/DNA molar ratios as low as 1:2, the MeCP2/nucleosome ratio in unsorted brain nuclei (60).
Methylated nucleosomes have a stronger affinity for MeCP2 than unmethylated nucleosomes.
In vivo, MeCP2 is most likely to encounter DNA in a nucleosomal context. We therefore determined the stoichiometry of MeCP2 binding to 207-bp nucleosomes centered on the strong “601” nucleosome positioning sequence (50, 63). Since the number of MeCP2 molecules bound to a nucleosome cannot be reliably quantitated by conventional EM, we used scanning transmission electron microscopy (STEM) (67), which provides direct mass values for individual particles. The mean mass for mononucleosomes was 249 kDa (standard deviation [SD], 38; standard error [SE], 4.1; n = 86), (Fig. 3a), close to the predicted mass of 235 kDa. After incubation with 2 molecules of MeCP2 per nucleosome, the mean mass of methylated nucleosomes increased to 338 kDa (SD, 59; SE, 3.0; n = 324) (Fig. 3c), indicating that the majority of nucleosomes had gained two 52-kDa MeCP2 molecules. Unmethylated nucleosomes also showed an increase in mass, but only to 292 kDa (SD, 37; SE, 3.5; n = 112), consistent with the idea that most nucleosomes contain a single MeCP2 (Fig. 3b).
FIG. 3.

STEM mass analysis of the interaction between 207-bp nucleosomes and MeCP2. (a) Nucleosomes without MeCP2. (b, c) Unmethylated (b) and methylated (c) nucleosomes treated with 2 MeCP2 molecules per nucleosome. Peaks of the Gaussian fits correspond to nucleosomes alone (a) and nucleosomes with one (b) and two (c) bound MeCP2 molecules. Unpaired t tests indicate that the differences between the populations are highly significant (P < 10e−10).
We previously showed that methylated nucleosome core particles lacking linker DNA fail to bind MeCP2 (50). Thus, in a nucleosomal context, MeCP2 binds primarily to linker DNA and methylation favors a stoichiometry of 2 MeCP2 molecules per nucleosome. The nucleosomal substrate used here has a cluster of CpGs on the entering linker DNA close to the nucleosomal dyad, and this appears to be the most likely site for binding of the first MeCP2 molecule to methylated nucleosomes (50). The nucleosomal-dyad-specific binding is likely to occur in unmethylated nucleosomes as well, promoted by linker geometry at the dyad and interaction between the MeCP2 C terminus and histone H3 (50). The second binding of MeCP2 to methylated nucleosome is also likely to occur on the entering linker DNA, which carries a high density of methyl CpGs (50) and thus would favor cooperative dimerization. In unmethylated nucleosomes, the second binding to the nucleosomal linker DNA would not be favored.
MeCP2 binding to nucleosomes produces an architectural motif at the nucleosomal dyad similar to that induced by histone H1.
To probe the early stages in chromatin compaction by MeCP2, not accessible in highly condensed 12-nucleosomal arrays, which obscures nucleosome orientational information as well as linker DNA trajectory (24), we imaged tetramers under salt conditions that induce limited compaction (see Materials and Methods). Tetranucleosomes without MeCP2 appeared as an open beaded chain of 4 nucleosomes separated by linker DNA (Fig. 4a). Incubation with MeCP2 (2 molecules per nucleosome) induced extensive compaction (Fig. 4b and c), with many showing the “bow tie” configuration seen in tetranucleosome crystals (53), resulting from the basic zigzag architecture. The same motif predominated in frozen hydrated samples (not shown).
FIG. 4.

MeCP2 compacts tetranucleosomes. (a) Tetranucleosomes in 50 mM NaCl exhibit the expected open beaded-string configuration. (b and c) In the presence of 2 MeCP2 molecules per nucleosome, the tetranucleosomes fold into a compact “bow tie” conformation similar to that observed in compact tetranucleosome crystals (53). (d, e, f, and g) Models derived from stereo pair images as viewed from 2 directions support the hypothesis that MeCP2 induces compaction primarily through zigzag folding. (a and b) Positive-stain TEM dark field. (c) Negative-stain TEM. (d to g) Models based on stereo images of negatively stained preparations. Scales = 10 nm.
Analysis of tilt pairs of the negatively stained and cryo images provided the 3-D coordinates of the nucleosome centers, edges, and linker DNA entry/exit points. Solid models constructed from these data (Fig. 4d to g) allowed the linker entry/exit angle between consecutive nucleosomes to be computed (5). In the absence of MeCP2, the angle was dependent on the salt concentration in which the tetranucleosomes were fixed. In 50 mM salt, the mean angle was large and highly variable (106° [SD, 44°; SE, 9°]), whereas in 150 mM salt, the mean angle decreased dramatically and was more consistent (26° [SD, 12°; SE, 9°]) (5). In the presence of MeCP2 and in 50 mM NaCl, the angle (26° [SD, 9°; SE, 7°]) was similar to that seen in untreated tetranucleosomes in 150 mM NaCl. Importantly, the MeCP2-induced architecture favored a close apposition of the entering and exiting linker DNA segments producing the “stem” motif observed in oligonucleosomes compacted with the linker histone (4).
MeCP2 is more potent in displacing H1 from nucleosomes than vice versa.
On the basis of the similarity of the chromatin architecture induced and the MNase protection conferred on nucleosomal linkers (50), it is likely that MeCP2 and H1 share a nucleosomal binding site and, if so, might compete for chromatin binding. Earlier work by Nan et al. (48) showed that the addition of MeCP2 to methylated minichromosomes resulted in H1 depletion. However, the use of minichromosomes reconstituted in a Xenopus egg extract precluded determination of binding dynamics in the context of the nucleosomal dyad, and a direct role for MeCP2 in competitive exclusion of H1 was not established. In the present study, we use fluorescence anisotropy to address the bidirectional competition between MeCp2 and H1 for nucleosome binding in a ternary system where the sole components are 172-bp nucleosomes with symmetrical linkers, MeCP2 and H1.
MeCP2 and H10 (a well-characterized H1 variant) were fluorescently (TMR) labeled under conditions that preserve functionality (see Materials and Methods), and changes in the fluorescence anisotropy of the labeled protein observed upon binding to an equimolar amount of methylated 172-bp chromatosome-length nucleosomes were recorded. In our fluorescence anisotropy experiments, the maximum increase in the anisotropy of labeled MeCP2 was achieved upon incubation with an equimolar amount of methylated 172-bp nucleosomes (not shown). In contrast, methylated complete (207-bp) nucleosomes bind two MeCP2 molecules (Fig. 3). To study the competitive exclusion of one protein by the other, the binary complexes were incubated with increasing amounts of the alternate unlabeled protein (Fig. 5a to c). As expected, when allowed to interact with methylated nucleosomes, both MeCP2-TMR and H10-TMR showed marked increases in anisotropy. Importantly, upon incubation with unlabeled protein, there was a progressive decrease in anisotropy, with the decay profile clearly showing a much stronger displacement of H10 by MeCP2 than the reverse (Fig. 5c). Moreover, incubation of the binary complex with unlabeled H10 or MeCP2 did not result in a further increase in anisotropy, which would be expected if both proteins coexisted on the same nucleosome. This supports the hypothesis that H10 and MeCP2 share a nucleosome-binding site and that a 172-bp nucleosome with equal-sized linkers occupied by H10 cannot be cooccupied by MeCP2 and vice versa.
FIG. 5.

MeCP2 and histone H1 compete for nucleosome-binding sites in vitro. (a) Fluorescence anisotropy of 10 nM TMR-labeled H10 (circles) by itself (lower short arrow), upon incubation with an equimolar amount of 172-bp (the approximate chromatosome length) nucleosomes (upper long arrow), and upon incubation of the binary complex with increasing amounts of unlabeled MeCP2. (b) Same as for panel a, but with TMR-MeCP2 (squares) incubated with increasing amounts of unlabeled H10. (c) Normalized version of data shown in panels a and b. At an equimolar input of unlabeled MeCP2, ∼70% labeled H10 is excluded from mononucleosomes, whereas with unlabeled H10, ∼30% of labeled MeCP2 is competed out. Error bars represent SEM. Data are based on 3 separate acquisitions for each experiment type.
MeCP2 and histone H1 compete in vivo.
Given the unique environment of the nucleus, it was important to determine whether MeCP2 and H1 compete for chromatin binding sites in vivo, since such competition is likely to be important in MeCP2 function. Both proteins have similar intranuclear mobilities, as determined by fluorescence recovery after photobleaching (FRAP) (31, 32, 36, 46), suggesting that their interaction could be usefully studied using this technique. One protein was expressed as a GFP fusion, and changes in FRAP kinetics upon microinjection of the second protein were monitored (11).
We first established the conditions under which microinjection of fluorescently labeled MeCP2 or H10 into the cytoplasm of mouse fibroblasts resulted in complete uptake into the nucleus and had no obvious deleterious effects on cell viability or intranuclear distribution (Fig. 6). Injection of cells with buffer containing 0.5 mg/ml Texas Red-labeled dextran showed that microinjection itself did not affect the viability or morphology of the nuclei (Fig. 6c) or alter the FRAP kinetics of H10-GFP (Fig. 7c and d and Table 3). Microinjected cells survived during the 15-h observation period and divided successfully (the number of H10-Texas Red-injected cells increased by ∼75%), suggesting that H10 microinjection was not toxic. When cells expressing H10-GFP (46) were injected with unlabeled H10, there was a substantial increase in the fluorescence recovery rate compared to the level for uninjected cells, but the ratio of the mobile fractions to the immobile fractions was little changed (Fig. 7 and Table 3). H10-GFP-expressing cells were then microinjected with TMR-labeled MeCP2. The two proteins were strongly colocalized, especially in the pericentromeric heterochromatin (PHC) identified by Hoechst staining (Fig. 6b). Similar results were obtained with the converse experiment, in which cells expressing MeCP2-GFP were injected with TMR-labeled H10 (Fig. 6a). Having established standard conditions, we recorded FRAP data from euchromatic and heterochromatic regions of H10-GFP cells that were either uninjected, mock injected, or injected with MeCP2. Also, the converse experiment, in which MeCP2-GFP-expressing cells were injected with H10, was carried out. From these data, the time for 50% recovery (T50) and the proportions of mobile (readily exchangeable) and immobile (exchangeable over much longer time periods) protein fractions were determined.
FIG. 6.

Microinjection does not affect general cellular health and allows visualization of the overlapping distribution of MeCP2 and H10. (a) Fluorescence imaging indicates that microinjected TMR-H10 (red) colocalizes with MeCP2-GFP (green), especially in PHC (merged image at right). Scale bar = 5 μm. (b) Images of H10-GFP-expressing nuclei indicate that H10-GFP (green) and microinjected TMR-MeCP2 (red) colocalize particularly well in PHC (merged image). Hoechst staining (cyan, far right) shows a very similar distribution. (c) Microinjected cells can be identified by the red fluorescence of Texas Red-dextran in the cytoplasm. Microinjection does not alter the nuclear morphology or the fluorescence of H10-GFP (green). Scale bar = 20 μm.
FIG. 7.

Microinjection of MeCP2 accelerates the FRAP kinetics of H10-GFP in heterochromatin and euchromatin. (a and b) Confocal images of cells expressing H10-GFP, where a 1-μm radius spot of EU (a) or HC (b) was bleached and the recovery monitored over time. From left to right are prebleaching, bleaching, and postbleaching images. (c and d) FRAP profiles of H10-GFP in 1-μm-radius bleached spots in EU (c) and HC (d) in uninjected (□) and mock-injected (⋄) cells. FRAP curves are also shown for H10-GFP cells injected with H10 (⋆), MeCP2 (▵), and transiently transfected with MeCP2-RFP plasmid (○). Error bars represent SEM. FRAP profiles show the mean values for 20 or more different nuclei.
TABLE 3.
Quantitative FRAP analysis of MeCP2-GFP-, H10-GFP-, and HP1α-GFP-expressing cells under different experimental conditions reveals dynamic interplay between MeCP2 and H10a
| Cell type | Expt type | Mean ± SE |
|||||
|---|---|---|---|---|---|---|---|
|
T50 (s) |
If (%) |
Mf (%) |
|||||
| EU | HC | EU | HC | EU | HC | ||
| H10-GFP | Control | 37 ± 3 | 40 ± 3 | 12 ± 3 | 26 ± 3 | 88 ± 3 | 75 ± 3 |
| H10-GFP | Texas Red-dextran MI | 37 ± 2 | 40 ± 2 | 14 ± 3 | 24 ± 2 | 864 ± 3 | 76 ± 2 |
| H10-GFP | H10 MI | NA | 15 ± 2 | NA | 24 ± 2 | NA | 77 ± 2 |
| H10-GFP | MeCP2 MI | 17 ± 2 | 18 ± 12 | 18 ± 3 | 32 ± 2 | 82 ± 3 | 68 ± 2 |
| H10-GFP | MeCP2-RFPb | 16 ± 1 | 17 ± 2 | 18 ± 2 | 30 ± 2 | 82 ± 2 | 71 ± 2 |
| MeCP2-GFP | Control | NA | 29 ± 2 | NA | 22 ± 2 | NA | 79 ± 2 |
| MeCP2-GFP | H10 MI (250 μM) | NA | 17 ± 2 | NA | 46 ± 4 | NA | 54 ± 4 |
| MeCP2-GFP | H10 MI (500 μM) | NA | 11 ± 2 | NA | 69 ± 2 | NA | 31 ± 2 |
| MeCP2-GFP | Plus 5-Aza-dC | NA | 21 ± 2 | NA | 15 ± 2 | NA | 85 ± 2 |
| HP1α-GFP | Control | NA | 3.5 ± 0.15 | NA | 7 ± 2 | NA | 93 ± 2 |
| HP1α-GFP | H10 MI (250 μM) | NA | 3.6 ± 0.15 | NA | 8.5 ± 2.5 | NA | 91.5 ± 2.5 |
MI, protein microinjection; If, immobile fraction; Mf, mobile fraction; T50, time required for 50% FRAP; NA, not applicable. For each experiment type, the T50, If, and Mf values are based on FRAP data from at least 20 different nuclei.
Transient transfection.
Injection of MeCP2 into cells expressing H10-GFP reduced the mean T50 in euchromatin (EU) from 37 ± 3 to 17 ± 2 s and that in heterochromatin (HC) from 40 ± 3 to 17.6 ± 2 s (Fig. 7 and Table 3). There were also small decreases in the mobile fraction both in EU and in HC (Fig. 7 and Table 3). Similar results were obtained when H10-GFP cells were transiently transfected with a MeCP2-mRFP-expressing plasmid (Fig. 7 and Table 3). This suggests that MeCP2 competes strongly for binding sites with the mobile fraction of H10 but relatively weakly with the tightly bound H10 fraction. This difference in competitive efficiency for two different pools of H10 will lead to a relative enrichment of tightly bound H10 in comparison with the level for the mobile fraction. Under equilibrium conditions, in an uninjected H10-GFP cell, the fluorescence at any spot is the sum of the fluorescences from the immobile and mobile fractions. Upon injection of MeCP2, some of these weakly bound sites will be invaded by MeCP2 in accordance with the law of mass action. This will create a larger free pool of H10 and H10-GFP, and there will be fewer binding sites, thus increasing the FRAP recovery rate. However, as a result of the relatively weak competition by MeCP2 for sites tightly occupied by the immobile fraction of H10 and the strong competition for sites weakly occupied by the mobile fraction of H10, once equilibrium is reached, the fluorescence at any spot will be composed of a relatively large proportion of tightly bound H10-GFP, and upon photobleaching, the extent of recovery will be lower than that in uninjected cells.
To examine the reverse situation, we injected (250 μM) recombinant H10 into cells stably expressing MeCP2-GFP (Fig. 8a and b and Table 3). This resulted in a more rapid recovery for HC (T50, 17 ± 2 s) than in uninjected cells (T50, 29 ± 2 s). The low fluorescence intensity of MeCP2-GFP at EU in these uninduced cells precluded effective FRAP analysis of MeCP2 binding to EU. Doubling the concentration of the injected H10 (from 250 μM and 500 μM) produced a further reduction in T50 from 17 ± 2 s to 11 ± 2 s. The use of exogenous H10 also resulted in an increase in the immobile fraction of MeCP2 from 22% ± 2% to 46% ± 4% and at 500 μM elicited an additional increase in the immobile fraction to 69% ± 2%. Thus, there is a striking difference between H10 and MeCP2 in chromatin binding, with the immobile fraction of MeCP2 showing a much greater increase when challenged with H10, suggesting a much less effective exclusion of tightly bound MeCP2 by H10 than vice versa (Fig. 8a and b and Table 3).
FIG. 8.

FRAP kinetics of MeCP2-GFP in pericentromeric heterochromatin is regulated by H1 concentration and the methylation status of chromatin. (a and b) Microinjection of H10 accelerates MeCP2-GFP FRAP in HC. (a) FRAP at a 1-μm-radius heterochromatic spot in a cell expressing GFP-tagged MeCP2. From left to right are prebleaching, bleaching, and postbleaching images. (b) FRAP profiles of GFP-tagged MeCP2 in HC in control cells (red) and cells injected with 250 μM (blue), and 500 μM (brown) H10. Error bars represent SEM. The FRAP profiles show the mean values for 20 or more different nuclei. Scale bar = 5 μm. (c to h) Treatment with 5-Aza-dC reduces DNA methylation and MeCP2 binding in PHC. (c and d) Immunodetection of methyl cytosine in MeCP2-GFP-expressing cells reveals that upon 5-Aza-dC treatment there is a marked loss of methylation in PHC (d) compared to the level for untreated cells (c). Scale bar = 5 μm. (e) Fluorescence recovery profile of MeCP2 in HC of nuclei treated with 5-Aza-dC. FRAP profiles show the mean values for 20 or more different nuclei. FRAP was performed in nuclei in which the fluorescence intensities in HC were at least half of the values for untreated nuclei. The effect of demethylation on MeCP2 binding kinetics is therefore underestimated. Error bars represent SEM. (f) MeCP2-GFP fluorescence intensity in PHC of cells treated with 5-Aza-dC (n = 300) or dimethyl sulfoxide (DMSO) only (n = 351). Nuclei in which GFP fluorescence of PHC foci was absent are not included. (g) Distribution of anti-5-methyl cytosine antibody in nuclei of cells treated with 5-Aza-dC (n = 300) or DMSO only (n = 300). (h) The Hoechst fluorescence intensity in PHC foci of cells treated with 5-Aza-dC (n = 400) is similar to the level for the DMSO-only control (n = 351).
To test whether the competitive exclusion of MeCP2 by H10 is specific or is an effect of the increased abundance of H1 in microinjected cells, we studied the effect of H1 on the binding dynamics of heterochromatin protein 1 (HP1). HP1, unlike MeCP2 or H1, does not bind to the nucleosome in a dyad-specific manner but rather binds to methylated lysine-9 in histone H3 (16). Thus, it was unlikely that HP1 would be competitively excluded from chromatin by H1 unless the competition was nonspecific. H10 was microinjected into cells stably expressing HP1α-GFP, the dynamics of which have been extensively characterized (16). This did not alter the FRAP kinetics of HP1 (Fig. 9 and Table 3), suggesting that H1-induced exclusion of MeCP2 from chromatin binding sites is not the result of nonspecific disturbance of nuclear dynamics.
FIG. 9.

Microinjection of H10 does not alter HP1α-GFP FRAP kinetics in heterochromatin after photobleaching. (a to c) FRAP kinetics at a 1-μm-radius heterochromatic spot in a cell expressing HP1α-GFP. Scale bar = 5 μm. From left to right are prebleaching, bleaching, and postbleaching images. (d) FRAP profiles of HP1α-GFP in HC in uninjected cells (red) as well as cells injected with 250 μM H10 (green). Error bars represent SEM. FRAP profiles show the mean values for 20 or more different nuclei.
Given the methylation-induced differences in MeCP2 binding to DNA in vitro (Table 2 and Fig. 2c), it was important to examine the effect of CpG methylation in vivo. For this, we grew MeCP2-GFP cells in 5-Aza-dC. This led to a strong (65%) depletion of methylation as measured with a methylation-specific antibody (Fig. 8c, d, and g) and a concomitant reduction (∼57%) of MeCP2-GFP fluorescence in PHC (Fig. 8f) but no change in the pattern or strength of Hoechst fluorescence (Fig. 8h). The FRAP kinetics of MeCP2 in 5-Aza-dC-treated cells (Table 3) showed a moderate decrease in T50 from 29 ± 2 s to 21 ± 2 s and a decrease in the immobile fraction (22% ± 2% to 15% ± 2%) compared to the level for untreated cells. The 5-Aza-dC treatment of cells, which provides a way to emulate the in vitro binding to unmethylated DNA, shows that, in our system, the loss of methylation has a dramatic effect on MeCP2 distribution and suggests that in vitro binding features are closed mirrored in vivo.
DISCUSSION
This work has revealed important new features of the interaction between MeCP2 and its DNA and chromatin substrates. First, MeCP2 binding to DNA is cooperative in nature and exhibits a monomer-dimer equilibrium which is modulated by DNA length, the density of methyl CpG clusters, and the presence of nearby (A/T)n repeats. MeCP2 is strictly monomeric in solution (1), indicating that dimer formation is DNA dependent and may be related to the marked disorder-to-order transition that occurs when MeCP2 binds to methylated DNA (25, 26). This suggests a role for DNA as both the substrate and the allosteric effector in its interaction with MeCP2 (35), a property that has been demonstrated for several other DNA binding proteins (45, 56, 69). The structure acquisition and conformational changes of MeCP2 that accompany DNA binding could also facilitate the protein-protein interactions required for dimer formation. Since the MeCP2 domains NTD, ID, MBD, TRD, and CTDα have been shown to either bind DNA autonomously or regulate MBD-dependent binding (26), disruption of any of these domains is likely to affect overall binding. However, the complete abolition of cooperative binding in the R294X mutant suggests that the CTDβ domain, which lacks a DNA binding site (and is deleted in the R294X mutant), mediates interprotein association, perhaps through the 4 molecular recognition features (MoRFs) present in this region (26). MeCP2 is ∼60% unstructured and is highly enriched in MoRFs (26), which are potential sites for intermolecular interactions (47). Further, the dimeric binding mode exhibited by MeCP2 would increase local MoRF density, creating additional binding surfaces for recruitment of transcriptional regulators. In this respect, it is important to note that some promoters known to bind MeCP2 exhibit a high density of methylated CpG (44, 66). This would favor cooperative dimerization and increase the likelihood of forming large repressive complexes through polyvalent associations. Our results thus provide a mechanistic basis for the phenomenon of methyl-CpG tracking by MeCP2 in the genome (60). MeCP2 fails to bind to nucleosome core particles that lack linker DNA (51) but, as shown here, does bind free DNA in a length-dependent manner. Thus, binding to chromatin will depend on the availability of nucleosomal linker DNA or nucleosome-free regions. DNA length-dependent cooperative interactions have been shown to be instrumental in other systems, such as that involving nonhomologous end-joining of DNA by the DNA repair protein Ku (43).
We show that MeCP2-induced conformational changes in the nucleosome are strikingly similar to those induced by histone H1 (Fig. 4) (5, 10) and promote local compaction. In vivo, this will impose constraints on nucleosomal mobility, consistent with the proposed transcription-inhibitory role of MeCP2. While the persistence length of DNA is much greater than that of the nucleosomal linker DNA, force extension studies have shown that in the context of chromatin, DNA persistence length is greatly reduced, partly due to the charge neutralization of the core nucleosomal sequence and partly due to transient contacts between the highly charged histone N-terminal tails and linker DNA (18). MeCP2, like H1, binds near the nucleosomal dyad and promotes a zigzag fiber geometry. However, with MeCP2, this effect is more pronounced, possibly due to the combination of increased positive-charge density compared to the level for H1, multiple DNA/chromatin binding domains (26), and interaction with the N-terminal region of histone H3 (50). The effect will also be enhanced by the dimeric binding mode of MeCP2.
Earlier work has shown that high salt concentrations (particularly divalent cations) can induce zigzag architecture in in vitro reconstituted arrays (20, 53). The similarity in the chromatin architectures induced by H1, MeCP2, and mono/divalent cations suggests that at a fundamental level, the geometry of the 30-nm chromatin fiber is largely induced by charge neutralization of linker DNA (17). Indeed, H1 binding reduces the electrostatic free energy of DNA by displacing bound cations, which also explains why native (H1-containing) chromatin is more prone to salt-induced folding than chromatin depleted of H1 (7). It has been suggested that chromatin exists in a dynamic equilibrium of folded and unfolded states, where the primary determinants of folding are screening of DNA charges (3) and internucleosomal interactions (27). Our new results suggest that MeCP2 has a role in modulating this equilibrium.
Importantly, our studies on the competition between MeCP2 and H1 reveal that MeCP2 displaces H1 from methylated nucleosomes more strongly than vice versa (Fig. 4a, b, and c). Although the amounts of H1 differ between cell types (22, 23, 70) and the expression level of MeCP2 depends on both cell type and cell age (21), in general H1 is more abundant than MeCP2. Moreover, the number of MeCP2 molecules appears to be at least an order of magnitude lower than the number of methyl CpGs in a typical diploid nucleus (48). Under equilibrium conditions, MeCP2 will be distributed between unmethylated and methylated sites, thus leaving a large fraction of methylated CpGs vacant. H1 is likely to compete more successfully with MeCP2 bound to unmethylated sites and thus lead to relative enrichment of MeCP2 at methylated sites. Our data showing MeCP2 depletion from PHC upon blocking of DNA methylation (Fig. 8c and d), together with the accelerated chromatin binding kinetics under these conditions (Fig. 8e and Table 3), clearly demonstrate that stereospecific binding to methyl-CpG accounts for the increased residence time of MeCP2 in PHC and also for the fraction tightly bound to chromatin. This further suggests that in methylase deficient cells (49), increases in the levels of H1 are likely to result in more-efficient removal of MeCP2 from chromatin than increases in cells having an intact methylome.
In contrast to the low levels in most nuclei, MeCP2 abundance in mature neuronal nuclei closely approximates the number of nucleosomes as well as the number of methyl-CpGs (60). Interestingly, a recent report indicates that histone H1, which is present at a ratio of approximately one molecule per two nucleosomes in neurons, undergoes a 2-fold elevation in expression level in cells lacking MeCP2. The suggestion that MeCP2 could potentially act as an alternate linker histone in neurons (60) is consistent with our data on the similarities between MeCp2 and H1 binding to chromatin. We suggest that a complex competitive equilibrium between MeCP2 and H1 for nucleosome/chromatin binding exists in vitro and in vivo. In addition to H1, other competing chromatin binding proteins will also affect this H1-MeCP2 dynamic equilibrium (11, 12).
Thus, our observations underscore the importance of understanding chromatin binding by nuclear proteins in the context of a shifting equilibrium where the relative abundance of each component will not only effect its cognate targets but also the targets of its competitors (68). The pleiotropic effects of MeCP2 mutations, ranging from neurodevelopmental symptoms in RTT to the association of MeCP2 with various types of cancer in other cells (26), may be understood in the context of these competitive equilibria.
Acknowledgments
We thank Joseph Wall and Martha Simon of the Brookhaven National Laboratory STEM Facility for carrying out the mass measurements and assisting with the data analysis, David Brown, University of Mississippi Medical Center, Jackson, MS, for the H10-ACA plasmid and the H10-GFP BALB/c 3T3 cell line, Carolyn Schanen, Department of Biological Sciences, University of Delaware, Newark, DE, and Asmita Kumar, Nemours Biomedical Research, Alfred I. duPont Hospital for Children, Wilmington, DE, for BALB/c 3T3 cells expressing MeCP2-GFP, Christina Cardoso, Max Delbruck Center for Molecular Medicine, Germany, for the MeCP2-mRFP overexpression plasmid, Sergei Grigoryev, Pennsylvania State University School of Medicine, Hershey, PA, for the 601-1 and 601-4 plasmids, Tom Misteli, National Cancer Institute, NIH, Bethesda, MD, for CHO cells expressing HP1α-GFP, and Yuri L. Lyubchenko, Department of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE, for AFM facilities and advice.
This work was supported by the Rett Syndrome Research Foundation and NIH GM070897 (C.L.W.), NSF 0701892 (Yuri L. Lyubchenko), and the Nebraska Research Initiative (L.S.S. and Yuri L. Lyubchenko).
Footnotes
Published ahead of print on 2 August 2010.
REFERENCES
- 1.Adams, V. H., S. J. McBryant, P. A. Wade, C. L. Woodcock, and J. C. Hansen. 2007. Intrinsic disorder and autonomous region function in the multifunctional nuclear protein, MeCP2. J. Biol. Chem. 282:15057-15064. [DOI] [PubMed] [Google Scholar]
- 2.Amir, R. E., I. B. den Veyber, M. Wan, C. Q. Tran, U. Francke, and H. Y. Zoghbi. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23:185-188. [DOI] [PubMed] [Google Scholar]
- 3.Arya, G., and T. Schlick. 2006. Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci. U. S. A. 103:16236-16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bednar, J., R. A. Horowitz, J. Dubochet, and C. L. Woodcock. 1995. Chromatin conformation and salt-induced compaction: three-dimensional structural information from cryoelectron microscopy. J. Cell Biol. 131:1365-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bednar, J., R. A. Horowitz, S. A. Grigoryev, L. M. Carruthers, J. C. Hansen, A. J. Koster, and C. L. Woodcock. 1998. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. U. S. A. 95:14173-14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bird, A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16:6-21. [DOI] [PubMed] [Google Scholar]
- 7.Blank, T. A., and P. B. Becker. 1995. Electrostatic mechanism of nucleosome spacing. J. Mol. Biol. 252:305-313. [DOI] [PubMed] [Google Scholar]
- 8.Boyes, J., and A. Bird. 1991. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell 64:1123-1134. [DOI] [PubMed] [Google Scholar]
- 9.Bulinski, J. C., D. J. Odde, B. J. Howell, T. D. Salmon, and C. M. Waterman-Storer. 2001. Rapid dynamics of the microtubule binding of ensconsin in vivo. J. Cell Sci. 114:3885-3897. [DOI] [PubMed] [Google Scholar]
- 10.Carruthers, L. M., J. Bednar, C. L. Woodcock, and J. C. Hansen. 1998. Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry 37:14776-14787. [DOI] [PubMed] [Google Scholar]
- 11.Catez, F., D. T. Brown, T. Misteli, and M. Bustin. 2002. Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep. 3:760-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catez, F., T. Ueda, and M. Bustin. 2006. Determinants of histone H1 mobility and chromatin binding in living cells. Nat. Struct. Mol. Biol. 13:305-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cavaluzzi, M. J., and P. N. Borer. 2004. Revised UV extinction coefficients for nucleoside-5′-monophosphates and unpaired DNA and RNA. Nucleic Acids Res. 32:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chadwick, L. H., and P. A. Wade. 2007. MeCP2 in Rett syndrome: transcriptional repressor or chromatin architectural protein. Curr. Opin. Genet. Dev. 17:1-5. [DOI] [PubMed] [Google Scholar]
- 15.Chen, W. G., Q. Chang, Y. Lin, A. Meissner, A. E. West, E. C. Griffith, R. Jaenisch, and M. E. Greenberg. 2003. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302:885-889. [DOI] [PubMed] [Google Scholar]
- 16.Cheutin, T., A. J. McNairn, T. Jenuwein, D. M. Gilbert, P. B. Singh, and T. Misteli. 2003. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299:721-725. [DOI] [PubMed] [Google Scholar]
- 17.Clark, D. J., and T. Kimura. 1990. Electrostatic mechanism of chromatin folding. J. Mol. Biol. 211:883-896. [DOI] [PubMed] [Google Scholar]
- 18.Cui, Y., and C. Bustamante. 2000. Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc. Natl. Acad. Sci. U. S. A. 97:127-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhasarathy, A., and P. A. Wade. 2008. The MBD protein family-reading an epigenetic mark? Mutat. Res. 647:39-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dorigo, B., T. Schalch, A. Kulangara, S. Duda, R. R. Schroeder, and T. J. Richmond. 2004. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 306:1571-1573. [DOI] [PubMed] [Google Scholar]
- 21.Dragich, J. M., Y. H. Kim, A. P. Arnold, and N. C. Schanen. 2007. Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. J. Comp. Neurol. 501:526-542. [DOI] [PubMed] [Google Scholar]
- 22.Fan, Y., T. Nikitina, E. M. Morin-Kensicki, J. Zhao, T. R. Magnuson, C. L. Woodcock, and A. I. Skoultchi. 2003. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol. Cell. Biol. 23:4559-4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan, Y., T. Nikitina, J. Zhao, T. J. Fleury, R. Bhattacharyya, E. E. Bouhassira, A. Stein, C. L. Woodcock, and A. I. Skoultchi. 2005. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123:1199-1212. [DOI] [PubMed] [Google Scholar]
- 24.Georgel, P. T., R. A. Horowitz-Scherer, N. Adkins, C. L. Woodcock, P. A. Wade, and J. C. Hansen. 2003. Chromatin compaction by human MeCP2—assembly of novel secondary structures in the absence of DNA methylation. J. Biol. Chem. 278:32181-32188. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh, R. P., R. A. Horowitz-Scherer, T. Nikitina, L. M. Gierasch, and C. L. Woodcock. 2008. Rett syndrome-causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J. Biol. Chem. 283:20523-20534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh, R. P., T. Nikitina, R. A. Horowitz-Scherer, L. M. Gierasch, V. N. Uversky, K. Hite, J. C. Hansen, and C. L. Woodcock. 2010. Unique physical properties and interactions of the domains of methylated DNA binding protein 2 (MeCP2). Biochemistry 49:4395-4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grigoryev, S. A., G. Arya, S. Correll, C. L. Woodcock, and T. Schlick. 2009. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl. Acad. Sci. U. S. A. 106:13317-13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho, K. L., I. W. McNae, L. Schmiedeberg, R. J. Klose, A. P. Bird, and M. D. Walkinshaw. 2008. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol. Cell 29:525-531. [DOI] [PubMed] [Google Scholar]
- 29.Horike, S., S. Cai, M. Miyano, J. F. Cheng, and T. Kohwi-Shigematsu. 2004. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37:31-40. [DOI] [PubMed] [Google Scholar]
- 30.Kar, S. R., J. Lebowitz, S. Blume, K. B. Taylor, and L. M. Hall. 2001. SmtB-DNA and protein-protein interactions in the formation of the cyanobacterial metallothionein repression complex: Zn2+ does not dissociate the protein-DNA complex in vitro. Biochemistry 40:13378-13389. [DOI] [PubMed] [Google Scholar]
- 31.Klose, R. J., S. A. Sarraf, L. Schmiedeberg, S. M. McDermott, I. Stancheva, and A. P. Bird. 2005. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 19:667-678. [DOI] [PubMed] [Google Scholar]
- 32.Kumar. A., S. Kamboj, B. M. Malone, S. Kudo, J. L. Twiss, K. Czymmek, J. M. LaSalle, and N. C. Schanen. 2008. Analysis of protein regions and Rett syndrome mutations indicate that multiple regions influence chromatin-binding dynamics of the chromatin-associated protein MECP2 in vivo. J. Cell Sci. 121:1128-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p. 90-125. In S. E. Harding, A. J. Rowe, and J. C. Horton (ed.), Analytical ultracentrifugation in biochemistry and polymer science. Royal Society of Chemistry, Cambridge, United Kingdom.
- 34.Lebowitz, J., M. S. Lewis, and P. Schuck. 2002. Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11:2067-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lefstin, J. A., and K. R. Yamamoto. 1998. Allosteric effects of DNA on transcriptional regulators. Nature 392:885-888. [DOI] [PubMed] [Google Scholar]
- 36.Lever, M. A., J. P. Th'ng, X. Sun, and M. J. Hendzel. 2000. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature 408:873-876. [DOI] [PubMed] [Google Scholar]
- 37.Levine, A., G. L. Cantoni, and A. Razin. 1991. Inhibition of promoter activity by methylation: possible involvement of protein mediators. Proc. Natl. Acad. Sci. U. S. A. 88:6515-6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu, X., and J. C. Hansen. 2004. Identification of specific functional subdomains within the linker histone H10 C-terminal domain. J. Biol. Chem. 279:8701-8707. [DOI] [PubMed] [Google Scholar]
- 39.Luger, K., A. W. Mäder, R. K. Richmond, D. F. Sargent, and T. J. Richmond. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251-260. [DOI] [PubMed] [Google Scholar]
- 40.Lushnikov, A. Y., V. N. Potaman, E. A. Oussatcheva, R. R. Sinden, and Y. L. Lyubchenko. 2006. DNA strand arrangement within the SfiI-DNA complex: atomic force microscopy analysis. Biochemistry 45:152-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyubchenko, Y. L., and L. S. Shlyakhtenko. 2009. AFM for analysis of structure and dynamics of DNA and protein-DNA complexes. Methods 47:206-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyubchenko, Y. L., L. S. Shlyakhtenko, and A. A. Gall. 2009. Atomic force microscopy imaging and probing of DNA, proteins, and protein DNA complexes: silatrane surface chemistry. Methods Mol. Biol. 543:337-351. [DOI] [PubMed] [Google Scholar]
- 43.Ma, Y., and M. R. Lieber. 2001. DNA length-dependent cooperative interactions in the binding of Ku to DNA. Biochemistry 40:9638-9646. [DOI] [PubMed] [Google Scholar]
- 44.Martinowich, K., D. Hattori, H. Wu, S. Fouse, F. He, Y. Hu, G. Fan, and Y. E. Sun. 2003. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302:890-893. [DOI] [PubMed] [Google Scholar]
- 45.Meijsing, S. H., M. A. Pufall, A. Y. So, D. L. Bates, L. Chen, and K. R. Yamamoto. 2009. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 324:407-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Misteli, T., A. Gunjan, R. Hock, M. Bustin, and D. T. Brown. 2000. Dynamic binding of histone H1 to chromatin in living cells. Nature 408:877-881. [DOI] [PubMed] [Google Scholar]
- 47.Mohan, A., C. J. Oldfield, P. Radivojac, V. Vacic, M. S. Cortese, A. K. Dunker, and V. N. Uversky. 2006. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 362:1043-1059. [DOI] [PubMed] [Google Scholar]
- 48.Nan, X., F. J. Campoy, and A. Bird. 1997. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88:471-481. [DOI] [PubMed] [Google Scholar]
- 49.Nan, X., P. Tate, E. Li, and A. Bird. 1996. DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell. Biol. 16:414-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nikitina, T. N., R. P. Ghosh, R. A. Horowitz-Scherer, J. C. Hansen, S. A. Grigoryev, and C. L. Woodcock. 2007. MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 282:28237-28245. [DOI] [PubMed] [Google Scholar]
- 51.Nikitina, T. N., X. Shi, R. P. Ghosh, R. A. Horowitz-Scherer, J. C. Hansen, and C. L. Woodcock. 2007. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol. Cell. Biol. 27:864-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phair, R. D., and T. Misteli. 2000. High mobility of proteins in the mammalian cell nucleus. Nature 404:604-609. [DOI] [PubMed] [Google Scholar]
- 53.Schalch, T., S. Duda, D. F. Sargent, and T. J. Richmond. 2005. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 436:138-141. [DOI] [PubMed] [Google Scholar]
- 54.Schuck, P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78:1606-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schuck, P. 2003. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320:104-124. [DOI] [PubMed] [Google Scholar]
- 56.Scully, K. M., E. M. Jacobson, K. Jepsen, V. Lunyak, H. Viadiu, C. Carrière, D. W. Rose, F. Hooshmand, A. K. Aggarwal, and M. G. Rosenfeld. 2000. Allosteric effects of Pit-1 DNA sites on long-term repression in cell type specification. Science 290:1127-1131. [DOI] [PubMed] [Google Scholar]
- 57.Shabazian, M. D., and H. Zoghbi. 2002. Rett syndrome and MeCP2: linking epigenetics and neuronal function. Am. J. Hum. Genet. 71:1259-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shlyakhtenko, L. S., J. Gilmore, A. Portillo, G. Tamulaitis, V. Siksnys, and Y. L. Lyubchenko. 2007. Direct visualization of the EcoRII-DNA triple synaptic complex by atomic force microscopy. Biochemistry 46:11128-11136. [DOI] [PubMed] [Google Scholar]
- 59.Shlyakhtenko, L. S., A. Y. Lushnikov, and Y. L. Lyubchenko. 2009. Dynamics of nucleosomes revealed by time-lapse atomic force microscopy. Biochemistry 48:7842-7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skene, P. J., R. S. Illingworth, S. Webb, A. R. Kerr, K. D. James, D. J. Turner, R. Andrews, and A. P. Bird. 2010. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37:457-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith, C. L., R. Horowitz-Scherer, J. F. Flanagan, C. L. Woodcock, and C. L. Peterson. 2003. Structural analysis of the yeast SWI/SNF chromatin remodeling complex. Nat. Struct. Mol. Biol. 10:141-145. [DOI] [PubMed] [Google Scholar]
- 62.Sprague, B. L., R. L. Pego, D. A. Stavreva., and J. G. McNally. 2004. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 86:3473-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thåström, A., L. M. Bingham, and J. Widom. 2004. Nucleosomal locations of dominant DNA sequence motifs for histone-DNA interactions and nucleosome positioning. J. Mol. Biol. 338:695-709. [DOI] [PubMed] [Google Scholar]
- 64.Ucci, J. W., and J. L. Cole. 2004. Global analysis of non-specific protein-nucleic interactions by sedimentation equilibrium. Biophys. Chem. 108:127-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vistica, J., J. Dam., A. Balbo, E. Yikilmaz, R. A. Mariuzza, T. A. Rouault, and P. Schuck. 2004. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326:234-256. [DOI] [PubMed] [Google Scholar]
- 66.Wade, P. A. 2004. Dynamic regulation of DNA methylation coupled transcriptional repression: BDNF regulation by MeCP2. Bioessays 26:217-220. [DOI] [PubMed] [Google Scholar]
- 67.Wall, J. S., J. F. Hainfeld, and M. Simon. 1998. STEM of nuclear structures. Methods Cell Biol. 53:139-164. [DOI] [PubMed] [Google Scholar]
- 68.Wasson, T., and A. J. Hartemink. 2009. An ensemble model of competitive multi-factor binding of the genome. Genome Res. 19:2101-2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weinberg, R. L., D. B. Veprintsev, and A. R. Fersht. 2004. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 341:1145-1159. [DOI] [PubMed] [Google Scholar]
- 70.Woodcock, C. L., A. I. Skoultchi, and Y. Fan. 2006. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 14:17-25. [DOI] [PubMed] [Google Scholar]
- 71.Yasui, D. H., S. Peddada, M. C. Bieda, R. O. Vallero, A. Hogart, R. P. Nagarajan, K. N. Thatcher, P. J. Farnham, and J. M. Lasalle. 2007. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. U. S. A. 104:19416-19421. [DOI] [PMC free article] [PubMed] [Google Scholar]