

Abstract
The discovery of the Toll-like receptors (TLRs) and their importance in the regulation of host responses to infection raised attention to the complex interplay between viral gene products and the host innate immune responses in determining the outcome of virus infection. Robust inflammatory cytokine responses are observed in herpes simplex virus (HSV)-infected animals and cells. Our studies have demonstrated that Toll-like receptor 2 (TLR2) activation by HSV results in NF-κB activation with concomitant inflammatory cytokine production and that TLR2 activation plays a critical role in HSV-induced pathology and mortality. Here we demonstrate that the HSV-1 immediate-early ICP0 protein reduces the TLR2-mediated inflammatory response to HSV 1 (HSV-1) infection. Expression of ICP0 alone is sufficient to block TLR2-driven responses to both viral and nonviral ligands at or downstream of the MyD88 adaptor and upstream of p65. ICP0 alone can also reduce the levels of MyD88 and Mal (TIRAP). In HSV-infected cells, the E3 ligase function of ICP0 and cellular proteasomal activity are required for the inhibitory activity. Our results argue for a model in which ICP0 promotes the degradation of TLR adaptor molecules and inhibition of the inflammatory response, much as it inhibits the interferon response by sequestration and degradation of interferon regulatory factor 3 (IRF-3).
Innate immune activation is the first critical step in the cascade of immunologic events that culminate in the targeted destruction of invading pathogens. Through their recognition of a broad spectrum of evolutionarily conserved pathogenic motifs, Toll-like receptors (TLRs) are key activators of the innate immune system. To date, 11 mammalian TLRs have been identified (26, 61). All TLRs signal through an evolutionarily conserved Toll/interleukin 1 (IL-1) receptor (TIR) domain, which recruits essential adaptor proteins such as MyD88 (myeloid differentiation factor 88), Mal (MyD88 adaptor-like protein), TIRAP (TIR domain-containing adaptor protein), TRIF (Toll/IL-1 receptor domain-containing adaptor-inducing beta interferon), and/or TRAM (TRIF-related adaptor molecule) to initiate downstream signaling (48). Of particular importance for herpes simplex virus 1 and 2 (HSV-1 and HSV-2) recognition are Toll-like receptor 2 (TLR2) and TLR9. TLR2 is located on cell surfaces and recognizes peptidoglycans, bacterial lipoproteins, and viral proteins, while TLR9 is located within endosomes and recognizes double-stranded DNA (dsDNA) (32, 38). Signaling through TLR2/TIR/MyD88/Mal activates the transcription factor NF-κB (nuclear factor κB), promoting production of proinflammatory cytokines IL-1β, IL-6, IL-8, IL-12, and monocyte chemotactic peptide 1 (MCP-1) (1, 25). In contrast, signaling through TLR9/TIR/MyD88 signaling activates interferon regulatory factors 3 and 7 (IRF-3 and IRF-7) that drive alpha/beta interferon (IFN-α/β) production (15, 34, 38). Collectively, these responses limit HSV replication, promote host cell apoptosis, and recruit and activate macrophages, dendritic cells, neutrophils, and other leukocytes of both innate and adaptive lineages at the site of infection.
HSV-1 and HSV-2 are members of the Herpesviridae family of viruses. These viruses are characterized by a large DNA genome, conserved virion structure and replication mechanisms, and more uniquely, the ability to establish lifelong latency in various cells of infected hosts (52). HSV carries genes that encode 5 immediate-early (IE) proteins, commonly identified by their infected-cell protein (ICP) number: ICP0, ICP4, ICP22, ICP27, and ICP47. ICP4 and ICP27 are essential gene products that activate expression of early and late viral gene products (54). ICP0 is a multifunctional protein that enhances replication, especially at low multiplicities of infection (MOIs), in part through its ability to block chromatin silencing of viral lytic genes (7, 19). ICP0 has an E3 ubiquitin ligase activity (21) that promotes degradation of certain host proteins, such as promyelocytic leukemia (PML) in nuclear bodies, SUMO-1, and the catalytic subunit of DNA protein kinase, the latter playing an important role in innate immune activation (3, 14). In vitro studies have demonstrated that HSV infection activates interferon signaling in various cell types, and ICP0 is critical for resistance to type 1 interferons (44). HSV is a weak interferon inducer but can strongly activate expression of interferon-stimulated genes (ISG) if viral protein synthesis is blocked (46), and the immediate-early ICP0 protein is required for this inhibitory effect (13). Furthermore, studies indicate HSV ICP0 interferes with IFN-α/β induction and signaling by limiting IRF-3 and IRF-7 activation (34, 42). Mechanistic studies have shown that ICP0 inhibits IFN-α/β production by sequestering phosphorylated IRF-3 away from host chromatin (43) and by reducing the levels of IRF-3 (42). HSV engages receptors that activate both IRF- and NF-κB-dependent gene expression. Here we demonstrate that in addition to inhibiting IFN responses, ICP0 also inhibits TLR2-driven inflammatory cytokine response following viral infection and does so by targeting the TLR2/NF-κB response pathway.
MATERIALS AND METHODS
Cell lines and viruses.
HEK293 cell lines expressing TLRs were generated as described previously (33). The wild-type (WT) HSV-1 KOS virus was propagated, and the titers of the virus were determined on Vero and U2OS cells. The HSV-1 ICP0 mutant viruses were propagated, and the titers of the viruses were determined on U20S cells. The HSV-1 KOS n212 virus contains a nonsense mutation that terminates translation of ICP0 at codon 212, while HSV-1 KOS 7134 virus has a lacZ expression cassette in place of the ICP0 gene (6). In 7134R virus, the lacZ cassette had been replaced with the wild-type ICP0 gene sequence. This virus was propagated, and the titers of the virus were determined on U2OS cells (6). The HSV-1 F ICP0 RING domain mutant vC116G/C156A virus contains the ICP0 cDNA in place of the full ICP0 sequence and has mutations in the cysteines at either end of the ICP0 RING domain (36). This virus was propagated, and the titers of the virus were determined in U20S cells. The WT control for this virus is the HSV-1 F vCPc0 virus, which also contains the ICP0 cDNA with a functional ICP0 RING domain (50). These two viruses were the kind gift of Saul Silverstein (College of Physicians and Surgeons, Columbia University, New York, NY) and were propagated, and the titers of the virus were determined on USOS cells. The HSV-1 ICP4 mutant n12 virus contains a nonsense mutation at codon 251 (9). This virus was propagated, and the titers of the virus were determined on the E5 complementing cell line. The HSV-1 ICP27 deletion mutant virus 5dl1.2 (40) was propagated, and the titers of the virus were determined on complementing V8-27 cells.
The wild-type HSV-1 F virus was propagated, and the titers of the virus were determined on Vero cells. The R7914 virus contains a D199A substitution within the ICP0 protein, which prevents the ICP0 protein from translocating out of the nucleus at late times postinfection. The R7916 mutant virus and the wild-type rescued virus R7915 were both propagated, and the titers of the viruses were determined on U2OS cells. All three viruses were the kind gift of Bernard Roizman (University of Chicago, Chicago, IL).
For all viruses, culture supernatant virus was concentrated by centrifugation onto a sucrose cushion, and the titers of the virus were determined by plaque assay as previously described. Genome copy number of the input virus was determined by quantitative real-time PCR (63).
IL-6 cytokine assays.
Wild-type C57BL/6J mice were purchased from Jackson Laboratories. TLR2 knockout mice were the kind gift of S. Akira (Osaka, Japan) and were back bred for >12 generations onto the C57BL/6 background in our facility. Back breeding was confirmed by satellite marker analysis (Charles River Laboratories). To isolate macrophages, mice were injected intraperitoneally (i.p.) with 4% thioglycolate, and peritoneal exudate cells (>90% macrophages) were harvested 4 days later. Macrophages were plated at 106 cells per well in 24-well plates and incubated with various HSV-1 viruses. Cytokine secretion into culture supernatants was measured 16 to 18 h later. IL-6 production was determined by enzyme-linked immunosorbent assay (ELISA) using the OptEIA kit (BD Pharmingen, San Diego, CA).
Luciferase and IL-8 cytokine assays.
H2.14.12 HEK293 cells stably expressing Toll-like receptor 2 (TLR2) and CD14 were plated in 96-well plates at a density of 2.5 × 104 cells/well. Twenty-four hours later, the cells were transfected with 100 ng of NF-κB-driven firefly luciferase reporter plasmid and 20 ng of HSV thymidine kinase (HSV-TK) promoter-driven Renilla luciferase plasmid using GeneJuice transfection reagent (Novagen), and cultured overnight. These cells were then infected with the indicated HSV-1 viruses at various multiplicities of infection (MOIs). Zymosan and Pam3CSK4 (100 ng/ml) were used as TLR2 stimulants, and IL-1β (100 ng/ml) was used as a positive control. Uninfected cells were included as a negative control. Cells were infected for 6 h before being analyzed for luciferase production and 16 to 18 h before being analyzed for IL-8 production (see below).
For MyD88-, Mal-, and p65-driven NF-κB studies, HEK293T cells were transfected with 100 ng of NF-κB-driven firefly luciferase reporter plasmid, 20 ng of HSV-TK promoter-driven Renilla luciferase plasmid, 50 ng of MyD88, Mal, or p65 plasmid, and 80 ng of DNA consisting of either empty plasmid alone (vector control), an IκB mutant (IκB superrepressor) control inhibitor plasmid, or increasing quantities of a plasmid containing the HSV-1 ICP0 gene. Transfected cells were incubated for 24 h at 37°C in 5% CO2 before being tested for luciferase and IL-8 production.
To determine luciferase expression, cells were lysed in 50 μl of passive lysis buffer (Promega), and firefly and Renilla luciferase levels were measured using the Dual-Glo luciferase assay system (Promega). Results are presented in relative luciferase units (RLU) where the NF-κB-dependent firefly luciferase activity is normalized to NF-κB-independent Renilla luciferase activity. In some experiments, an HEK cell line (SZ10) stably expressing the NF-κB luciferase reporter gene as well as TLR2 and CD14 were used for reporter studies (66). Luciferase activity in NF-κB stable cells was determined using Steady-Glo (Promega) reagent and normalized by analyzing proteins of the lysates with a MicroBCA protein assay kit. The results are shown as the means ± standard deviations (SDs) of six wells and are representative of at least three independent experiments.
To determine IL-8 production, cell culture supernatants were collected, and the levels of IL-8 were measured by an ELISA using the BD OptEIA kit (San Diego, CA).
Western blots.
HEK293T cells were cultured in 24-well plates (Corning) at 1 × 105 cells/well. Cells were transfected with ICP0, Mal, MyD88, or pUC plasmids as indicated, using GeneJuice (Novagen) transfection reagent according to the manufacturer's specifications. Following overnight incubation at 37°C and 5% CO2, cells were lysed in 200 μl of passive lysis buffer (Promega), supernatants were boiled in Laemmli sample buffer (Bio-Rad), and proteins were resolved by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were blocked in a 5% milk solution and probed with horseradish peroxidase (HRP)-conjugated anti-Flag M2 monoclonal antibody (catalog no. A8592; Sigma). Blots were developed with enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham). Blots were stripped with Restore Western blot stripping buffer (Pierce) and reprobed with rabbit anti-beta-actin antibody (ab8227; Abcam) followed by HRP-conjugated goat anti-rabbit IgG (heavy and light) antibody (ab6721; Abcam) and ECL detection to confirm equal loading of lanes.
Statistical analysis.
Data were analyzed using Student's t test. P values of ≤0.05 were considered significant.
RESULTS
Roles of immediate-early gene products in TLR2 signaling.
In previous studies, we showed that HSV-1 wild-type (WT) strain KOS infection activates TLR2 signaling in human 293 cells expressing TLR2 and in primary mouse macrophages (31, 32). As a first step in the analysis of the roles of HSV gene products in TLR2 signaling, we assessed the roles of viral IE gene products in HSV-1-activated NF-κB and inflammatory cytokine responses. We used WT HSV-1 KOS and mutant strains of virus to infect murine macrophages. Infection with WT KOS virus induced IL-6 expression and secretion in a dose-dependent manner (Fig. 1). In contrast, HSV-1 5dl1.2 virus, which has a deletion in the ICP27 (UL54) gene, showed decreased ability to induce IL-6, suggesting that ICP27 or one of the gene products that it induces augments TLR2 signaling. Similarly, infection with n12 virus, which has a nonsense mutation in the IE ICP4 gene, resulted in reduced TLR2 signaling (Fig. 1). However, cells infected with the n212 virus, which has a nonsense mutation in the ICP0 gene, showed elevated IL-6 secretion, suggesting that ICP0 inhibits this process. It should be noted that KOS virus induced lower levels of cytokines than n212 virus did when infections were based on either viral particles (genome copy number) (Fig. 1A) or infectious virus (multiplicity of infection [MOI]) (Fig. 1B). Therefore, potential differences in the particle-to-PFU ratio of the different viruses could not explain the differences in the phenotypes. The phenotype of increased TLR2 signal was observed with the ICP0 mutants at low and high MOIs (Fig. 1), indicating that the function of ICP0 involved is different from its promotion of viral replication, which is required only at low MOIs (55, 60).
FIG. 1.

The immediate-early gene ICP0 inhibits virus-induced cytokine production in HSV-1-infected macrophages. Various doses of wild-type (WT) or mutant HSV-1 viruses were used to infect murine peritoneal macrophages. Culture supernatants from infected macrophages were collected 16 h later, and levels of secreted IL-6 were determined by ELISA. The values are means ± standard deviations (SDs) (error bars). (A) Wild-type macrophages infected with WT KOS virus, an ICP0 gene nonsense mutant (n212), an ICP4 gene nonsense mutant (n12), or an ICP27 gene deletion mutant (5dl1.2). Viral doses were based on the numbers of genomes as defined by real-time PCR. (B) Comparison of the cytokine response of wild-type, TLR4-deficient (TLR4 −/−) and TLR2-deficient (TLR2 −/−) macrophages following infection with WT KOS virus or an ICP0 gene nonsense mutant (n212). Viral doses were based on PFU per cell (MOI). The values are means plus standard deviations (SDs) (error bars).
To confirm that the elevated signaling seen with n212 virus was exerted through TLR2, we tested the ability of the n212 ICP0 mutant virus to activate IL-6 expression in TLR2−/− cells (Fig. 1B). Macrophages from TLR2−/− or congenic C57BL/6 mice were infected with n212 virus or with WT KOS virus. IL-6 expression induced by either WT KOS or n212 virus was much higher in the C57BL/6 cells than in the TLR2-deficient cells (Fig. 1B), confirming that the bulk of IL-6 expression with both viruses was via TLR2 stimulation. The low but detectable activation of IL-6 expression in n212-infected TLR2−/− cells suggested that mechanisms other than TLR2 can induce IL-6, and these mechanisms were most evident when ICP0 was not expressed.
To confirm that the ICP0 defect was responsible for the increased IL-6 secretion observed with n212 mutant virus, we tested another ICP0-deficient mutant virus, HSV-1 strain KOS 7134, which has an inactive ICP0 gene due to a lacZ expression cassette insertion (6). We compared virus strain 7134 with a rescued viral strain, 7134R, in which the WT ICP0 gene had been restored by homologous recombination. We transfected TLR2-HEK cells (that stably express TLR2 and CD14 [33]) with an NF-κB-driven firefly luciferase gene and a control TK Renilla luciferase gene and then infected with 7134 or 7134R virus (Fig. 2). Induction of NF-κB signaling was measured by the expression of firefly luciferase. The 7134 ICP0-negative mutant virus activated NF-κB signaling to higher levels than the 7134R rescued ICP0-positive virus (Fig. 2). Similarly, the n212 ICP0 nonsense mutant virus induced NF-κB signaling to higher levels than the WT KOS virus did (Fig. 2). Therefore, two independent ICP0-defective viruses (n212 and 7134) activated cytokine or NF-κB signaling to a greater extent than either the parental or restored WT ICP0-expressing viruses (KOS and 7134R) did, arguing that the phenotype was due to the ICP0 defect. In general, reduced cytokine levels correlated with ICP0 expression (Table 1).
FIG. 2.

ICP0 expression restores the cytokine inhibitory activity of HSV mutant virus. HEK293 cells were transfected with TLR2, CD14, an NF-κB-driven firefly luciferase reporter gene, and a thymidine kinase (TK) promoter-driven Renilla luciferase reporter gene. Twenty-four hours later, the cells were infected with 7134 or n212 (two ICP0-deficent viral strains), the ICP0-rescued virus (7134R), or WT KOS. Luciferase activity was measured 6 h after viral infection and is expressed as firefly luciferase normalized to Renilla luciferase (or relative light units [RLU]). The values are means plus SDs (error bars). Values that are statistically significantly different (P ≤ 0.05) Student's t test are indicated by a bracket and an asterisk.
TABLE 1.
HSV strains and the macrophage cytokine response
| HSV-1 strain | Genotype | DNA replication | ICP0 expression | Peritoneal macrophage cytokine responsea |
|---|---|---|---|---|
| KOS | Wild-type | Yes | Yes | ++ |
| n212 | ICP0 nonsense | Yes | No | ++++ |
| 5dl1.2 | ICP27 deletion | Yes | Yes | + |
| n12 | ICP4 nonsense | No | Yes | − |
| d106 | ICP4, ICP22, ICP27, ICP47− | No | Yes | − |
| 7134 | ICP0 deletion (lacZ replaces ICP0) | Yes | No | ++++ |
| 7134R | ICP0 (wild type) (rescue of strain 7134, ICP0 replaces lacZ) | Yes | Yes | ++ |
The peritoneal macrophage cytokine response is shown as a quartile cytokine response (e.g., IL-6 secretion at an MOI of 10) as follows: ++++, ≥9 ng/ml; +++, 6 to 9 ng/ml; ++, 3 to 6 ng/ml; +, 0.5 to 3 ng/ml; −, <0.5 ng/ml.
ICP0 expression resulted in reduced cytokine responses to all HSV strains tested (Table 1). Virus replication was not necessary for cytokine responses to HSV, but nonreplicating ICP0-expressing viruses, (n12 and d106) were very weak cytokine inducers, indicating that ICP0 could actively suppress the cytokine response in the absence of DNA replication (Table 1).
Inhibition of TLR2 signaling by HSV-1 ICP0.
Our results with mutant viruses indicated that ICP0 was required for inhibition of inflammatory cytokine secretion and NF-κB activation. To determine whether ICP0 was sufficient to inhibit TLR2 activity, we examined the ability of ICP0 expressed from a transfected plasmid to affect TLR2 signaling. TLR2-HEK cells were transfected with an NF-κB firefly luciferase and TK Renilla luciferase plasmids and an ICP0 gene expression vector plasmid or an empty plasmid vector as a control prior to incubation with TLR2 agonists. ICP0 expression did not affect basal levels of NF-κB activation or IL-8 secretion from TLR2-HEK cells (Fig. 3). Expression of ICP0 suppressed TLR2-dependent NF-κB responses induced by zymosan (Fig. 3A) and IL-8 induced by Pam3CSK4 (Fig. 3B), two defined TLR2 ligands. ICP0 inhibition of TLR2 activation was dose dependent as a function of the amount of ICP0 gene plasmid that was transfected. Although ICP0 inhibited Pam3CSK4 responses, it had little effect on the response to IL-1β (Fig. 3B), arguing that ICP0 did not nonspecifically inhibit all signaling pathways. Therefore, ICP0 appeared to be sufficient to inhibit TLR2 signaling. Expression of ICP0 also reduced the NF-κB response to HSV-1 n212 to the level of WT KOS virus (Fig. 3A), and expression of ICP0 in WT KOS virus-infected cells further reduced NF-κB signaling (Fig. 3A). As a control, expression of HSV-1 ICP8 did not affect TLR2 signaling (M. Horn and D. M. Knipe, unpublished results), arguing that the effect was specific for ICP0.
FIG. 3.

ICP0 inhibits TLR2 signaling. (A) TLR2-HEK cells were transfected with an NF-κB luciferase reporter plasmid and TK Renilla luciferase reporter plasmid plus either control vector or ICP0 plasmid. The cells were then stimulated with zymosan (TLR2 ligand) or infected with WT KOS virus or n212 ICP0-deficient virus. Luciferase activity was measured 6 h later in cell lysates. (B) TLR2-HEK cells were transfected with various amounts of ICP0 plasmid. The cells were then stimulated with Pam3CSK4 (TLR2 ligand) or IL-1β (non-TLR ligand). IL-8 production was measured 24 h later in culture supernatants by an ELISA. The values are means plus SDs (error bars). Values that are statistically significantly different (P ≤ 0.05) by Student's t test are indicated by a bracket and an asterisk.
ICP0 effects on MyD88 and Mal.
TLR2 signals by engaging the downstream adaptors MyD88 and Mal. MyD88/Mal activation triggers degradation of IκB, resulting in the processing and release of the p65 subunit of NF-κB. After release from IκB, nuclear translocation of p50-p65 heterodimers occurs, leading to the activation of NF-κB-driven genes, including those of inflammatory cytokines. Overexpression of MyD88, Mal, or p65 in HEK293T cells is sufficient to drive NF-κB signaling (16, 17). Therefore, we examined the effects of ICP0 directly on these adaptors by overexpression of each of the adaptors in the presence or absence of ICP0. MyD88-driven IL-8 secretion was inhibited by ICP0 expression in a dose-dependent manner (Fig. 4 A). Using a reporter gene, we found that NF-κB activation in response to either MyD88 or Mal was inhibited by ICP0 in a dose-dependent manner (Fig. 4B). Interestingly, p65-driven NF-κB activity, which bypasses MyD88/Mal and IκB, was unaffected by ICP0 expression (Fig. 4B), arguing that the effect of ICP0 is stage specific and that ICP0 acts upstream of p65-NF-κB promoter binding. In contrast to ICP0, the mutant IκB (superrepressor), IκB-SR, blocked signaling by p65, as well as that by MyD88 and Mal (Fig. 4B). These results argued that ICP0 inhibits MyD88 and Mal signaling upstream of IκB.
FIG. 4.

ICP0 blocks MyD88- and Mal-dependent signaling. (A) HEK293T cells were transfected either with pcDNA vector control DNA or with various amounts of ICP0 plasmid. The cells were cotransfected with a MyD88 plasmid to drive NF-κB activation and IL-8 secretion. IL-8 secretion into culture supernatants was measured 16 h later by an ELISA. (B) HEK239T cells were transfected with NF-κB luciferase reporter plasmid and various amounts of ICP0 plasmid or a dominant-negative mutant IκB (IκB superrepressor [IKB-SR]). MyD88, Mal, or p65 were cotransfected to drive NF-κB, and luciferase activity was measured at 6 h. The values are means ± SDs (error bars).
ICP0 is known to target certain cellular proteins for proteasome degradation, thereby eliminating their activity. We expressed ICP0 with MyD88 or Mal in the presence or absence of protease inhibitor and measured NF-κB signaling. The addition of a proteasome inhibitor was sufficient to maintain high-level NF-κB signaling in cells transfected with MyD88 and Mal plasmids, even at high concentrations of ICP0, suggesting that ICP0 may target MyD88 and Mal for degradation via the proteasome (Fig. 5 A and B). p65-driven NF-κB expression, which bypasses MyD88 or Mal, was unaffected by either ICP0 or the presence of proteasome inhibitor (Fig. 5C). Similarly, NF-κB activity driven by TRIF or TRAM, two adaptor proteins not associated with TLR2 signaling, was unaffected either by the addition of ICP0 or by the addition of proteasome inhibitor (Fig. 5D and E), thus suggesting that ICP0 specifically targets the TLR2 signaling pathway via MyD88 and Mal proteins in a proteasome-dependent manner.
FIG. 5.

Role of proteasomal activity in ICP0 inhibition of TLR2 signaling. HEK293T cells were transfected with NF-κB luciferase reporter plasmid and various amounts of ICP0 plasmid or a dominant-negative mutant IκB in the presence or absence (−) of protease inhibitor (P.I.) clasto-lactacytin β-lactone. MyD88 (A), Mal (B), p65 (C), TRIF (D), TRAM (E), or pcDNA (vector control) plasmids were cotransfected to drive NF-κB signaling, and luciferase activity was measured at 6 h. The values are means ± SDs (error bars).
To further assess the effects of ICP0 on MyD88 and Mal signaling, we examined MyD88 and Mal protein levels when coexpressed with ICP0. We observed that ICP0 expression reduced levels of both MyD88 and Mal (Fig. 6). In contrast, IκB-SR had a modest effect on MyD88 and Mal protein levels compared to ICP0, even at the highest levels tested. Actin levels were unaffected by ICP0 expression (Fig. 6). Because ICP0 usually increases expression of all cotransfected genes, these results suggest that ICP0 targets MyD88 and Mal specifically for degradation. Thus, ICP0 can modulate the innate immune signaling cascade very early during infection by reducing the levels of MyD88 and Mal (Fig. 6).
FIG. 6.
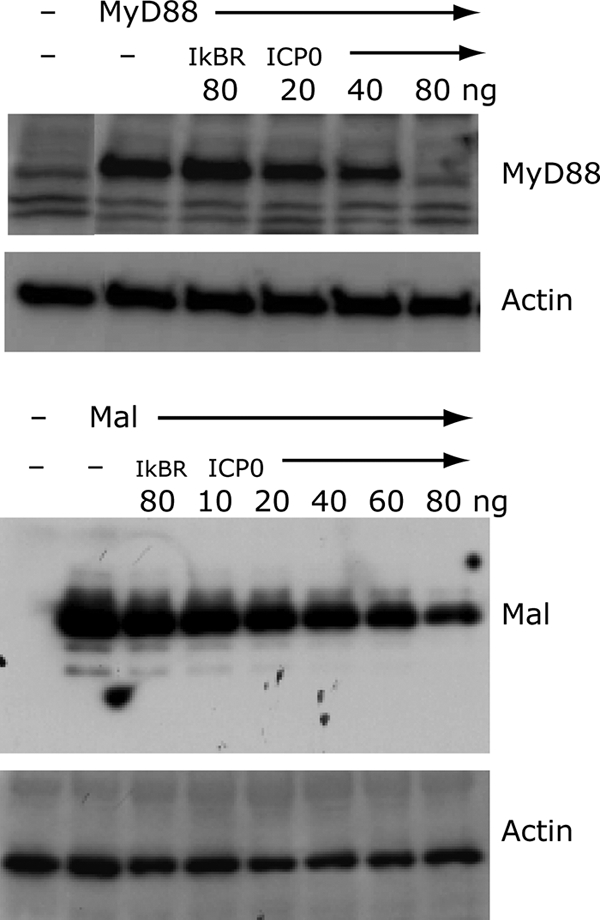
MyD88 and Mal levels in the presence of ICP0. HEK239T cells were transfected with plasmids containing genes encoding MyD88 or Mal along with various amounts of ICP0 plasmid or a dominant-negative mutant IκB (IκB superrepressor [IkBR]). Twenty-four hours later, lysates were prepared and analyzed by Western blotting for MyD88, Mal, and actin protein expression levels.
Role of the ICP0 E3 ubiquitin ligase activity.
ICP0 has an E3 ubiquitin ligase activity which is required for promotion of degradation of specific proteins in infected cells (3, 21). To further test the idea that ICP0 promotes the degradation of MyD88 and Mal, we examined the ability of an HSV mutant virus strain, VC116G/C156A, which has defects in the ICP0 RING finger domain and associated E3 ligase activity (36), to regulate TLR2 signaling upon infection of HEK293 cells stably transformed with TLR2 and the NF-κB-driven firefly luciferase gene. The VC116G/C156A mutant virus activated NF-κB reporter gene expression (Fig. 7 A) and IL-8 secretion (Fig. 7B) to higher levels than the parental virus vCPc0 expressing the wild-type ICP0 protein. Due to the lack of the RING domain-associated E3 ligase, the mutant ICP0 protein is expressed at levels higher than the vCPc0 ICP0 (Horn and Knipe, unpublished). Thus, the RING finger and likely the associated E3 ligase activity are required for ICP0 inhibition of NF-κB activation, consistent with the hypothesis that ICP0 inhibits signaling through targeting adaptor or other cellular proteins for degradation.
FIG. 7.

Functional RING domain of ICP0 is required to inhibit TLR2 signaling. HEK293 cells stably expressing TLR2, CD14, and an NF-κB-driven firefly luciferase reporter gene were infected with various MOIs of an ICP0 RING domain mutant virus, vC116G/C156A, or the control WT virus, vCPc0 which contains a functional RING domain in the ICP0 protein. (A) Luciferase activity was measured at 6 h postinfection. (B) IL-8 production was measured by an ELISA at 16 h postinfection. The values are means ± SDs (error bars).
Role of the ICP0 nuclear export function.
ICP0 localizes first to the nucleus at early times postinfection (29) but then localizes to the cytoplasm at later times postinfection (28). Because the TLR2 signaling adaptors are localized in the cytoplasm, ICP0 may need to be localized from the nucleus to the cytoplasm to inhibit TLR2 signaling. To determine whether the nuclear export function of ICP0 was needed for its ability to inhibit TLR2 signaling, we tested the ability of the HSV-1 R7914 mutant virus, which exhibits defective nuclear export, or the R7915 rescued virus (37) to induce TLR2 signaling. HEK293 cells expressing TLR2 and stably transformed with an NF-κB-driven firefly luciferase reporter gene showed induction of NF-κB signaling when infected with WT F virus (Fig. 8). The HSV-1 R1656 ICP0 mutant virus induced higher levels of luciferase, consistent with the results above. Cells infected with the R7914 mutant virus, in which ICP0 remains in the nucleus (37, 62; A. L. van Lint and D. M. Knipe, unpublished results) showed low levels of luciferase expression, similar to that of the R7915 rescued virus (Fig. 8). Therefore, nuclear export of ICP0 was not needed for its inhibition of the TLR2 signaling process.
FIG. 8.

HSV expressing an ICP0 molecule defective for nuclear export can suppress TLR2 signaling. HEK293 cells stably expressing TLR2, CD14, and an NF-κB-driven firefly luciferase reporter gene were incubated with zymosan or infected with the wild-type virus F, the R1656 ICP0 mutant virus, the ICP0 cytoplasmic translocation mutant R7914, or the rescued virus R7915 at an MOI of 200. Luciferase production was measured at 6 h postinfection. The values are means plus SDs (error bars).
DISCUSSION
In this study, we have shown that the immediate-early protein ICP0 encoded by HSV-1 can inhibit the TLR2-dependent activation of NF-κB signaling. We have further demonstrated that this blockade occurs at an early stage in the signaling cascade, possibly via reduction in the levels of the MyD88 and Mal adaptor proteins. We have shown previously that ICP0 is sufficient to cause (i) the sequestration of interferon regulatory factor 3 (IRF-3) away from host chromatin and (ii) the accelerated degradation of activated IRF-3 (42, 43). The activation of NF-κB and IRF-3 and IRF-7, whether through TLR-dependent or -independent pathways, has the potential to play a major role in the immune response to virus infection. Activation of NF-κB leads to upregulation of a variety of proinflammatory cytokines and chemokines, while activation of IRF-3 and IRF-7 leads to the induction of the type I interferon genes and the acquisition of a potent antiviral state within an infected cell and its local environment. Thus, both major innate pathways are important targets for immune evasion by HSV through its ICP0 gene product.
Functions of ICP0.
ICP0 has two general types of functions which may be conducted in tandem (20): (i) stimulation of gene expression and viral replication, which is essential at low MOIs (60) and (ii) inhibition of host cell responses and innate responses. During the course of infection, ICP0 localizes into the nucleus and disrupts nuclear domain 10 (ND-10) bodies followed by blocking of host chromatin silencing mechanisms that block the transition from immediate-early to early gene expression. At high MOIs, viral replication seems to circumvent the requirements for these ICP0 functions because replication is not dependent on ICP0 at high MOIs. Inhibition of TLR2 signaling by ICP0 is independent of the MOI; therefore, this function is at least partially distinct from the replication functions of ICP0, because no other viral function can substitute. Consistent with this, the infection conditions used in these studies showed no replication defect for the ICP0 mutants (van Lint and Knipe, unpublished). Inhibition of TLR2 signaling by ICP0 requires the RING finger domain, so the simplest explanation is that ICP0 uses its E3 ligase activity to promote the degradation of TLR2 signaling molecules.
Mechanism of inhibition.
Our results raise the possibility that ICP0 may exert its inhibitory effects on TLR2 signaling through the degradation of MyD88 and Mal and other adaptor or signaling molecules, but we do not yet know the mechanism by which this degradation occurs. ICP0 may be directly involved in that it has E3 ubiquitin ligase activity associated with its RING finger domain (3) and HUL-1 domain (22). This is further supported by previous findings that both MyD88 and Mal are targets of proteasome-dependent degradation (39, 45). We suggest that ICP0 specifically targets MyD88 and Mal for degradation via a proteasome-dependent pathway. ICP0 has been shown to promote the degradation of a number of proteins, including PML, sumoylated SP100, the catalytic subunit of DNA-dependent protein kinase (DNA-PK) (reviewed in reference 54), and more recently the membrane-bound and cytoplasmic CD83 (30). It has long been a puzzle how ICP0 can promote the degradation of such a variety of proteins, including both nuclear and cytoplasmic proteins, the latter group now possibly including the TLR adaptor proteins, MyD88 and Mal.
Although ICP0 is a nuclear protein at early times postinfection, studies have shown that ICP0 is translocated out of the nucleus as early as 5 to 9 h postinfection (37); thus, this exported ICP0 could be affecting TLR2 signaling steps. However, we observed that R7914 ICP0, which is largely retained in the nucleus (37, 62; A. L. van Lint and D. M. Knipe, results not shown), can repress TLR2 signaling to levels equivalent to WT ICP0. In addition, ICP0 expressed from a transfected plasmid localizes largely in the nucleus but can repress TLR2 signaling. Several possible explanations could explain how nuclear ICP0 might inhibit TLR2 cytoplasmic signaling events. (i) Newly made ICP0 could directly affect cytoplasmic complexes prior to or as it is being transported to the nucleus. (ii) Small amounts of ICP0 that are present in the cytoplasm are still sufficient to effect the inhibition of TLR2 signaling. (iii) Nuclear ICP0 inhibits the cytoplasmic signaling events indirectly via effects on the levels of other cellular proteins. Further studies are needed to define how this nuclear protein can affect cytoplasmic signaling proteins.
While this article was being prepared, Daubeuf et al. (8) reported that HSV ICP0 inhibits TLR responses but by a different mechanism, through recruitment of USP7 from the nucleus to the cytoplasm. A number of differences are apparent between that study and the current study. Daubeuf et al. (8) reported that a RING finger mutant virus could inhibit TLR signaling, while we found that a RING mutant virus does not inhibit TLR2. Daubeuf et al. reported that ICP0 promoted the export of USP7 from the nucleus to the cytoplasm, while we observed that a mutant ICP0 that was not exported from the nucleus to the cytoplasm could still inhibit TLR2 signaling. In fact, Daubeuf et al. also observed that ICP0 promotes the degradation of USP7 as observed previously (3). Therefore, the effect of ICP0 that they observed may be via degradation of USP7 and other cellular proteins regulating signaling similar to what we found in our studies. Nevertheless, differences in host cells and viral strains could explain at least some of the observed differences.
Comparison with other viral effects.
This proposed mechanism of action appears to be novel among the viruses currently known to block NF-κB activation. Several viruses, including two members of the poxvirus family, inhibit the activation of NF-κB by interfering with the degradation of its inhibitor molecule IκB. Vaccinia virus carries genes that encode two proteins, K1 and N1, which prevent IκB degradation and inhibit the function of the IκB kinase (IKK) complex, respectively (12, 58). Molluscum contagiosum virus and hepatitis C virus each carry a gene that encodes a protein that interferes with the IKK complex, thereby preventing the degradation of IκB (27, 47). Interestingly, vaccinia virus carries genes that encode a second set of proteins, A46 and A52, which also act to prevent activation of NF-κB. A52 is thought to associate with IL-1 receptor-associated kinase 2 (IRAK2) and tumor necrosis factor receptor-associated factor 6 (TRAF6), disrupting any signaling complexes which contain these proteins and thus blocking the signaling cascade from multiple TLRs, particularly TLR3 (4, 24). A46 contains a TIR domain and interacts with MyD88 and several other adaptor proteins to inhibit MyD88-dependent NF-κB activation through a number of TLRs (59). Finally, proteins encoded by genes in influenza virus and human cytomegalovirus (HCMV) have also been shown to inhibit NF-κB activation, although the mechanism of action of these proteins remains to be determined (5, 18, 49, 64). Thus, HSV ICP0 blocks TLR signaling at an early stage in the pathway by a unique mechanism.
HSV and NF-κB activity.
The relationship between HSV infection and the host NF-κB innate response is quite complex because while HSV infection activates NF-κB and this activity is required for full viral yields (51), NF-κB signaling promotes inflammatory cytokine expression, which would reduce viral infection. NF-κB signaling is induced by both TLR2-dependent (32, 56) and TLR2-independent (35) pathways in HSV-infected cells, and different HSV isolates can vary in their TLR-inducing activity (56). This may reflect differences in HSV gene expression, including ICP0 (D. M. Knipe, unpublished). HSV infection activates NF-κB by non-TLR2 mechanisms in two waves, an early wave that does not require HSV replication and a later wave that requires viral replication (2). The early wave involves the UL37 virion tegument protein (35) and possibly virion glycoprotein D (41). UL37 activates NF-κB signaling through interaction with TRAF6 (35), which is downstream of the MyD88/Mal adaptors that we hypothesize are targeted by the immediate-early ICP0 protein. Thus, HSV may transiently activate NF-κB to start its infection optimally and then inhibit the major part of the innate response using viral gene products expressed during infection, like ICP0, as shown here.
Our studies suggest that ICP0 does not interfere with IκB degradation, i.e., ICP0 does not inhibit NF-κB activation through the IL-1 receptor (IL-1R). This fits with evidence showing that during HSV-1 infection, NF-κB is recruited to the ICP0 promoter, in a move that is thought to help promote viral gene expression (2). If this were indeed the case, it would be counterproductive for ICP0 to interfere with IκB degradation. It has been well documented that HSV-1 infection induces activation of NF-κB. It was initially reported that ICP0 from HSV-1, along with BICP0 from bovine herpesvirus 1, induces activation of NF-κB by degrading IκBα (11). However, a recent study, using a variety of mutant viruses, demonstrated that, in HSV-infected cells, ICP27 was responsible for IκB degradation and subsequent NF-κB activation (23). Therefore, it is possible to imagine a situation where ICP27 or other viral molecules lead to TLR-independent NF-κB activation through downstream signaling molecules that the virus then appropriates for its own use, leading to targeting of activated NF-κB to the ICP0 gene promoter to help promote viral gene expression. Meanwhile, another IE protein, ICP0, is able to block TLR2-dependent NF-κB activation, and the proinflammatory response that this would induce in an infected cell, enabling the viral infection to progress relatively unencumbered.
This apparent conflict of NF-κB activation and suppression within a single viral infection is seen not only during HSV-1 infection but also in other infections. A similar situation is thought to occur during HCMV infection, where studies have demonstrated that HCMV infection activates NF-κB (53) and proposed that the virus then uses NF-κB to help promote viral gene expression (10), while other work, discussed above, indicates that HCMV carries a gene(s) that encodes at least one protein that can block NF-κB activation (5). Hepatitis C virus (HCV) also provides a conflicting story, with studies showing that the HCV core protein can inhibit NF-κB activation in macrophages (27) but can lead to NF-κB activation in hepatocytes (57, 65). Further studies of the mechanisms by which HSV activates and inhibits NF-κB signaling should elucidate the process by which this virus utilizes the host response for its own replication but evades its immune response manifestations.
Acknowledgments
This work was supported by grants from the National Institutes of Health: RO1 AI51405 (to E.A.K.-J.), PO1 AI083215 (to E.A.K.-J., K.A.F., D.M.K., and R.W.F.), and RO1 GM63244 and RO1 AI39576 (to R.W.F.).
We thank Bernard Roizman, Saul Silverstein, and Neal DeLuca for generously providing mutant and wild-type HSV strains. We thank Leisa Mandell and Jayashree Paranjape for excellent technical and administrative assistance.
Footnotes
Published ahead of print on 4 August 2010.
REFERENCES
- 1.Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2:675-680. [DOI] [PubMed] [Google Scholar]
- 2.Amici, C., A. Rossi, A. Costanzo, S. Ciafre, B. Marinari, M. Balsamo, M. Levrero, and M. G. Santoro. 2006. Herpes simplex virus disrupts NF-kappaB regulation by blocking its recruitment on the IkappaBalpha promoter and directing the factor on viral genes. J. Biol. Chem. 281:7110-7117. [DOI] [PubMed] [Google Scholar]
- 3.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain as ubiquitin E3 ligases in vitro. J. Virol. 76:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowie, A., E. Kiss-Toth, J. A. Symons, G. L. Smith, S. K. Dower, and L. A. O'Neill. 2000. A46R and A52R from vaccinia virus are antagonists of host IL-1 and Toll-like receptor signaling. Proc. Natl. Acad. Sci. U. S. A. 97:10162-10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browne, E. P., and T. Shenk. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. U. S. A. 100:11439-11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai, W. Z., and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 63:4579-4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cliffe, A. R., and D. M. Knipe. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 82:12030-12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daubeuf, S., D. Singh, Y. Tan, H. Liu, H. J. Federoff, W. J. Bowers, and K. Tolba. 2009. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 113:3264-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLuca, N. D., and P. A. Schaffer. 1987. Activities of herpes simplex virus type 1 (HSV-1) ICP4 genes specifying nonsense polypeptides. Nucleic Acids Res. 15:4491-4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeMeritt, I. B., L. E. Milford, and A. D. Yurochko. 2004. Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J. Virol. 78:4498-4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diao, L., B. Zhang, J. Fan, X. Gao, S. Sun, K. Yang, D. Xin, N. Jin, Y. Geng, and C. Wang. 2005. Herpes virus proteins ICP0 and BICP0 can activate NF-kappaB by catalyzing IkappaBalpha ubiquitination. Cell Signal. 17:217-229. [DOI] [PubMed] [Google Scholar]
- 12.DiPerna, G., J. Stack, A. G. Bowie, A. Boyd, G. Kotwal, Z. Zhang, S. Arvikar, E. Latz, K. A. Fitzgerald, and W. L. Marshall. 2004. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by Toll-like receptors. J. Biol. Chem. 279:36570-36578. [DOI] [PubMed] [Google Scholar]
- 13.Eidson, K. M., W. E. Hobbs, B. J. Manning, P. Carlson, and N. A. DeLuca. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 76:2180-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Everett, R., P. O'Hare, D. O'Rourke, P. Barlow, and A. Orr. 1995. Point mutations in the herpes simplex virus type 1 Vmw110 RING finger helix affect activation of gene expression, viral growth, and interaction with PML-containing nuclear structures. J. Virol. 69:7339-7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finberg, R. W., D. M. Knipe, and E. A. Kurt-Jones. 2005. Herpes simplex virus and Toll-like receptors. Viral Immunol. 18:457-465. [DOI] [PubMed] [Google Scholar]
- 16.Fitzgerald, K. A., E. M. Palsson-McDermott, A. G. Bowie, C. A. Jefferies, A. S. Mansell, G. Brady, E. Brint, A. Dunne, P. Gray, M. T. Harte, D. McMurray, D. E. Smith, J. E. Sims, T. A. Bird, and L. A. O'Neill. 2001. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413:78-83. [DOI] [PubMed] [Google Scholar]
- 17.Fitzgerald, K. A., D. C. Rowe, B. J. Barnes, D. R. Caffrey, A. Visintin, E. Latz, B. Monks, P. M. Pitha, and D. T. Golenbock. 2003. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 198:1043-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gon, Y., Y. Asai, S. Hashimoto, K. Mizumura, I. Jibiki, T. Machino, C. Ra, and T. Horie. 2004. A20 inhibits Toll-like receptor 2- and 4-mediated interleukin-8 synthesis in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 31:330-336. [DOI] [PubMed] [Google Scholar]
- 19.Gu, H., and B. Roizman. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A. 104:17134-17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu, H., and B. Roizman. 2009. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J. Virol. 83:181-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagglund, R., and B. Roizman. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 78:2169-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagglund, R., C. Van Sant, P. Lopez, and B. Roizman. 2002. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci., U. S. A. 99:631-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hargett, D., S. Rice, and S. L. Bachenheimer. 2006. Herpes simplex virus type 1 ICP27-dependent activation of NF{kappa}B. J. Virol. 80:10565-10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harte, M. T., I. R. Haga, G. Maloney, P. Gray, P. C. Reading, N. W. Bartlett, G. L. Smith, A. Bowie, and L. A. O'Neill. 2003. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 197:343-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horng, T., G. M. Barton, R. A. Flavell, and R. Medzhitov. 2002. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 420:329-333. [DOI] [PubMed] [Google Scholar]
- 26.Imler, J. L., and L. Zheng. 2004. Biology of Toll receptors: lessons from insects and mammals. J. Leukoc. Biol. 75:18-26. [DOI] [PubMed] [Google Scholar]
- 27.Joo, M., Y. S. Hahn, M. Kwon, R. T. Sadikot, T. S. Blackwell, and J. W. Christman. 2005. Hepatitis C virus core protein suppresses NF-kappaB activation and cyclooxygenase-2 expression by direct interaction with IkappaB kinase beta. J. Virol. 79:7648-7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawaguchi, Y., R. Bruni, and B. Roizman. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J. Virol. 71:1019-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knipe, D. M., and J. L. Smith. 1986. A mutant herpesvirus protein leads to a block in nuclear localization of other viral proteins. Mol. Cell. Biol. 6:2371-2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kummer, M., N. M. Turza, P. Muhl-Zurbes, M. Lechmann, C. Boutell, R. S. Coffin, R. D. Everett, A. Steinkasserer, and A. T. Prechtel. 2007. Herpes simplex virus type 1 induces CD83 degradation in mature dendritic cells with immediate-early kinetics via the cellular proteasome. J. Virol. 81:6326-6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurt-Jones, E. A., J. Belko, C. Yu, P. E. Newburger, J. Wang, M. Chan, D. M. Knipe, and R. W. Finberg. 2005. The role of Toll-like receptors in herpes simplex infection in neonates. J. Infect. Dis. 191:746-748. [DOI] [PubMed] [Google Scholar]
- 32.Kurt-Jones, E. A., M. Chan, S. Zhou, J. Wang, G. Reed, R. Bronson, M. M. Arnold, D. M. Knipe, and R. W. Finberg. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. U. S. A. 101:1315-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurt-Jones, E. A., L. Mandell, C. Whitney, A. Padgett, K. Gosselin, P. E. Newburger, and R. W. Finberg. 2002. Role of Toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood 100:1860-1868. [PubMed] [Google Scholar]
- 34.Lin, R., R. S. Noyce, S. E. Collins, R. D. Everett, and K. L. Mossman. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 78:1675-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu, X., K. Fitzgerald, E. Kurt-Jones, R. Finberg, and D. M. Knipe. 2008. Herpesvirus tegument protein activates NF-kappaB signaling through the TRAF6 adaptor protein. Proc. Natl. Acad. Sci. U. S. A. 105:11335-11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lium, E. K., and S. Silverstein. 1997. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. J. Virol. 71:8602-8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez, P., C. Van Sant, and B. Roizman. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol. 75:3832-3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mansell, A., R. Smith, S. L. Doyle, P. Gray, J. E. Fenner, P. J. Crack, S. E. Nicholson, D. J. Hilton, L. A. O'Neill, and P. J. Hertzog. 2006. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat. Immunol. 7:148-155. [DOI] [PubMed] [Google Scholar]
- 40.McCarthy, A. M., L. McMahan, and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP27 deletion mutants exhibit altered patterns of transcription and are DNA deficient. J. Virol. 63:18-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medici, M. A., M. T. Sciortino, D. Perri, C. Amici, E. Avitabile, M. Ciotti, E. Balestrieri, E. De Smaele, G. Franzoso, and A. Mastino. 2003. Protection by herpes simplex virus glycoprotein D against Fas-mediated apoptosis: role of nuclear factor kappaB. J. Biol. Chem. 278:36059-36067. [DOI] [PubMed] [Google Scholar]
- 42.Melroe, G., N. DeLuca, and D. M. Knipe. 2004. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 78:8411-8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melroe, G. T., L. Silva, P. A. Schaffer, and D. M. Knipe. 2007. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: potential role in blocking IFN-beta induction. Virology 360:305-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mossman, K. L., H. A. Saffran, and J. R. Smiley. 2000. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 74:2052-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naiki, Y., K. S. Michelsen, W. Zhang, S. Chen, T. M. Doherty, and M. Arditi. 2005. Transforming growth factor-beta differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J. Biol. Chem. 280:5491-5495. [DOI] [PubMed] [Google Scholar]
- 46.Nicholl, M. J., L. H. Robinson, and C. M. Preston. 2000. Activation of cellular interferon-responsive genes after infection of human cells with herpes simplex virus type 1. J. Gen. Virol. 81:2215-2218. [DOI] [PubMed] [Google Scholar]
- 47.Nichols, D. B., and J. L. Shisler. 2006. The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor alpha-induced NF-kappaB activation via inhibition of I kappa kinase complex formation. J. Virol. 80:578-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Neill, L. A. 2006. How Toll-like receptors signal: what we know and what we don't know. Curr. Opin. Immunol. 18:3-9. [DOI] [PubMed] [Google Scholar]
- 49.Onose, A., S. Hashimoto, S. Hayashi, S. Maruoka, F. Kumasawa, K. Mizumura, I. Jibiki, K. Matsumoto, Y. Gon, T. Kobayashi, N. Takahashi, Y. Shibata, Y. Abiko, T. Shibata, K. Shimizu, and T. Horie. 2006. An inhibitory effect of A20 on NF-kappaB activation in airway epithelium upon influenza virus infection. Eur. J. Pharmacol. 541:198-204. [DOI] [PubMed] [Google Scholar]
- 50.Panagiotidis, C. A., E. K. Lium, and S. J. Silverstein. 1997. Physical and functional interactions between herpes simplex virus immediate-early proteins ICP4 and ICP27. J. Virol. 71:1547-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel, A., J. Hanson, R. I. McLean, J. Olgiate, M. Hilton, W. E. Miller, and S. L. Bachenheimer. 1998. Herpes simplex virus type 1 induction of persistent NF-κΒ nuclear translocation increases the efficiency of virus replication. Virology 247:212-222. [DOI] [PubMed] [Google Scholar]
- 52.Pellett, P. E., and B. Roizman. 2007. The family Herpesviridae: a brief introduction, p. 2479-2500. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 2. Wolters Kluwer/Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 53.Poole, E., C. A. King, J. H. Sinclair, and A. Alcami. 2006. The UL144 gene product of human cytomegalovirus activates NFkappaB via a TRAF6-dependent mechanism. EMBO J. 25:4390-4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roizman, B., D. M. Knipe, and R. J. Whitley. 2007. Herpes simplex virus, p. 2501-2602. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 2. Wolters Kluwer/Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 55.Sacks, W. R., and P. A. Schaffer. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 61:829-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sato, A., M. M. Linehan, and A. Iwasaki. 2006. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 103:17343-17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato, Y., J. Kato, R. Takimoto, K. Takada, Y. Kawano, K. Miyanishi, M. Kobune, Y. Sato, T. Takayama, T. Matunaga, and Y. Niitsu. 2006. Hepatitis C virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor alpha expression via activation of nuclear factor-kappaB. Gut 55:1801-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shisler, J. L., and X. L. Jin. 2004. The vaccinia virus K1L gene product inhibits host NF-kappaB activation by preventing IkappaBalpha degradation. J. Virol. 78:3553-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stack, J., I. R. Haga, M. Schroder, N. W. Bartlett, G. Maloney, P. C. Reading, K. A. Fitzgerald, G. L. Smith, and A. G. Bowie. 2005. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 201:1007-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571-2585. [DOI] [PubMed] [Google Scholar]
- 61.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335-376. [DOI] [PubMed] [Google Scholar]
- 62.Van Sant, C., P. Lopez, S. J. Advani, and B. Roizman. 2001. Role of cyclin D3 in the biology of herpes simplex virus 1 ICP0. J. Virol. 75:1888-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang, Q.-Y., C. Zhou, K. E. Johnson, R. C. Colgrove, D. M. Coen, and D. M. Knipe. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. U. S. A. 102:16055-16059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang, X., M. Li, H. Zheng, T. Muster, P. Palese, A. A. Beg, and A. Garcia-Sastre. 2000. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 74:11566-11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waris, G., K. D. Tardif, and A. Siddiqui. 2002. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem. Pharmacol. 64:1425-1430. [DOI] [PubMed] [Google Scholar]
- 66.Zhou, S., A. M. Cerny, G. Bowen, M. Chan, D. M. Knipe, E. A. Kurt-Jones, and R. W. Finberg. 22 July 2010. Discovery of a novel TLR2 signaling inhibitor with anti-viral activity. Antiviral Res. doi: 10.1016/j.antiviral.2010.06.011. [Epub ahead of print.] [DOI] [PMC free article] [PubMed]