

Abstract
The association between HLA-B*2705 and the immune control of human immunodeficiency virus type 1 (HIV-1) has previously been linked to the targeting of the HLA-B*2705-restricted Gag epitope KRWIILGLNK (KK10) by CD8+ T cells. In order to better define the mechanisms of the HLA-B*2705 immune control of HIV, we first characterized the CD8+ T-cell responses of nine highly active antiretroviral therapy (HAART)-naïve B*2705-positive subjects. Unexpectedly, we observed a strong response to an HLA-B*2705-restricted Pol epitope, KRKGGIGGY (KY9), in 8/9 subjects. The magnitude of the KY9 response was only marginally lower than that of the KK10-specific response (median, 695 versus 867 spot-forming cells [SFC]/million peripheral blood mononuclear cells [PBMCs]; not significant [NS]), and viral escape mutants were observed in both KY9 and KK10, resulting from selection pressure driven by the respective CD8+ T-cell response. By comparing inhibitions of viral replication by CD8+ T cells specific for the Gag KK10, Pol KY9, and Vpr VL9 HLA-B*2705-restricted epitopes, we observed a consistent hierarchy of antiviral efficacy (Gag KK10 > Pol KY9 > Vpr VL9). This hierarchy was associated with early recognition of HIV-1-infected cells, within 6 h of infection, by KK10- and KY9-specific CD8+ T cells but not until 18 h postinfection by VL9-specific CD8+ T cells. There was no association between antiviral efficacy and proliferative capacity, cytotoxicity, polyfunctionality, or T-cell receptor (TCR) avidity. These data are consistent with previous studies indicating an important role for the B*2705-Gag KK10 response in the control of HIV but also suggest a previously unrecognized role played by the subdominant Pol-specific KY9 response in HLA-B*2705-mediated control of HIV and that the recognition of HIV-infected cells by CD8+ T cells early in the viral life cycle may be important for viral containment in HIV-infected individuals.
Current human immunodeficiency virus (HIV) vaccine strategies are focused on emulating the protective effect observed for HIV-infected individuals carrying alleles such as B*2705 by inducing the virus-specific CD8+ T-cell responses that are thought to be responsible for delaying or preventing disease progression. Understanding why such alleles confer protection facilitates a rational approach to vaccine design. It has been hypothesized that the slow progression to AIDS exhibited by HLA-B*2705-positive (HLA-B*2705+) HIV-infected individuals is due to the immunodominant B*27-restricted CD8+ T-cell response toward the p24 Gag epitope KRWIILGLNK (KK10) (Gag residues 263 to 272). Escape from this epitope typically occurs late in infection and is associated with rapid progression to AIDS (14, 16). The commonly selected mutation R264K abrogates CD8+ T-cell recognition but also confers a substantial fitness cost to the virus, and the selection of compensatory mutations is required to restore viral replicative capacity (19, 29, 30). This has prompted the hypothesis that CD8+ T-cell responses that can drive escape mutations that reduce viral fitness are a contributing factor in the immune control of HIV, either by promoting the outgrowth of a viral quasispecies with a lower replicative capacity or by delaying the selection of escape mutations, both of which may slow the onset of AIDS (11, 21, 25).
To better understand how CD8+ T cells can be most effective against HIV, recent studies have directly assessed the antiviral activity of CD8+ T cells via the viral suppression of HIV-infected CD4+ T cells during coculture. Such studies indicated that Gag-specific CD8+ T cells have a higher potency for viral suppression than Env-specific CD8+ T cells (10), supporting previous data indicating that broad CD8+ T-cell targeting of Gag epitopes was associated a with lower viral set point and, hence, slower progression to AIDS (20). A recent study of simian immunodeficiency virus (SIV) suggested that the protective effect of Gag-specific CD8+ T cells is mediated by the early presentation of Gag epitopes, processed from the viral Gag protein from incoming virions during infection, which can sensitize target cells for lysis by Gag-specific CD8+ T cells within 6 h of infection (26, 27). In addition, it was proposed previously that the ability of CD8+ T cells to secrete multiple cytokines may also be an important correlate of immune protection (6), and a further recent study demonstrated a more polyfunctional cytokine profile of Gag-specific B*2705-KK10 CD8+ T-cell responses than those of other HIV-specific CD8+ T-cell responses (1). The ability of CD8+ T cells to proliferate in response to the cognate epitope peptide has also been associated with immune control (1, 12). Other studies demonstrated the importance of lytic granule loading of CD8+ T cells for the effective elimination of HIV-infected cells (6, 22). However, the induction of a Gag KK10-specific CD8+ T-cell vaccine response in a B*2705-positive vaccinee did not protect against rapid progression following subsequent HIV-1 infection (5). This anecdotal case suggests the possibility that HLA-B*2705-associated immune control of HIV-1 may not be dependent on the Gag KK10-specific CD8+ T-cell response alone.
Since current vaccine strategies hope to induce a protective effect, such as that observed for HLA-B*2705+ HIV-infected individuals, the study of the functional and phenotypic characteristics of B*2705-specific CD8+ T cells provides an opportunity to redefine the proposed correlates of immune protection essential for rational vaccine design. In this study we analyze three different specificities of HLA-B*2705-restricted CD8+ T cells from chronically HIV-infected individuals in order to directly compare antiviral activity with potential correlates of immune protection, including the kinetics of viral inhibition, cytokine profile, granzyme production, proliferative capacity, and cytotoxicity.
MATERIALS AND METHODS
Study subjects.
Nine antiretroviral therapy-naïve HLA-B*2705-positive study subjects with chronic HIV infection were recruited from the Royal Berkshire Hospital, Reading, United Kingdom; Northampton General Hospital, Northampton, United Kingdom; Wycombe Hospital, High Wycombe, United Kingdom; and Churchill Hospital, Oxford, United Kingdom. This study was approved by the Oxford Research Ethics Committee, and all subjects gave formal written consent. High-resolution HLA typing was undertaken with genomic DNA. We examined HLA and HIV integrase sequence data from 582 chronically HIV-infected, antiretroviral-naïve individuals from the British Columbia HAART Observational Medical Evaluation and Research (HOMER) cohort (97.5% HIV-1 subtype B) (9). In addition, integrase sequences were determined for nine HLA B*27+ HIV subtype B-infected subjects who were recruited during acute/early infection and were analyzed for incidence and the time course of escape. These patients were recruited a median of 72 days following the estimated date of infection, and the median follow-up time was 36 months (8).
ELISPOT assay.
Gamma interferon (IFN-γ) enzyme-linked immunospot (ELISPOT) assays were performed as previously described (3), using optimally defined epitopes and 18-mer overlapping peptides (OLP) with the number of input cells/well ranging from 30,000 to 100,000. The number of specific spot-forming cells (SFC) was calculated by subtracting the mean number of spots in the negative-control wells from the number of spots counted in each well. The magnitude of epitope-specific responses was calculated as SFC per million cells. The B*2705 peptides screened were as follows: KK10 (KRWIILGLNK) for Gag p24, GY10 (GRRGWEALKY) for Env gp160, VL9 (VRHFPRIWL) for Vpr, KV10 (KRQDILDLWV) for Nef, IK9 (IRLRPGGKK) for Gag p17, and KY9 (KRKGGIGGY) for Pol integrase (Int).
HLA restriction.
Determination of HLA restriction was performed as previously described (18). Briefly, a panel of Epstein-Barr virus (EBV)-transformed B-cell lines (BCLs) was matched through individual HLA class I alleles with the effector CD8+ T cells being tested. The peptide of interest and a control mock peptide were independently incubated with each BCL for 60 min at 37°C, washed four times in phosphate-buffered saline (PBS), and incubated with the effector cells for 5 h in the presence of brefeldin A (10 μg/ml). Cells were stained for surface and intracellular markers using antibodies against CD8 (Alexa Fluor 750), CD3 (PE-Texas red), CD19 (Pacific Blue), and macrophage inflammatory protein 1β (MIP-1β) (fluorescein isothiocyanate [FITC]) and then acquired by fluorescence-activated cell sorter (FACS) analysis (BD LSRII). The optimal epitope was defined by an IFN-γ ELISPOT assay using truncations of the predicted epitope as previously described (18).
KY9 escape variant recognition.
B*2705-associated viral polymorphisms located within the Pol epitope KY9 were analyzed for their effect on epitope recognition by KY9-specific CD8+ T cells. HLA-matched BCLs were peptide pulsed for 1 h with log10 dilutions of peptide. BCLs were then washed four times with PBS and incubated with effector CD8+ T cells at a ratio of 1:1 in the presence of CD107a antibodies (phycoerythrin [PE]), brefeldin A (10 μg/ml), and Golgi Stop (BD Golgi Stop protein transport inhibitor) for 6.5 h at 37°C in a 5% CO2 incubator. Cells were then harvested and stained for intracellular and extracellular antibodies, including MIP-1β (FITC), CD8 (Alexa Fluor 750), CD3 (Alexa Fluor 700), CD19 (Pacific Blue), and the Live/Dead marker (Pacific Blue) and then acquired immediately by FACS analysis (BD LSRII). This was performed using KY9-specific CD8+ T cells from three different individuals.
T-cell receptor (TCR) avidity and MHC-I peptide binding affinity.
The functional avidity of epitope-specific CD8+ T cells was investigated by log10 serial peptide titrations in an ELISPOT assay and defined as the peptide concentration that resulted in 50% maximum (50% inhibitory concentration [IC50]) IFN-γ production, calculated with Graphpad Prism software, version 5.0a. Major histocompatibility complex class I (MHC-I) peptide binding affinity was quantified as previously described (17).
Cell lines.
The HIV-1-permissive U937 cell line transfected with the HLA-B*2705 gene was a gift from Paul Bowness (Oxford). The cells were cultured in RPMI medium supplemented with 10% fetal calf serum (FCS) (HyClone), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.5 mg/ml Geneticin and split every 2 to 3 days. Tetramer-enriched CD8+ T-cell lines were cultured by sorting tetramer-positive cells from peripheral blood mononuclear cells (PBMCs) by magnetic separation using anti-PE microbeads (Miltenyi Biotec). The flowthrough fraction was peptide pulsed (20 μg/ml) for 1 h at 37°C, irradiated (30 Gy), washed once in 1× PBS, and cultured with the tetramer-positive fraction at a ratio of 100:1 in H10-10 medium (RMPI medium supplemented with 10% human serum, 10% natural T-cell growth factor [TCGF; Helvetica Healthcare], 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin). Every 10 days following the initial setup, the tetramer-positive cells were fed by using peptide-pulsed, irradiated, autologous PBMCs or BCLs at a ratio of 1:1 and irradiated feeder PBMCs from three healthy HIV-seronegative donors. Tetramer-positive CD8+ T-cell lines of >97% specificity were consistently observed. Purified PBMCs were isolated from whole blood of HIV-infected individuals by Ficoll-Hypaque density gradient centrifugation and cryopreserved until required.
Virus preparation.
Viral stocks were generated by electroporation cotransfection of a plasmid carrying the 5′ half of the HIV-1 NL43 strain (p83-2) and a plasmid carrying the 3′ half of the genome of HIV-1 NL43 (p83-10eGFP) (25, 33) in MT4 cells. The cell-free supernatant was harvested before peak infection (∼85%), as measured by the percentage of green fluorescent protein-positive (GFP+) cells, and stored at −80°C. The 50% tissue culture infective dose (TCID50) was determined for each viral stock on MT4 cells by using a method described previously by Sacha et al. (27).
Twenty-four-hour viral inhibition assay.
Viral inhibition during the first 24 h after infection was assessed using a previously described method (27). Viral stocks of HIV-1 were purified through a 20% sucrose cushion immediately prior to use. The purified virus was magnetized with ViroMag beads (Magnetofection ViroMag R/L; Ozbiosciences), and U937 target cells were synchronously infected, according to the manufacturer's instructions, at a multiplicity of infection (MOI) of 1. Briefly, U937 target cells were incubated with the magnetized virus for 15 min in the presence of a magnetic field and then washed three times in PBS. The synchronously infected U937 target cells (1 × 106 cells) were then cocultured with tetramer-enriched CD8+ T cells (>97% specific) at an effector/target (E/T) ratio of 1:4 in ∼1 ml of medium at 37°C. At specific time points, cells were harvested, stained with antibodies specific for CD4 (allophycocyanin [APC]) and CD8 (peridinin chlorophyll protein [PerCP]), and then fixed in 2% formaldehyde. Cells from all time points were processed together to stain for intracellular Gag p24 (PE). Cell data were acquired immediately by FACS analysis (BD FACSCalibur). Each CD8+ T-cell effector tested was run in quadruplicate and repeated at least twice in independent experiments.
Seven-day viral inhibition assay.
Viral inhibition was assessed by using a coculture 7- to 10-day assay as previously described (35). Briefly, U937 target cells were infected with HIV-1 at an MOI of 0.02 TCID50/cell for 4 h at 37°C, washed four times with PBS, and resuspended in H10-10 medium (RMPI medium supplemented with 10% human serum, 10% [Helvetica Healthcare], 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin) and plated at 1 × 106 cells per well in a 24-well plate. Effector cells were then added at a ratio of 1:100 or as otherwise specified to a total of 2 ml of medium. On days 1, 3, 5, and 7, 1 ml of coculture supernatant was removed and replaced with fresh medium. The removed supernatant was stored at −80°C for later p24 antigen quantitation by a standard quantitative enzyme-linked immunosorbent assay (ELISA) (commercial kit) (Innotest HIV antigen monoclonal antibody [MAb]; Innogenetics). Each well was run in triplicate. The use of modified U937 monocyte target cells in the virus inhibition assays was done according to a protocol described previously (35), with the MOI and target cell numbers selected to provide the optimal viral growth rate and the E/T ratio selected to maximally resolve differential antiviral effects of the respective antigen-specific CD8+ T cells.
CFSE proliferation assay.
CFSE (carboxyfluorescein succinimidyl ester) proliferation assays were performed on tetramer-specific CD8+ T cells using a previously described method (34). Briefly, cells were resuspended in prewarmed PBS with 0.1% bovine serum albumin (BSA), and CFSE was added to a final concentration of 10 μM. Cells were incubated at 37°C for 10 min, quenched with 10 ml ice-cold R10 medium, incubated for 5 min on ice, and then washed three times in fresh R10 medium. Autologous BCLs were peptide pulsed (20 μg/ml) with specific and mock peptides independently for 1 h at 37°C, irradiated (30 Gy), washed once in 1× PBS, and cocultured with the CFSE-labeled cells at a ratio of 1:1 in H10 medium. The cultures were fed twice during the following week, and on day 7, the cells were harvested and stained with surface antibodies against CD38 (APC), CD3 (Alexa Fluor 700), CD8 (Alexa Fluor 750), CD19 (Pacific Blue), and the Live/Dead marker (Pacific Blue). Cell data were immediately acquired by FACS analysis (BD LSRII).
Cell stimulation/preparation for polyfunctional assessment and effector phenotype assessment.
PBMCs from HIV-infected B*2705-positive subjects were thawed and rested overnight at 37°C in R10 medium. For stimulation for polyfunctional assessments, 0.5 × 106 to 1 × 106 PBMCs or tetramer-enriched CD8+ T cells were incubated in 200 μl R10 medium in the presence of 2 μg/ml of specific peptide, 15 μl of anti-CD107a antibodies, brefeldin A (10 μg/ml), Golgi Stop (BD Golgi Stop protein transport inhibitor), CD49d, and CD28 (1 μg/ml) (BD Biosciences) for 6.5 h at 37°C in a 5% CO2 incubator. Negative controls were obtained in the absence of peptide. For effector phenotype assessments, between 1 × 106 and 2 × 106 PBMCs or tetramer-enriched CD8+ T cells were stained with pretitrated concentrations of tetramers (conjugated to APC) for 20 min at room temperature (RT). Negative controls were obtained by using mismatched tetramers and isotype controls. Cells were stained with antibodies for surface markers and intracellular markers and then immediately acquired by FACS analysis (BD LSRII).
Flow cytometric cytotoxicity assay.
Matched (HLA-A 0101/0301, HLA-B 2705/5701, and HLA-C 0202/0602) and mismatched (HLA-A 3001/0201, HLA-B 4201/3501, and HLA-C 0401/1701) B-cell targets were labeled with Cell Tracker violet (catalog number C10094; Invitrogen) to final concentrations of 5 μM and 0.32 μM, respectively, according to the manufacturer's protocol. Briefly, cells were resuspended in R10 medium supplemented with Cell Tracker violet dye and incubated at 37°C for 30 min. The supernatant was then replaced with fresh R10 medium and incubated for a further 30 min at 37°C. Finally, the cells were washed twice in R10 medium and pulsed with 50 μg/ml peptide for 1 h at 37°C in 5% CO2. After incubation, B-cell target cells were washed four times in R10 medium, cocultured in triplicate with effector tetramer-specific CD8+ T cells titrated at E/T ratios ranging from 2:1 to 1:40, and incubated for 5 h at 37°C in 5% CO2. After incubation, cells were washed with PBS and stained with anti-human CD3 (ECD), CD8 (Alexa Fluor750), and CD19 (APC) surface antibodies and immediately acquired by FACS analysis (BD LSRII). Using FlowJo software, version 8.8.6, target cells were gated in the Pacific Blue channel for high (5 μM) and intermediate (0.32 μM) violet to establish a ratio between HLA-matched and HLA-mismatched target cells by real cell counts. CD8+ effector cells were gated separately in the APC-Cy7 channel to count the actual number of effecter cells added and thereby obtain a more precise ratio of effector cells to target cells present within the same sample. Wells with no effector CD8+ T cells added served as a control for the expected ratio (ER) between HLA-matched and HLA-mismatched target cells. All E/T ratios were determined in triplicate. Percent specific killing was calculated by using the following formula: percent killing = {[(ER × number of mismatched cells) − number of matched cells]/(ER × number of mismatched cells)} × 100%.
Antibody staining.
Cell surface staining with antibodies for extracellular markers was performed at room temperature for 20 min. The cells were washed in PBS and then fixed in 2% paraformaldehyde or Fix/Perm buffer (BD Biosciences). Intracellular antibody staining was then performed at room temperature for 20 min by using the saponin-containing buffer Perm/Wash (BD biosciences). Cells were washed in PBS and resuspended in a stabilizing fixative (BD biosciences) before acquisition by FACS analysis.
Flow cytometry.
Soluble biotinylated monomeric peptide MHC-I (pMHCI) proteins were produced as described above. Multimerization was performed by the gradual addition of Extravidin-R-PE (Sigma) or streptavidin-APC (Caltag Laboratories) to aliquots to a total pMHCI/streptavidin molar ratio of 4:1. Specifically, pMHCI tetramers for B*2705-KK10, B*2705-KY9, and B*2705-VL9 were synthesized and tested on CBA beads (BD) coated with anti-HLA antibodies (gift from Paul Klenerman). Directly conjugated and unconjugated antibodies used were CD19 (Pacific Blue), human CD3 (Pacific Orange), mouse anti-human CD4 (Qdot 605) clone 53.3, mouse anti-human CD8 (Qdot 655), and a Live/Dead fixable violet dead cell stain kit (all from Invitrogen); CD14 (Pacific Blue) (Caltag Laboratories); anti-human CD8a (APC-Alexa Fluor 750) clone RPA-T8 and anti-human CD127 (PE-Cy5) clone eBioRDR5 (from eBioscience); anti-human interleukin-2 (IL-2) (APC), mouse anti-human granzyme B (FITC), mouse anti-human CD107a (PE), and tumor necrosis factor (TNF) (Alex Fluor 700) clone MAb11 (all from BD biosciences); CD3 (ECD) (from Beckman Coulter); IFN (PE-Cy7) clone 45.B3, CD3 (Alexa Fluor 700) clone UCHT1, CD107 (PE), and mouse IgG1 isotype control (FITC) (all from BD Pharmingen); mouse IgG1 isotype control (PE) and anti-hMIP1b/CCL4 (FITC) (from R&D Systems); CD38 (APC), CD8 (PerCP), and CD4 (APC) (SK3) (from Becton Dickinson); perforin (PE) clone B-D48 (from Diaclone); anti-human CD45RA (Alex Fluor 700) clone H100 (from Biolegend); and HIV Gag p24 (PE) clone KC57-RD1 (from Coulter Clone). Cells were analyzed on a standard FACS instrument (LSRII equipped with blue, red, and violet lasers; BD Biosciences) or on a FACSCalibur instrument as stated above. The data files were analyzed by using FlowJo software (version 8.2; TreeStar, Inc.). Multifunctional data were analyzed with PESTLE software (version 1.6.2) and SPICE software (version 4.2.3; obtained from M. Roederer, National Institutes of Health, Bethesda, MD).
Statistical analysis.
Calculations of the mean viral slope were determined with Excel (Microsoft) by using the LOGEST function and were converted to natural logs for data shown in Fig. 5 and Table 1. The variation in mean viral inhibition by the effector cells, determined by calculating the effect on the slope of viral growth, was evaluated by using a Student's t test. The statistically significant variation in the mean viral inhibition in the 24-h assay was determined by analysis of variance (ANOVA) and Dunnett's multiple-comparison posttest. All statistical analyses were performed by using Prism 5.0a software (GraphPad).
TABLE 1.
Inhibitory effect of B*2705-specific cytotoxic T lymphocytes on viral spreada
| CTL specificity | E/T ratio of 1:100 |
E/T ratio of 1:1,000 |
||
|---|---|---|---|---|
| Viral spread (mean ln slope ± SEM) | Significance (P value) compared to no CTL | Viral spread (mean ln slope ± SEM) | Significance (P value) compared to no CTL | |
| No CTL | 1.08 ± 0.02 | 1.40 ± 0.06 | ||
| Mismatch | 1.09 ± 0.04 | NS | 1.43 ± 0.009 | NS |
| Gag KK10 | 0.650 ± 0.04 | <0.001 | 1.10 ± 0.02 | <0.01 |
| Pol KY9 | 0.793 ± 0.007 | <0.001 | 1.23 ± 0.05 | NS |
| Vpr VL9 | 0.923 ± 0.06 | NS | 1.24 ± 0.04 | NS |
P values were calculated by using ANOVA and Bonferroni's multiple-comparison test (NS, not significant). ln slope is the natural log of the slope taken during the exponential growth phase. CTL, cytotoxic T lymphocytes.
RESULTS
Definition of a B*2705-restricted Pol epitope, KRKGGIGGY.
B*2705-positive HIV-infected subjects characteristically produce a strong and sustained dominant CD8+ T-cell response to the p24 Gag epitope KRWIILGLNK (KK10) (Gag residues 263 to 272) during chronic infection (16). In an initial analysis of the other CD8+ T-cell responses generated by HIV-infected B*2705-positive subjects, we observed that the B*2705-restricted epitope in Pol integrase, KRKGGIGGY (KY9) (Pol Int residues 901 to 909), is also frequently targeted in B*2705-positive subjects with chronic HIV infection (Fig. 1 and data not shown). Although not tested using individual optimal epitope peptides, IFN-γ ELISPOT responses to two additional HLA-B*27-restricted epitopes, identified in a recent study via a prediction program (28), ARRNRRRW (Rev residues 37 to 45) and NRWQVMIVW (Vif residues 3 to 11), were detected using an overlapping peptide panel. We here formally confirmed the KY9 epitope to be an optimal HLA-B*2705-restricted epitope, consistent with a recent study which predicted this epitope and also demonstrated a high frequency of responders (28).
FIG. 1.

Magnitude of B*2705 epitope-specific CD8+ T-cell responses and restriction of a B*2705 Pol epitope. (A) OLP and B*2705 optimal peptide IFN-γ ELISPOT responses from subject R039. OLP 265 contains the B*2705 peptide binding motif: Arg at P2 and a hydrophobic or positively charged residue at the C terminus. (B) Peptide-pulsed BCLs, matched at each HLA allele to the HLA type of the donor subject, were incubated with a KY9-specific CD8+ T-cell line cultured from PBMCs from the donor subject. Only the HLA-matched BCLs and the autologous BCLs elicited a response measured by the production of MIP-1β by intracellular cytokine staining. (C) Truncations of the predicted epitope were titrated in serial log dilutions in an IFN-γ ELISPOT assay and run in triplicates. Error bars show standard errors of the means (SEM). The epitope with the lowest 50% effective concentration (EC50) value is confirmed as the optimal epitope. (D) PBMCs from nine HIV-infected B*2705-positive subjects were screened by IFN-γ ELISPOT assay for the recognition of six known B*2705 epitopes (see Materials and Methods). Responses of >80 SFC per million cells are shown. (E) Median IFN-γ SFC per million PBMCs for KK10 (n = 9) and KY9 (n = 9) were compared. A Student's t test was performed (P = 0.438).
To begin to assess the contribution of KK10, KY9, and other HLA-B*2705-restricted epitopes to the well-described immune control displayed by B*2705-positive HIV-infected subjects, we tested the recognition of KK10, KY9, and four other previously described B*2705-restricted HIV-1 epitopes (see Materials and Methods), initially by an IFN-γ ELISPOT assay, in nine subjects expressing HLA-B*2705. These nine subjects were antiretroviral therapy naïve, had a median viral load of 10,000 copies per ml, and had median CD4 cell counts of 490 cells/mm3 (data not shown). In contrast to previous studies that have emphasized the immunodominance of the KK10 epitope (16), KK10 was targeted no more frequently than KY9 (by 8/9 subjects in each case) (Fig. 1D) and overall not at a significantly greater magnitude (median, 867 versus 695 SFC/million cells, respectively; P = 0.438 by a Student's t test) (Fig. 1E). The Vpr epitope VRHFPRIWL (VL9) (Vpr residues 31 to 30) was recognized by 45% of subjects (n = 4), and the p17 Gag epitope IRLRPGGKK (IK9) (Gag residues 19 to 27) was recognized by 33% of subjects (n = 3). The other two B*2705-restricted epitopes studied, GRRGWEALKY (GY10) (Env gp160) and KRQDILDLWV (KV10) (Nef), were recognized by none of the nine study subjects tested here.
B*2705-KY9-specific CD8+ T cells select viral escape mutations.
To determine whether KY9-specific CD8+ T-cell responses in B*2705-positive HIV-infected individuals impose sufficient pressure on the virus to drive the selection of escape mutants, we analyzed the viral sequences in the region of this epitope (Pol HXB2 residues 901 to 909) from the previously described HOMER cohort (n = 582) (Fig. 2). We found that viral polymorphisms at positions 2, 3, and 8 of the KY9 epitope (HXB2) (Pol residues 902, 903, and 908) were statistically significantly enriched in the B*2705-positive individuals compared to the B*2705-negative individuals (Fig. 2A). Furthermore, these viral polymorphisms all significantly reduced epitope recognition (Fig. 2B). The marked increase in the dissociation constant (Kd) resulting from the R902K and G908E substitutions (at positions 2 and 8 of the KY9 epitope) indicates >100-fold and >7-fold decreases in variant epitope binding, respectively, to HLA-B*2705 compared to the wild type (Fig. 2C).
FIG. 2.

KY9 escape mutations. (A) DNA sequences from 557 chronically clade B-infected subjects were analyzed for the presence of amino acid variations located within the newly restricted B*2705 Pol integrase epitope KY9. (B) KY9-specific CD8+ T cells were tested for recognition by intracellular cytokine staining of peptide-pulsed HLA-matched BCLs using the KY9 variants. Data shown in the graph are representative of three independent experiments using CD8+ T cells cultured from three different subjects. (C) Kd values (nM) show the binding affinity of the KY9 peptide and variants for the B*2705 molecule. (D) Longitudinal sequences from nine acutely clade B-infected subjects were analyzed by using a Kaplan-Meier plot to estimate the escape rate of B*2705-associated escape mutations located in Gag KK10 and Pol KY9.
Since the KK10 R264X mutation has been linked with the precipitation of progression to AIDS in B*2705-positive subjects, we compared the kinetics of selection of KY9 and KK10 escape mutations (Fig. 2D). A longitudinal analysis of HIV Gag and Pol sequences from nine B*2705-positive acutely infected individuals from a previously described cohort (8) demonstrated the escape rates at the sites described above within KK10 (at Gag position 264 [Gag-264]and Gag-268) and KY9 (at Pol-902, Pol-903, and Pol-908), respectively. As described above, the L268X KK10 variant has a high escape rate, with all subjects here showing mutations by 2 years postinfection. The G908E KY9 variant typically emerged soon after the R264X KK10 mutation, which arises late (14, 16), although in one instance, the selection of G908E preceded that of R264X. These sequence data suggest therefore that both the KY9 and the KK10 responses are imposing selection pressure on the virus at the same time, indicating that KY9 may contribute to the HLA-B*2705-mediated immune control of HIV.
Gag- and Pol-specific CD8+ T cells can inhibit HIV-infected targets earlier than Vpr-specific CD8+ T cells.
To further assess the contribution of the KK10- and KY9-specific CD8+ T-cell responses to HLA-B*2705-mediated control of viral replication, we utilized in vitro viral inhibition assays. We sought to evaluate the contribution of the most commonly targeted B*2705 epitopes in the subjects studied, Gag KK10, the Pol epitope KY9, and the Vpr epitope VL9, to immune control in B*2705-positive HIV-infected individuals by comparing the antiviral activities of epitope-specific CD8+ T cells. Peptide-MHC-I recombinant tetramers were used to generate antigen-specific CD8+ T-cell populations (>97% epitope specific in all cases [data not shown]) for B*2705-KK10 (Gag), B*2705-KY9 (Pol), and B*2705-VL9 (Vpr), respectively, derived from five B*2705-positive HIV-infected individuals. We then investigated how rapidly these CD8+ T cells could eliminate synchronized HIV-infected U937 target cells transfected with HLA-B*2705 by measuring the reduction of Gag p24+ cells at 3- to 6-h intervals in the first 24 h of infection, analyzing CD8+ T cells from three different study subjects in each case (Fig. 3). An illustration of the gating strategy is shown in Fig. 3A. The Gag KK10- and Pol KY9-specific CD8+ T cells could recognize and eliminate infected cells early after infection, significantly reducing the percentage of p24+ cells, compared to the control, virus-infected, HLA-B*2705-expressing target cells with no CD8+ T cells added, by the first 6 h postinfection (mean reduction ± standard deviation [SD] of 26.3% ± 9.6% and 26.5% ± 16.8%, respectively) (Fig. 4). The Vpr VL9-specific CD8+ T cells, however, had no significant effect at 6 or 12 h postinfection, and elimination of infected cells was not detected until 18 h postinfection (mean reduction ± SD of 20.16% ± 15.38%).
FIG. 3.

Gag- and Pol-specific CD8+ T cells elicit antiviral activity by 6 h postinfection. U937 cells transfected with HLA-B*2705 were synchronously infected with HIV-1 NL4-3GFP by Magnetofection. Gag- and Pol-specific tetramer-enriched CD8+ T cells can recognize and eliminate HIV-infected cells by 6 h postinfection. Vpr-specific tetramer-enriched CD8+ T cells recognize and eliminate HIV-infected cells after 18 h postinfection. (A) Gating strategy to show productively infected (p24+ CD4+/−) cells. By 24 h, the infected cells downregulated CD4. (B to E) Percentages of infected U937 cells, as measured by intracellular p24 at 3- to 6-h intervals in the presence and absence of tetramer-enriched CD8+ T cells from the same individual as indicated in figure. D also shows HLA-mismatched, KF11-specific, tetramer-enriched CD8+ T cells from subject R039. All time points were run in quadruplicates, and error bars show SEM.
FIG. 4.

Gag-, Pol-, and Vpr-specific CD8+ T cells display a consistent hierarchy of antiviral activity. U937 cells transfected with HLA-B*2705 were synchronously infected with HIV-1 NL4-3GFP by Magnetofection. Gag-, Pol-, and Vpr-specific tetramer-enriched CD8+ T cells from three different B*2705+ subjects were cocultured with infected cells at an E/T ratio of 1:4. Inhibition was calculated as the percentage of p24+ cells in the presence of tetramer-enriched CD8+ T cells relative to that in the presence of no tetramer-enriched CD8+ T cells. Inhibitions from Gag (KK10)-, Pol (KY9)-, and Vpr (VL9)-specific CD8+ T cells from three different individuals were grouped together according to time point and CD8+ T-cell protein specificity. Data represent quadruplicates from four independent experiments for Gag-, Pol-, and Vpr-specific CD8+ T cells from three individuals. Error bars show standard deviations. Statistical analysis was performed by using one-way analysis of variance and Dunnett's multiple-comparison posttest, comparing all columns to data for no CD8+ T cells. At the 24-h time point (E), statistical analysis was performed, comparing all columns to data for mismatch CD8+ T cells due to nonspecific inhibition by mismatched B*5701-KF11-specific CD8+ T cells. Statistical significance was observed despite nonspecific inhibition. At 24 h, gating on GFP+ cells was possible, to calculate the level of infection compared to that with no CD8+ T cells (F). *, P < 0.01; **, P < 0.001; ***, P < 0.0001; NS, not significant.
To confirm that the elimination of virus-infected B*2705-expressing U937 target cells was MHC-I dependent, we performed all experiments with an HLA-mismatched control HLA-B*5701 KAFSPEVIPMF (KF11) (Gag residues 162 to 172)-specific CD8+ T-cell population (Fig. 3D and 4). We observed no non-HLA-restricted inhibition at the time points shown, with the notable exception of 24 h postinfection (Fig. 4E). However, this nonspecific inhibition was not seen when the frequency of GFP+ cells was examined (Fig. 4F). These data represent four independent experiments using tetramer-enriched epitope-specific CD8+ T cells from three separate patients, with each one run in quadruplicate during each experiment. The data consistently show that the Gag KK10- and Pol KY9-specific CD8+ T cells elicit antiviral activity earlier than Vpr VL9-specific CD8+ T cells. Despite this temporal difference in viral inhibition, the Gag KK10-, Pol KY9-, and Vpr-VL9-specific CD8+ T cells all displayed potent antiviral activity by 24 h postinfection (mean reductions ± SD of 70.1% ± 3.4%, 58.2% ± 8.4%, and 58.5% ± 11.6%, respectively) (Fig. 4E), with the Gag KK10-specific CD8+ T cells eliciting the strongest effect.
Gag KK10-specific CD8+ T cells display more-potent antiviral activity.
We further tested the antiviral activity of the B*2705-specific CD8+ T cells in a 7-day viral inhibition assay designed to approximate more closely the in vivo situation of a low infectious dose, in this case with supernatant p24 antigen monitored as a marker of viral spread. Viral spread was determined by best-fit analysis with an exponential curve of the slope of p24 measurements taken during the exponential growth phase (7). These data, as described above, demonstrate a hierarchy of potency in which the KK10-specific CD8+ T cells are significantly more effective than the Pol KY9-specific CD8+ T cells, and the Vpr VL9-specific CD8+ T cells are the least effective (Table 1 and Fig. 5). However, all three CD8+ T-cell specificities inhibited viral replication at the higher E/T ratios tested, with a dose-dependent effect (Table 1, Fig. 5, and data not shown). This hierarchy mirrors the temporal inhibition observed for the 24-h inhibition assay, suggesting that early inhibition events indeed have longer-term effects on viral dissemination.
FIG. 5.

Gag-, Pol-, and Vpr-specific CD8+ T cells were tested for their effects on viral spread in a 7-day viral inhibition assay. (A and B) U937 cells were infected at an MOI of 0.02 with HIV-1 NL4-3 and cocultured with tetramer-enriched CD8+ T cells at an effector-to-target (E/T) ratio of 1:100 (A and C) and 1:1,000 (B and D). The supernatant was harvested every 2 to 3 days, and the p24 level was quantified by ELISA. Vpr VL9-specific CD8+ T cells from subject R035, Pol KY9-specific CD8+ T cells from subject R039, Gag KK10-specific CD8+ T cells from subject R039, and the HLA-mismatched Gag KF11-specific CD8+ T cells from subject R039 were tested. Each was run in triplicate. The shaded area shows data points taken from within the exponential growth phase used to calculate the slope of viral spread. Error bars show SEM. (C and D) The natural log slope of viral spread was calculated for each cell line as described in Materials and Methods. The mean values ± SEM are shown. Results were compared by using ANOVA and Bonferroni's multiple-comparison test. *, P < 0.01; **, P < 0.001; ***, P < 0.0001. Mean viral spread ± SEM and P values are shown in Table 1.
B*2705-restricted CD8+ T cells display a polyfunctional profile irrespective of epitope specificity.
We next investigated whether the observed differences in viral inhibition between the Gag KK10-, Pol KY9-, and Vpr VL9-specific tetramer-purified CD8+ T cells might result from the differential expression of certain chemokines or cytokines. We assessed the abilities of these CD8+ T cells to produce the effector cytokines and chemokines IFN-γ, TNF-α, MIP-1β, and IL-2 and to release cytotoxic factors by monitoring the expression of the degranulation marker CD107a upon antigen stimulation. We assessed both ex vivo B*2705-restricted epitope-specific PBMCs from HIV-infected subjects (n = 18 responses; n = 7 subjects) (Fig. 6 A) and the tetramer-purified epitope-specific CD8+ T cells expanded in culture (n = 9 responses; n = 5 subjects) (Fig. 6B). Neither ex vivo nor tetramer-purified Gag KK10-specific CD8+ T cells exhibited superiority in functional profile (Fig. 6A and B). Indeed, there were no more multifunctional cells (>3 functions) in the Gag KK10-specific CD8+ T cells analyzed than in Pol KY9- and Vpr VL9-specific CD8+ T cells. Similar findings were made whether ex vivo epitope-specific PBMCs or tetramer-purified CD8+ T cells were evaluated. In addition, proliferation data from the tetramer-purified CD8+ T cells showed that B*2705-restricted CD8+ T cells of different epitope specificities exhibited comparable proliferation profiles. Indeed, all epitope-specific CD8+ T cells displayed a high degree of proliferation upon antigen stimulation, with a mean for divided cells of 81.2% (range, 70.0% to 92.3%) (data not shown).
FIG. 6.

Functional assessment of B*2705-restricted epitope-specific CD8+ T cells. B*2705-specific CD8+ T cells ex vivo (A) and B*2705-specific tetramer-enriched CD8+ T cells (B) were tested for effector function in response to stimulation with a specific peptide. Pie charts represent background-adjusted multifunctional behavior (one to five functions; CD107a, TNF-α, MIP-1β, IFN-γ, and IL-2) of tetramer-enriched B2705-KK10-specific CD8+ T cells (n = 3) and B2705-KK10-specific CD8+ T cells ex vivo (n = 7), tetramer-enriched B2705-KY9-specific CD8+ T cells (n = 3) and B2705-KY9-specific CD8+ T cells ex vivo (n = 7), and tetramer-enriched B2705-VL9-specific CD8+ T cells (n = 3) and B2705-VL9-specific CD8+ T cells ex vivo (n = 4). The complete combinations of responses for all tetramer-enriched CD8+ T cells (A) and ex vivo CD8+ T cells (B) are shown in the bar charts according to epitope specificity, with each bar showing the mean percentage of cells displaying a particular combination. The bar charts are grouped by number of functions (numbers under colored bars, which are matched by color to the pie charts).
Cytotoxic capacity of B*2705-restricted CD8+ T cells is independent of epitope specificity.
We further investigated the functional profile of B*2705-restricted CD8+ T cells by comparing the cytotoxic capacities of the Gag-, Pol-, and Vpr-specific tetramer-enriched CD8+ T cells. The direct-killing ability of tetramer-enriched epitope-specific CD8+ T cells (effector) was measured using a cytotoxicity assay, which uses peptide-pulsed BCLs (targets) to sensitize target cells for lysis. We observed no difference in the killing capacities of the tetramer-purified CD8+ T cells of different epitope specificities at any of the E/T ratios tested (Fig. 7 A). In view of a previous study that highlighted the importance of lytic granule content in determining CD8+ T-cell efficacy in HIV infection (22), we also determined the cytotoxic granule content of the different epitope-specific tetramer-purified CD8+ T cells by examining the proportion of cells expressing granzyme B and perforin. The mean fluorescence intensities (MFIs) of granzyme B and perforin of tetramer-purified CD8+ T cells from a total of five different subjects were compared, and no differences were observed between the different B*2705 epitope specificities (Fig. 7B to D). In addition, the tetramer-purified CD8+ T cells expressed the same effector memory (CD45RA− CCR7−) and intermediate differentiated (CD45RA− CD127−) phenotypes irrespective of specificity (data not shown). Further analysis of the B*2705 epitope-specific CD8+ T cells showed that, measured directly ex vivo, these CD8+ T cells express an effector memory phenotype (CD45RA− CCR7−) similar to that of the tetramer-purified cells (data not shown). Notably, all the B*2705 tetramer-specific CD8+ T cells measured directly ex vivo expressed low levels of perforin (data not shown). However, overall, no statistically significant differences were observed between the different epitope specificities for the expression of the memory and differentiation markers CD45RA, CCR7, and CD127 or the expression of granzyme B and perforin. Thus, we found no differences in the cytotoxic killing capacities of the Gag-, Pol-, and Vpr-specific tetramer-purified CD8+ T cells and, hence, no correlation between killing capacity and the observed hierarchy of inhibition of virally infected cells by these epitope-specific CD8+ T cells.
FIG. 7.

Assessment of direct-killing abilities and cytotoxic markers for B*2705 epitope-specific CD8+ T cells. (A) The direct-killing ability of tetramer-enriched B*2705 epitope-specific CD8+ T cells (effector) was measured by using a cytotoxicity assay, which uses peptide-pulsed BCLs (target) to sensitize target cells for lysis. Tetramer-enriched CD8+ T cells specific for Gag KK10 and Pol KY9 from three subjects and specific for Vpr VL9 from one subject were analyzed at different E/T ratios. (B) The mean fluorescence intensity (MFI) was used to determine the granzyme B and perforin contents of the different epitope-specific CD8+ T cells using a mismatched tetramer and isotype controls as the background. (C and D) Tetramer-enriched CD8+ T cells from different subjects, specific for Gag KK10 (n = 3), Pol KY9 (n = 4), and Vpr VL9 (n = 3), were analyzed for granzyme B and perforin content.
TCR binding avidity does not predict antiviral efficacy.
Finally, comparisons of the TCR binding avidities of the ex vivo and tetramer-enriched B*2705-restricted epitope-specific CD8+ T cells showed no significant differences between the Gag KK10-specific CD8+ T cells and the other B*2705 epitope-specific CD8+ T cells tested (Fig. 8). A comparison of the MHC-I binding affinities of the three B*2705-restricted epitope peptides studied here, Gag KK10, Pol KY9, and Vpr VL9, showed that the Gag KK10 peptide does not bind with substantially greater affinity to the B*2705 MHC-I molecule than the Vpr VL9 peptide (Kd of 13 nM versus 32 nM, respectively). However, we observed that the Pol KY9 peptide had a weaker binding affinity than both the Gag KK10 and Vpr VL9 peptides (Kd of 162 nM).
FIG. 8.
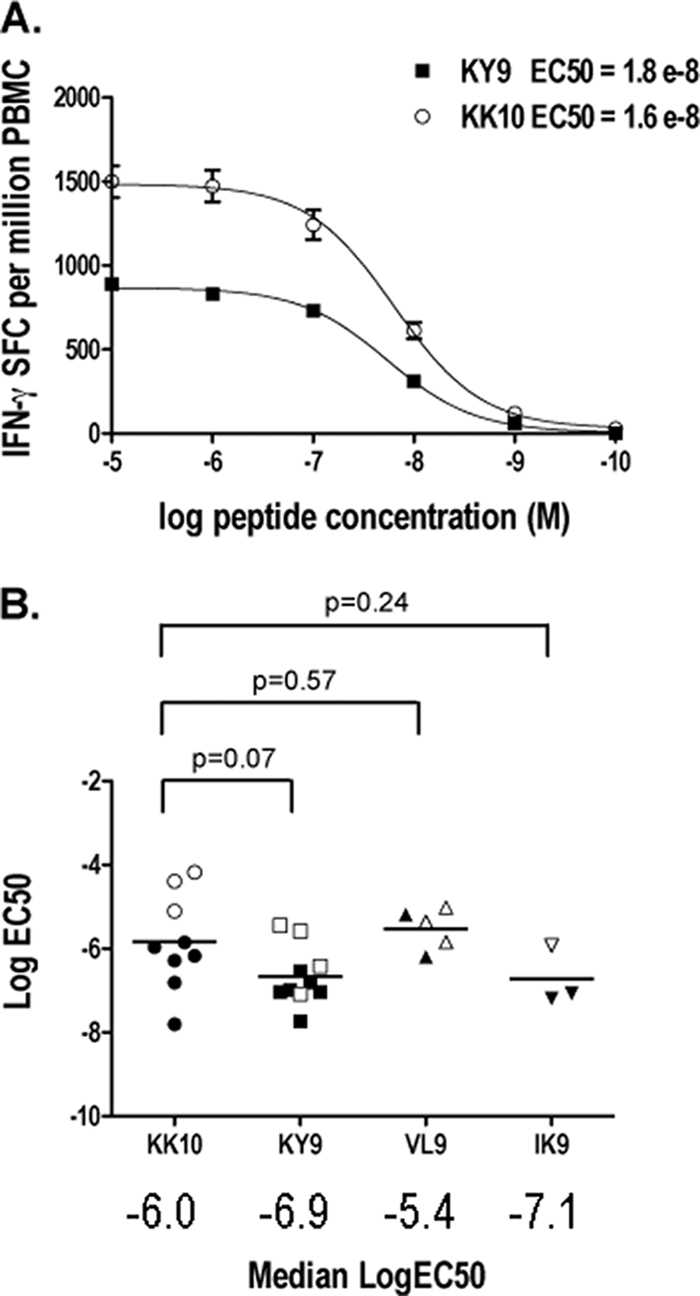
Avidity of B*2705 epitope-specific CD8+ T cells. (A) Functional avidities of B*2705 epitope-specific CD8+ T-cell responses within the same individual (subject 0145088RI) were compared. Serial log dilutions of specific peptides were titrated, in triplicate, in an IFN-γ ELISPOT assay with PBMCs or tetramer-enriched CD8+ T cells. Error bars are SEM. EC50 values were calculated by using Graphpad Prism software (version 5.0a). (B) PBMCs from six subjects were analyzed in total; six had KY9 responses, six had KK10 responses, two had IK9 responses, and two had VL9 responses (filled data points). Tetramer-enriched CD8+ T cells from five different subjects were analyzed (open data points). Log EC50 values for all subjects were plotted, and the median values were compared.
Thus, the superior inhibition of the Gag KK10-specific CD8+ T cells was not reflected by their polyfunctional cytokine profile, proliferation profile, or cytotoxic capacity or by their TCR binding avidity or MHC peptide binding affinity.
DISCUSSION
We focused on HLA-B*2705-restricted HIV-specific CD8+ T-cell responses to investigate further the association between HLA-B*2705 and slow progression in HIV infection and to test the hypothesis that qualitative differences exist between B*2705-restricted Gag-specific CD8+ T cells and non-Gag-specific CD8+ T cells in the control of viral replication. Unexpectedly, we observed frequent targeting of a newly restricted HLA-B*2705-restricted epitope in Pol (KY9) in B*2705-positive subjects. Escape mutants were selected in this epitope as well as in the well-studied Gag KK10 epitope, suggesting that the Pol KY9 response may contribute to the immune control that is associated with HLA-B*2705. By comparing the abilities of Gag KK10, Pol KY9, and Vpr VL9 CD8+ T cells to inhibit viral replication, we observed a consistent hierarchy of efficacy from Gag KK10-, Pol KY9-, and Vpr VL9-specific CD8+ T cells, respectively, in descending order. This was related to the kinetics of the CD8+ T-cell inhibition of virus-infected HLA-B*2705-expressing targets but not to any other marker of CD8+ T-cell functional activity measured in this study. These functions included the CD8+ T-cell proliferation capacity, cytokine profile, granzyme and perforin content, killing capacity, and TCR avidity. These data suggest that antiviral CD8+ T-cell efficacy is related not to these individual functions that do not differ in this study significantly by epitope specificity but to consistent epitope-specific differences in the kinetics of CD8+ T-cell-mediated viral inhibition.
These data are consistent with several previous cohort studies in which Gag-specific CD8+ T-cell activity, measured by an IFN-γ ELISPOT assay, was shown to be associated with a low viral set point (15, 20, 37) and also corroborate a recent study of Gag- versus Env-specific CD8+ T-cell-mediated virus neutralization in which Gag-specific CD8+ T cells elicited stronger antiviral activity (10). Analysis of multiple profiles of CD8+ T-cell activity in this study suggests a plausible mechanistic explanation for the observed superior antiviral capacity of Gag-specific CD8+ T cells. A previous study of SIV showed that Gag- and Pol-specific CD8+ T cells can recognize and eliminate SIV-infected targets cells by 6 h postinfection through the early presentation of Gag and Pol epitopes processed from the incoming virions (26, 27). These data from the SIV macaque model are consistent with the findings presented here, which show that Gag KK10- and Pol KY9-specific CD8+ T cells have a significant effect on viral inhibition as early as 6 h postinfection compared to VL9-specific CD8+ T cells, which did not elicit effective antiviral activity until 18 h postinfection.
Previous studies suggested that the highly functional HIV-specific CD8+ T cells, maintained by HIV nonprogressors, constitute a direct correlate of a successful immune response capable of eliciting viral control (6). However, we observed no difference in the polyfunctional profiles of Gag KK10-, Pol KY9-, and Vpr VL9-specific CD8+ T cells to account for the hierarchy of the efficacy of viral inhibition. These data are consistent with more recent studies suggesting that the polyfunctional profile of HIV-specific CD8+ T cells may be linked to antigen presentation and sensitivity and is not a direct correlate of protective immunity (2, 31).
In addition, the avidity of the different B*2705 epitope-specific CD8+ T cells did not correlate with effective viral suppression in this study, supporting previous studies that suggested that the suppression of HIV replication by CD8+ T cells depends more on antigen specificity than functional avidity (36). In support of recent studies of lytic granule loading (4, 22), we observed an increase in the granzyme and perforin contents of tetramer-enriched B*2705-specific CD8+ T cells cultured in vitro compared to granzyme and perforin levels measured directly from ex vivo CD8+ T cells (data not shown). However, although superior lytic granule loading was previously associated with CD8+ T-cell responses from nonprogressors (22), we did not observe any differences in granzyme and perforin contents between the epitope-specific B*2705-restricted CD8+ T cells studied here to explain the differences in effective viral suppression. Together, these data suggest that the different epitope-specific B*2705-restricted CD8+ T cells analyzed in this study have similar functional and cytotoxic killing capacities and that the observed differences in antiviral activity can be attributed to the different kinetics of epitope presentation, with the Gag KK10- and Pol KY9-specific CD8+ T cells eliciting earlier antiviral activity than Vpr VL9-specific CD8+ T cells.
Although consistent differences were observed in this study between the Gag KK10-, Pol KY9-, and Vpr VL9-specific CD8+ T cells in their ability to inhibit viral replication in vitro, the extent to which these differences are significant in vivo is unknown. Viral sequencing from longitudinal sampling may not allow escape within different B*27 epitopes to be distinguished temporally and, therefore, their individual impact to be assessed by viral load changes (Fig. 2D) (32). Also, it is problematic to demonstrate an effective individual CD8+ T-cell specificity in vivo because analyses are confounded by many other factors, including other HLA-B*27-restricted and non-B*27-restricted CD8+ T-cell responses and other non-CD8+ T-cell immune responses. In addition, interpretations of functional data from cultured CD8+ T cells must be undertaken with caution, since functional characteristics may change during culture. Indeed, where possible, assays were run in parallel using both ex vivo and cultured CD8+ T cells to highlight substantial functional changes. Also, highly purified (>98%) antigen-specific T-cell populations were used for these studies, in preference to the study of individual CD8+ T-cell clones that might arguably be less representative of the situation in vivo. Furthermore, viral inhibition monitored in this study in the U937 transformed monocytic cell line may not equate to that in primary HIV-infected CD4+ T cells. The observed differences in viral inhibition between the different epitope specificities, although remarkably consistent, and supporting previously reported observations (36), were relatively small, particularly between KK10 and KY9. On the basis of these experiments, it might not necessarily be the case that HLA-B*2705+ individuals would fail to contain HIV in the absence of a KK10-specific response, since the KY9-specific response appears itself to be relatively efficacious. Alternatively, the KK10- and KY9-specific responses may act in tandem to direct effective viral suppression in vivo, since both responses elicit early inhibitory activity in vitro and were present in comparable magnitudes and dominances when measured ex vivo in this cohort. A previous vaccine study using a recombinant canarypox virus vector expressing gp120, gp41, Gag, and protease successfully induced a strong “protective” B*2705-KK10-specific CD8+ T-cell response in a B*2705-positive uninfected individual. However, following subsequent infection, the vaccinee also developed rapid viral escape in the KK10 epitope and progressed to AIDS much more quickly than would have been expected for an unvaccinated B*2705-positive subject (5). The development of rapid viral escape in the context of a dominant viral epitope was previously reported for a TCR transgenic mouse model for lymphocytic choriomeningitis virus (LCMV) (24); however, a broader response prevented the selection of escape mutants (23), supporting the data presented in this study suggesting that the protective effect of HLA-B*2705 is not mediated through the KK10 epitope alone.
The lack of clear KK10 immunodominance observed in the study subjects evaluated here was unexpected. However, all these subjects were highly active antiretroviral therapy (HAART) naïve, and viral sequencing showed that none of the seven subjects analyzed had accumulated the R264K escape mutation and the associated S173A compensatory mutation, which have been shown to precede progression to AIDS in B*2705+ HIV-infected subjects (14, 16, 19). These subjects therefore appear representative of untreated B*2705+ HIV-infected subjects in chronic infection.
By comparing different epitope-specific CD8+ T cells from within the same subject and by comparing B*2705-restricted epitope-specific CD8+ T cells between subjects, we have controlled for factors such as viral load while also testing the consistency of our findings. However, it will be useful in further studies to extend this analysis to other HLA alleles and to include more Gag, Pol, and non-Gag/Pol epitopes for analysis to assess whether the findings in this study are consistent across a broad spectrum of HLA-restricted CD8+ T cells. It would be especially useful to study HLA alleles that direct dominant CD8+ T-cell responses against non-Gag epitopes and subdominant responses against Gag epitopes. A recent study indicated that viral control is due to the epitope targeted by CD8+ T-cell responses rather than the restricting HLA allele and that these are located mostly in conserved regions (13), consistent with the data presented here suggesting that Gag and Pol epitopes are beneficial. However, the protein relevance of these responses needs further evaluation, since only one epitope from each protein was analyzed in this study.
It has been suggested that B*27-KK10-specific CD8+ T cells are responsible for the long-term nonprogression of B*27+ patients. Here we have identified a previously unreported CD8+ T-cell response in B*27 patients that may also have a critical role in preventing the onset of AIDS. We have shown that polyfunctionality, proliferative capacity, TCR avidity, and killing capacity are not predictors of potent viral inhibition. Instead, we suggest that the early presentation of Gag and Pol epitopes gives subjects who make CD8+ T-cell responses against these epitopes a kinetic advantage, being able to eliminate virally infected cells before viral dissemination. Early presentation and the elimination of virally infected cells combined with epitopes from conserved regions in which escape mutations are not well tolerated may be the key to the long-term nonprogression observed for patients with the protective HLA-B*2705 allele.
Acknowledgments
This work is funded by grants from the National Institutes of Health (grant RO1AI46995) (P.J.R.G.), the Wellcome Trust (P.J.R.G.), and the UK Medical Research Council (R.P.P.). Z.B. is supported by a new investigator award from the Canadian Institutes of Health Research.
We are grateful for the support of Paul Bowness for the kind donation of HLA-B*2705-transfected U937 cells and Paul Klenerman and the NIHR biomedical research center program for support making tetramers. For access to the acute longitudinal data, we thank Bruce Walker, Martin Markowitz, and Heiko Jessen. We thank Richard Harrigan at the British Columbia Centre for Excellence in HIV/AIDS for data access and Jennifer Sela and Pamela Rosato for DNA sequence analysis.
We have no conflicting financial interests.
Footnotes
Published ahead of print on 4 August 2010.
REFERENCES
- 1.Almeida, J. R., D. A. Price, L. Papagno, Z. A. Arkoub, D. Sauce, E. Bornstein, T. E. Asher, A. Samri, A. Schnuriger, I. Theodorou, D. Costagliola, C. Rouzioux, H. Agut, A. G. Marcelin, D. Douek, B. Autran, and V. Appay. 2007. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 204:2473-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almeida, J. R., D. Sauce, D. A. Price, L. Papagno, S. Y. Shin, A. Moris, M. Larsen, G. Pancino, D. C. Douek, B. Autran, A. Saez-Cirion, and V. Appay. 2009. Antigen sensitivity is a major determinant of CD8+ T-cell polyfunctionality and HIV-suppressive activity. Blood 113:6351-6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altfeld, M. A., A. Trocha, R. L. Eldridge, E. S. Rosenberg, M. N. Phillips, M. M. Addo, R. P. Sekaly, S. A. Kalams, S. A. Burchett, K. McIntosh, B. D. Walker, and P. J. Goulder. 2000. Identification of dominant optimal HLA-B60- and HLA-B61-restricted cytotoxic T-lymphocyte (CTL) epitopes: rapid characterization of CTL responses by enzyme-linked immunospot assay. J. Virol. 74:8541-8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appay, V., P. R. Dunbar, M. Callan, P. Klenerman, G. M. Gillespie, L. Papagno, G. S. Ogg, A. King, F. Lechner, C. A. Spina, S. Little, D. V. Havlir, D. D. Richman, N. Gruener, G. Pape, A. Waters, P. Easterbrook, M. Salio, V. Cerundolo, A. J. McMichael, and S. L. Rowland-Jones. 2002. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8:379-385. [DOI] [PubMed] [Google Scholar]
- 5.Betts, M. R., B. Exley, D. A. Price, A. Bansal, Z. T. Camacho, V. Teaberry, S. M. West, D. R. Ambrozak, G. Tomaras, M. Roederer, J. M. Kilby, J. Tartaglia, R. Belshe, F. Gao, D. C. Douek, K. J. Weinhold, R. A. Koup, P. Goepfert, and G. Ferrari. 2005. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 102:4512-4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betts, M. R., M. C. Nason, S. M. West, S. C. De Rosa, S. A. Migueles, J. Abraham, M. M. Lederman, J. M. Benito, P. A. Goepfert, M. Connors, M. Roederer, and R. A. Koup. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781-4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brockman, M. A., A. Schneidewind, M. Lahaie, A. Schmidt, T. Miura, I. Desouza, F. Ryvkin, C. A. Derdeyn, S. Allen, E. Hunter, J. Mulenga, P. A. Goepfert, B. D. Walker, and T. M. Allen. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 81:12608-12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brumme, Z. L., C. J. Brumme, J. Carlson, H. Streeck, M. John, Q. Eichbaum, B. L. Block, B. Baker, C. Kadie, M. Markowitz, H. Jessen, A. D. Kelleher, E. Rosenberg, J. Kaldor, Y. Yuki, M. Carrington, T. M. Allen, S. Mallal, M. Altfeld, D. Heckerman, and B. D. Walker. 2008. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J. Virol. 82:9216-9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brumme, Z. L., M. John, J. M. Carlson, C. J. Brumme, D. Chan, M. A. Brockman, L. C. Swenson, I. Tao, S. Szeto, P. Rosato, J. Sela, C. M. Kadie, N. Frahm, C. Brander, D. W. Haas, S. A. Riddler, R. Haubrich, B. D. Walker, P. R. Harrigan, D. Heckerman, and S. Mallal. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4:e6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, H., A. Piechocka-Trocha, T. Miura, M. A. Brockman, B. D. Julg, B. M. Baker, A. C. Rothchild, B. L. Block, A. Schneidewind, T. Koibuchi, F. Pereyra, T. M. Allen, and B. D. Walker. 2009. Differential neutralization of human immunodeficiency virus (HIV) replication in autologous CD4 T cells by HIV-specific cytotoxic T lymphocytes. J. Virol. 83:3138-3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crawford, H., J. G. Prado, A. Leslie, S. Hue, I. Honeyborne, S. Reddy, M. van der Stok, Z. Mncube, C. Brander, C. Rousseau, J. I. Mullins, R. Kaslow, P. Goepfert, S. Allen, E. Hunter, J. Mulenga, P. Kiepiela, B. D. Walker, and P. J. Goulder. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81:8346-8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Day, C. L., P. Kiepiela, A. J. Leslie, M. van der Stok, K. Nair, N. Ismail, I. Honeyborne, H. Crawford, H. M. Coovadia, P. J. Goulder, B. D. Walker, and P. Klenerman. 2007. Proliferative capacity of epitope-specific CD8 T-cell responses is inversely related to viral load in chronic human immunodeficiency virus type 1 infection. J. Virol. 81:434-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinges, W. L., J. Richardt, D. Friedrich, E. Jalbert, Y. Liu, C. E. Stevens, J. Maenza, A. C. Collier, D. E. Geraghty, J. Smith, Z. Moodie, J. I. Mullins, M. J. McElrath, and H. Horton. 2010. Virus-specific CD8+ T-cell responses better define HIV disease progression than HLA genotype. J. Virol. 84:4461-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feeney, M. E., Y. Tang, K. A. Roosevelt, A. J. Leslie, K. McIntosh, N. Karthas, B. D. Walker, and P. J. Goulder. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927-8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geldmacher, C., J. R. Currier, E. Herrmann, A. Haule, E. Kuta, F. McCutchan, L. Njovu, S. Geis, O. Hoffmann, L. Maboko, C. Williamson, D. Birx, A. Meyerhans, J. Cox, and M. Hoelscher. 2007. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 81:2440-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goulder, P. J., R. E. Phillips, R. A. Colbert, S. McAdam, G. Ogg, M. A. Nowak, P. Giangrande, G. Luzzi, B. Morgan, A. Edwards, A. J. McMichael, and S. Rowland-Jones. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212-217. [DOI] [PubMed] [Google Scholar]
- 17.Harndahl, M., S. Justesen, K. Lamberth, G. Roder, M. Nielsen, and S. Buus. 2009. Peptide binding to HLA class I molecules: homogenous, high-throughput screening, and affinity assays. J. Biomol. Screen. 14:173-180. [DOI] [PubMed] [Google Scholar]
- 18.Honeyborne, I., A. Rathod, R. Buchli, D. Ramduth, E. Moodley, P. Rathnavalu, S. Chetty, C. Day, C. Brander, W. Hildebrand, B. D. Walker, P. Kiepiela, and P. J. Goulder. 2006. Motif inference reveals optimal CTL epitopes presented by HLA class I alleles highly prevalent in southern Africa. J. Immunol. 176:4699-4705. [DOI] [PubMed] [Google Scholar]
- 19.Kelleher, A. D., C. Long, E. C. Holmes, R. L. Allen, J. Wilson, C. Conlon, C. Workman, S. Shaunak, K. Olson, P. Goulder, C. Brander, G. Ogg, J. S. Sullivan, W. Dyer, I. Jones, A. J. McMichael, S. Rowland-Jones, and R. E. Phillips. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiepiela, P., K. Ngumbela, C. Thobakgale, D. Ramduth, I. Honeyborne, E. Moodley, S. Reddy, C. de Pierres, Z. Mncube, N. Mkhwanazi, K. Bishop, M. van der Stok, K. Nair, N. Khan, H. Crawford, R. Payne, A. Leslie, J. Prado, A. Prendergast, J. Frater, N. McCarthy, C. Brander, G. H. Learn, D. Nickle, C. Rousseau, H. Coovadia, J. I. Mullins, D. Heckerman, B. D. Walker, and P. Goulder. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46-53. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Picado, J., J. G. Prado, E. E. Fry, K. Pfafferott, A. Leslie, S. Chetty, C. Thobakgale, I. Honeyborne, H. Crawford, P. Matthews, T. Pillay, C. Rousseau, J. I. Mullins, C. Brander, B. D. Walker, D. I. Stuart, P. Kiepiela, and P. Goulder. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80:3617-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Migueles, S. A., C. M. Osborne, C. Royce, A. A. Compton, R. P. Joshi, K. A. Weeks, J. E. Rood, A. M. Berkley, J. B. Sacha, N. A. Cogliano-Shutta, M. Lloyd, G. Roby, R. Kwan, M. McLaughlin, S. Stallings, C. Rehm, M. A. O'Shea, J. Mican, B. Z. Packard, A. Komoriya, S. Palmer, A. P. Wiegand, F. Maldarelli, J. M. Coffin, J. W. Mellors, C. W. Hallahan, D. A. Follman, and M. Connors. 2008. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29:1009-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moskophidis, D., and R. M. Zinkernagel. 1995. Immunobiology of cytotoxic T-cell escape mutants of lymphocytic choriomeningitis virus. J. Virol. 69:2187-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pircher, H., D. Moskophidis, U. Rohrer, K. Burki, H. Hengartner, and R. M. Zinkernagel. 1990. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature 346:629-633. [DOI] [PubMed] [Google Scholar]
- 25.Prado, J. G., I. Honeyborne, I. Brierley, M. C. Puertas, J. Martinez-Picado, and P. J. Goulder. 2009. Functional consequences of human immunodeficiency virus escape from an HLA-B*13-restricted CD8+ T-cell epitope in p1 Gag protein. J. Virol. 83:1018-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sacha, J. B., C. Chung, E. G. Rakasz, S. P. Spencer, A. K. Jonas, A. T. Bean, W. Lee, B. J. Burwitz, J. J. Stephany, J. T. Loffredo, D. B. Allison, S. Adnan, A. Hoji, N. A. Wilson, T. C. Friedrich, J. D. Lifson, O. O. Yang, and D. I. Watkins. 2007. Gag-specific CD8+ T lymphocytes recognize infected cells before AIDS-virus integration and viral protein expression. J. Immunol. 178:2746-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sacha, J. B., C. Chung, J. Reed, A. K. Jonas, A. T. Bean, S. P. Spencer, W. Lee, L. Vojnov, R. Rudersdorf, T. C. Friedrich, N. A. Wilson, J. D. Lifson, and D. I. Watkins. 2007. Pol-specific CD8+ T cells recognize simian immunodeficiency virus-infected cells prior to Nef-mediated major histocompatibility complex class I downregulation. J. Virol. 81:11703-11712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schellens, I. M., C. Kesmir, F. Miedema, D. van Baarle, and J. A. Borghans. 2008. An unanticipated lack of consensus cytotoxic T lymphocyte epitopes in HIV-1 databases: the contribution of prediction programs. AIDS 22:33-37. [DOI] [PubMed] [Google Scholar]
- 29.Schneidewind, A., M. A. Brockman, J. Sidney, Y. E. Wang, H. Chen, T. J. Suscovich, B. Li, R. I. Adam, R. L. Allgaier, B. R. Mothe, T. Kuntzen, C. Oniangue-Ndza, A. Trocha, X. G. Yu, C. Brander, A. Sette, B. D. Walker, and T. M. Allen. 2008. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J. Virol. 82:5594-5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneidewind, A., M. A. Brockman, R. Yang, R. I. Adam, B. Li, S. Le Gall, C. R. Rinaldo, S. L. Craggs, R. L. Allgaier, K. A. Power, T. Kuntzen, C. S. Tung, M. X. LaBute, S. M. Mueller, T. Harrer, A. J. McMichael, P. J. Goulder, C. Aiken, C. Brander, A. D. Kelleher, and T. M. Allen. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 81:12382-12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Streeck, H., Z. L. Brumme, M. Anastario, K. W. Cohen, J. S. Jolin, A. Meier, C. J. Brumme, E. S. Rosenberg, G. Alter, T. M. Allen, B. D. Walker, and M. Altfeld. 2008. Antigen load and viral sequence diversification determine the functional profile of HIV-1-specific CD8+ T cells. PLoS Med. 5:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Streeck, H., B. Li, A. F. Poon, A. Schneidewind, A. D. Gladden, K. A. Power, D. Daskalakis, S. Bazner, R. Zuniga, C. Brander, E. S. Rosenberg, S. D. Frost, M. Altfeld, and T. M. Allen. 2008. Immune-driven recombination and loss of control after HIV superinfection. J. Exp. Med. 205:1789-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber, J., J. Weberova, M. Carobene, M. Mirza, J. Martinez-Picado, P. Kazanjian, and M. E. Quinones-Mateu. 2006. Use of a novel assay based on intact recombinant viruses expressing green (EGFP) or red (DsRed2) fluorescent proteins to examine the contribution of pol and env genes to overall HIV-1 replicative fitness. J. Virol. Methods 136:102-117. [DOI] [PubMed] [Google Scholar]
- 34.Weston, S. A., and C. R. Parish. 1990. New fluorescent dyes for lymphocyte migration studies. Analysis by flow cytometry and fluorescence microscopy. J. Immunol. Methods 133:87-97. [DOI] [PubMed] [Google Scholar]
- 35.Yang, O. O., S. A. Kalams, A. Trocha, H. Cao, A. Luster, R. P. Johnson, and B. D. Walker. 1997. Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: evidence for HLA class I-restricted triggering of cytolytic and noncytolytic mechanisms. J. Virol. 71:3120-3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang, O. O., P. T. Sarkis, A. Trocha, S. A. Kalams, R. P. Johnson, and B. D. Walker. 2003. Impacts of avidity and specificity on the antiviral efficiency of HIV-1-specific CTL. J. Immunol. 171:3718-3724. [DOI] [PubMed] [Google Scholar]
- 37.Zuniga, R., A. Lucchetti, P. Galvan, S. Sanchez, C. Sanchez, A. Hernandez, H. Sanchez, N. Frahm, C. H. Linde, H. S. Hewitt, W. Hildebrand, M. Altfeld, T. M. Allen, B. D. Walker, B. T. Korber, T. Leitner, J. Sanchez, and C. Brander. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 80:3122-3125. [DOI] [PMC free article] [PubMed] [Google Scholar]