

Abstract
The replication and transcription of influenza A virus are carried out by ribonucleoproteins (RNPs) containing each genomic RNA segment associated with nucleoprotein monomers and the heterotrimeric polymerase complex. These RNPs are responsible for virus transcription and replication in the infected cell nucleus. Here we have expressed, purified, and analyzed, structurally and functionally, for the first time, polymerase-RNA template complexes obtained after replication in vivo. These complexes were generated by the cotransfection of plasmids expressing the polymerase subunits and a genomic plasmid expressing a minimal template of positive or negative polarity. Their generation in vivo was strictly dependent on the polymerase activity; they contained mainly negative-polarity viral RNA (vRNA) and could transcribe and replicate in vitro. The three-dimensional structure of the monomeric polymerase-vRNA complexes was similar to that of the RNP-associated polymerase and distinct from that of the polymerase devoid of template. These results suggest that the interaction with the template is sufficient to induce a significant conformation switch in the polymerase complex.
The influenza A viruses are members of the family Orthomyxoviridae whose genome comprises eight single-stranded RNA segments of negative polarity. The extreme genetic and antigenic diversity and the segmented nature of their genome are the basis of the yearly epidemics and occasional pandemics of influenza respiratory disease. The influenza A viruses are at evolutionary equilibrium in their natural reservoir, which includes several wild avian species, and from this reservoir, viruses or viral genes can occasionally be transferred to new influenza virus strains and result in the infection of mammals, including humans (22, 68). Although fears have been generated regarding the generation of a new pandemic by highly pathogenic H5N1 viruses, which have caused sporadic cases in humans (50), a new H1N1 virus of swine origin produced worldwide outbreaks (41) and led the WHO to declare a pandemic situation (http://www.who.int/csr/disease/swineflu/4th_meeting_ihr/en/index.html).
The replication and transcription of the influenza A virus genome take place in the nucleus of infected cells and are carried out by macromolecular complexes called ribonucleoproteins (RNPs) that include each one of the virus RNA segments associated with monomers of the nucleoprotein (NP) and with the RNA-dependent RNA polymerase (reviewed in reference 12). This virus enzyme is formed by three subunits (PB1, PB2, and PA) and is responsible for both transcription and replication, as mutations in any of its subunits can lead to alterations in either process (3, 13, 14, 16, 17, 33, 51). For transcription, the virus polymerase recognizes cellular cap-containing newly synthesized cellular RNA polymerase II (Pol II) transcripts in a template-dependent manner (6, 31) and generates capped oligonucleotides that serve as primers to copy the RNP template (52, 54). The virus mRNAs are polyadenylated by the reiterative copy of an oligo(U) signal located close to the 5′ end of the genomic RNA (53, 56). In contrast to transcription, virus RNA replication in infected cells involves de novo initiation (8, 18) and proceeds via replication intermediates that are complementary full copies of the templates and that are encapsidated in the form of RNPs (cRNPs) (reviewed in reference 12). Although our understanding of virus RNA replication and transcription has improved in the last years, several alternatives are still considered potential mechanisms to explain the functional differences between virus RNA transcription and replication (reviewed in references 12, 40, and 47). It is clear that new viral proteins, specifically polymerase and NP, are required to allow the generation of progeny RNPs (7, 23, 38, 60, 67), and recently reported genetic experiments support a trans model for influenza virus RNA replication whereby a polymerase complex distinct from that present in the parental RNP is responsible for replicative RNA synthesis but not for viral transcription (26). In vitro RNA replication of short recombinant RNA templates can proceed in the absence of NP (21, 30, 43, 71), but NP improves the efficiency of replication (43) and is essential for elongation on long templates (21).
The three-dimensional (3D) structure of a biologically active recombinant RNP was reported previously (7, 36), providing basic information on the interaction of its various elements and serving as a framework for the establishment of a quasiatomic model of this RNA synthesis machine. Likewise, three-dimensional models for the polymerase complex have been reported for both the RNP-associated complex (1, 7) and a soluble polymerase devoid of template RNA (65). In addition, the atomic structures of specific polymerase domains have been solved, covering most of the PA subunit (9, 19, 45, 70), a large fraction of the PB2 subunit (17, 29, 63, 64), and the small sites of connection of PB1 with the other subunits (19, 45, 62). In an attempt to analyze the virus RNA replication process structurally and functionally, in this report we have generated influenza virus polymerase heterotrimers associated with a short model RNA template as a result of RNA replication in vivo. The three-dimensional structure of these polymerase-viral RNA (vRNA) complexes, determined by single-particle electron microscopy (EM) and image processing, indicates that the interaction with the template RNA is sufficient to induce a large conformational change in the soluble polymerase heterotrimer to adopt a structure similar to that of the polymerase present in a recombinant virus RNP.
MATERIALS AND METHODS
Biological materials.
The HEK293T (11) cell line was provided by J. C. de la Torre and cultivated as described previously (48). Plasmids pCPB1, pCPB2, and pCPA, expressing the PB1, PB2, and PA influenza virus proteins, respectively, were described previously (55). Antibodies specific for PB2 and PA were described previously (2, 46). Antiserum to the PB1 protein was generated in rabbit by using the C-terminal domain of PB1. Ni2+-nitrilotriacetic acid (NTA) agarose, Sephacryl S300, and 7 methyl-GTP-Sepharose 4B resins were obtained from Invitrogen, Sigma, and GE Healthcare, respectively.
In vitro mutagenesis and plasmid constructions.
Plasmid pCPB2-His was generated by swapping the C-terminal region of the gene from pCMVPB2His (17) into pCPB2. To generate plasmids pHHvRNA and pHHcRNA, two complementary oligonucleotides were inserted into BsmBI-digested plasmid pHH21 (42). They comprised the 5′- and 3′-terminal sequences of the NS segment, an internal sequence, and two BsmBI overhangs. Upon transfection, plasmids pHHvRNA and pHHcRNA generated vRNA and cRNA model templates, respectively (Table 1). The PB1 D445E mutant was generated by site-directed mutagenesis using the QuikChange system from Stratagene on plasmid pCPB1.
TABLE 1.
Sequences of RNA templates and probes used in this study
| Template or probe sequencea | Description |
|---|---|
| Template sequences | |
| 5′-AGUAGAAACAAGGAAAAAGAAAGAAAAGAAAAACCCUGCUUUUGCU-3′ | vRNA |
| 5′-AGCAAAAGCAGGGAAAAAGAAAGAAAAGAAAAACCUUGUUUCUACU-3′ | cRNA |
| Probe sequencesb | |
| 5′-CUUUUUCCUUGUUUCUACU-3′ | vRNA template |
| 5′-GAAAAACCUUGUUUCUACU-3′ | vRNA complement |
| 5′-CUUUUUCCCUGCUUUUGCU-3′ | cRNA template |
| 5′-GAAAAACCCUGCUUUUGCU-3′ | cRNA complement |
Underlining indicates the 5′- and 3′-terminal sequences of the NS RNA segment.
Detects.
Protein analyses.
Western blotting and silver staining of protein gels were carried out as described previously (25, 35). Discontinuous native gel electrophoresis was carried out essentially as described previously (44). In brief, samples were prepared in loading buffer (100 mM Tris-HCl, 40% glycerol, 0.5% Serva Blue G) and incubated for 10 min at room temperature. The samples were run at 15 V/cm and 4°C on 5 to 15% polyacrylamide gradient gels using a solution containing 100 mM Tris-histidine and 0.002% Serva Blue G (pH 8.0) as a cathode buffer and 100 mM Tris-HCl (pH 8.8) as an anode buffer. The molecular mass markers tyroglobulin (670 kDa), ferritin (440 kDa), and catalase (220 kDa) were used to verify that mobility did not deviate from the linear range.
Polymerase purification.
Cultures of HEK293T cells were transfected by the calcium phosphate method (69) using a mixture (for 107 cells) of plasmids pCPB1 (3 μg), pCPB2 (3 μg), and pCPA (1.5 μg). In some experiments pCPB1 D445E, pHHvRNA (12 μg), pHHcRNA (12 μg), or pCPB2-His was used. The cultures were incubated for 72 h at 32°C and collected in cold phosphate-buffered saline (PBS). Total cell extracts were prepared by incubation in 1.5 ml of buffer S1 (50 mM Tris-HCl, 100 mM NaCl, 5 mM EDTA, 0.2% Igepal, 1 mM dithiothreitol [DTT], 1 mM protease inhibitors [Incomplete; Roche] [pH 7.5]) during 2 h at 0°C with occasional vortexing. The extract was centrifuged for 30 min at 12,000 rpm and at 4°C. The supernatant was incubated with 60 μl of 7mGTP-Sepharose 4B for 2 h at 4°C in buffer S1 containing 1 μM 7mGTP. The resin was washed with 60 volumes of the same buffer and eluted stepwise with buffer S1 without EDTA and containing 140 μM 7mGTP. The eluted polymerase was diluted up to 3 ml with buffer N1 (50 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2, 0.5% Igepal, 20 mM imidazole, 10 mM 2-mercaptoethanol, 1 mM protease inhibitors [Incomplete; Roche] [pH 8.0]) and incubated with 30 μl of Ni2+-NTA agarose resin for 14 h at 4°C with agitation. The resin was washed with 100 volumes of the same buffer and eluted stepwise with buffer N2 (20 mM Tris-HCl, 100 mM KCl, 5 mM MgCl2, 0.5% Igepal, 200 mM imidazole, 10 mM 2ME). Aliquots of the purified polymerase were filtered on a 3-ml Sephacryl S300 column equilibrated with F1 buffer (50 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2, 0.5% Igepal, 5 mM 2ME) and previously calibrated with tyroglobulin (670 kDa), ferritin (440 kDa), and catalase (220 kDa).
RNA analyses.
For in vitro transcription, aliquots of purified polymerase were incubated in 20-μl reaction mixtures containing 50 mM Tris-HCl; 10 mM NaCl; 50 mM KCl; 5 mM MgCl2; 1 mM DTT; 1 mM (each) ATP, CTP, and UTP; 1 μM [α-32P]GTP (0.5 mCi/μmol); 10 μg/ml actinomycin D; and 1 U/μl human placental RNase inhibitor (pH 8.0). Either 100 μM ApG or 10 μg/ml β-globin mRNA was used as a primer, and incubation was performed for 60 min at 30°C. The reaction products were tricarboxylic acid (TCA) precipitated and quantitated with a PhosphorImager. Alternatively, they were phenol extracted, ethanol precipitated, and analyzed by electrophoresis on 15% polyacrylamide-6 M urea gels. For RNA hybridization, polymerase-associated RNA was extracted by incubation with 200 μg/ml proteinase K in a solution containing 50 mM Tris-HCl, 100 mM NaCl, 5 mM EDTA, and 1% SDS for 30 min at 37°C and phenol extraction. Aliquots of the RNAs were denatured for 15 min at 55°C in 10× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-7.5% formaldehyde and fixed on nylon filters by UV cross-linking. Hybridization was carried out overnight at 40°C in a solution containing 6× SSC, 0.5% SDS, 5× Denhardt's mixture, 20 to 27% formamide (depending on the probe), and 100 μg/ml single-stranded DNA. After washing, the filters were quantitated with a PhosphorImager. As probes, synthetic oligonucleotides specific for either positive- or negative-polarity templates or their complementary sequences (Table 1) were labeled with [γ-32P]ATP and polynucleotide kinase. As controls, various amounts of vRNA or cRNA templates, or their complements, were fixed on the hybridization filters.
Electron microscopy and 3D reconstruction of negatively stained polymerase-vRNA complexes.
Fractions from the RNA-polymerase complex obtained after gel filtration chromatography were applied directly onto carbon-coated grids and negatively stained with 2% uranyl formate. The preparations were observed with a Jeol 1230 electron microscope operating at 100 kV. Micrographs were recorded at a magnification of ×50,000 under low-dose conditions. These micrographs were digitized by using a Minolta Dimage Scan Multi Pro scanner, with the contrast transfer function corrected and downsampled to 4.2 Å/pixel. A total of 12,698 images corresponding to individual molecules of an RNA-polymerase complex were extracted. A reference-free two-dimensional (2D) analysis of this data set was performed by using EMAN (32), 2D maximum likelihood methods (59), and self-organizing maps (49). The alignment and comparison between reference-free averages and projections of the cryo-EM structure of the polymerase were performed by using commands found in EMAN and XMIPP (34, 61). 3D refinement of the data was performed by using angular-refinement methods and testing several initial references, including models obtained using common lines as well as previously reported structures of the polymerase (7), after low-pass filtering to 100 Å to reduce the model bias. The data were initially refined by using EMAN (32), and the model obtained was then subjected to further refinement by using 3D maximum likelihood methods (57). The resolution of the reconstruction was estimated to correspond to 25 Å using a cutoff of 0.3 of the correlation coefficient of the Fourier shell correlation (27 Å at an FSC of 0.5).
RESULTS
Aggregation of highly purified influenza virus RNA polymerase.
The structure of the isolated influenza virus RNA polymerase complex at low resolution was obtained by electron microscopy of stained samples and image processing (65). Since the major limitation of those studies was the small amount of sample available, we set out to develop a new purification strategy. The polymerase complex was expressed in human cells by the cotransfection of plasmids expressing the PA, PB1, and PB2 subunits, with the latter containing a C-terminal His tag. The first purification step made use of the capacity of the polymerase to recognize the cap structure (4, 17, 66). Total extracts of cells coexpressing all three polymerase subunits, or PA-containing heterodimers, were analyzed by chromatography over 7methyl-GTP-Sepharose, using Sepharose as the negative control. The binding to the resins was analyzed by Western blotting using subunit-specific antibodies and is presented in Fig. 1 A. As a positive control we used the eukaryotic initiation factor 4E (eIF4E) protein, the main cytoplasmic cap-binding protein. The retention of the polymerase in the cap resin was apparent, paralleled that of eIF4E, and was specific, since retention was not detected in the control resin (Fig. 1A). Retention was dependent on the presence of PB2, as expected, since the PB1-PA heterodimer did not bind (Fig. 1A). When PB2 and PA subunits were coexpressed, PB2 could also bind, albeit to a lesser extent, but PA was not retained, since the PB2-PA interaction is rather weak (20) (Fig. 1A). The conditions for binding to and elution from the cap resin were optimized to maximize the recovery of the polymerase and diminish the copurification of the eIF4F complex, and the eluted material was subjected to Ni2+-NTA agarose chromatography. A summary of the purification is presented in Fig. 1B, with Western blotting of the various fractions using anti-PA antibodies. The purified material contained all three polymerase subunits, as documented by Western blotting with subunit-specific antibodies (Fig. 1C). Furthermore, the polymerase subunits were the main protein components of the sample and were present in roughly equimolar amounts, as revealed by silver staining of the purified sample (Fig. 1D). Unfortunately, analysis of the purified polymerase by gel filtration on Sephacryl S300 indicated that most of the material was in a high-molecular-mass form that eluted close to the excluded volume (Fig. 1E). Such an elution profile is consistent with the formation of large polymerase aggregates (n > 3), in agreement with data from previous reports (10, 24, 25), and precluded their structural analysis.
FIG. 1.

Purification of the polymerase heterotrimer. (A) Various combinations of the polymerase subunits were expressed in HEK293T cells, as indicated at the top, and the extracts were analyzed by binding to 7mGTP-Sepharose. Aliquots of the input extracts (I) and the material bound to 7mGTP-Sepharose (7m) or bound to control Sepharose 4B resin (S) were analyzed by Western blotting using polymerase subunit-specific or eIF4E-specific antibodies. Results are shown for the analysis of the heterotrimer (Pol) or heterodimers (PB1 + PA and PB2 + PA). The positions of specific protein bands are indicated at the right, and the mobilities of molecular mass markers are shown at the left. (B) Analysis of the purification process by Western blotting with anti-PA antibodies. Aliquots of the purification fractions are presented (NB7m, not bound to 7mGTP-Sepharose; e7m1 to e7m4, elutions of the 7mGTP-Sepharose column; R7m, retained in the 7mGTP-Sepharose column; I2, input for the Ni-agarose column; en1 to en4, elutions of the Ni2+-agarose column; Rn, retained in the Ni2+-agarose column). The positions of the PA bands are indicated at the right, and the mobilities of the molecular mass markers are shown at the left. (C) Western blot analyses of the purified polymerase preparation (en) using extracts of virus-infected cells (Flu) as a control. The specificity of the antibodies used is indicated at the bottom. (D) The protein composition of two steps in the polymerase purification (e7m, 7mGTP-Sepharose eluates; en, Ni2+-agarose eluates) is shown after polyacrylamide gel electrophoresis and silver staining. The mobilities of molecular weight (MW) markers (in thousands) are shown on the left, and the positions of relevant bands are indicated on the right. (E) Gel filtration of the purified polymerase on Sephacryl S300. The Western blot analysis of the elution fractions is presented, using anti-PA antibodies. Numbers at the top indicate elution fractions. The position of the PA-specific band is shown at the right (Flu). The elution of molecular mass markers is indicated at the bottom (T, tyroglobulin; F, ferritin; C, catalase).
Generation of viral RNA polymerase-vRNA complexes.
As the purified polymerase preparations described above contain reduced amounts of associated host factors compared to those used for proteomic analyses (27), a possible explanation for the observed aggregation could be that the naked heterotrimer is able to self-interact, possibly through its PB1 or PB2 subunit (25). This is in contradistinction to the purified recombinant RNPs, which behave as monomeric complexes by gel filtration (7). Hence, we reasoned that a polymerase-vRNA complex, which would emulate the polymerase complex in the RNP, might remain monomeric and be amenable for structural analysis. Several groups, including ours (65), have generated polymerase-promoter complexes in vitro (12, 40, 47), but the efficiency of such a process is low. Therefore, we set out to generate such complexes in vivo by the coexpression of the polymerase subunits and a 46-nucleotide (nt)-long model vRNA template (Table 1). The purification process indicated above was again optimized by using the in vitro transcription activity as an assay, in an attempt to purify the polymerase-vRNA complexes. A summary of the new purification process is presented in Fig. 2 A, using Western blot analysis of the various fractions with PA-specific antibodies. The purified material contained all three polymerase subunits, as shown by Western blotting with subunit-specific antibodies (Fig. 2B). Analysis of the purified samples by polyacrylamide gel electrophoresis and silver staining revealed that the ratio among polymerase subunits was roughly equimolar, and these were the most abundant proteins in the sample (Fig. 2C, lane en). Similar preparations were obtained when a His-PA subunit was used instead of PB2-His (data not shown). To characterize the potential viral RNA associated with the purified polymerase, total RNA was isolated, dephosphorylated, and labeled with [γ-32P]ATP using polynucleotide kinase, and the labeled RNA was analyzed by denaturing polyacrylamide gel electrophoresis. As a control, the potential RNA associated with polymerase purified from cells transfected only with the subunit-expressing plasmids was analyzed in parallel. The results are presented in Fig. 2D and show that no RNA was detected in the control preparation (Fig. 2D, lane Pol), while a main band of the expected size was apparent in the polymerase-vRNA preparation (Fig. 2D, lane vPol, arrow). In addition, minor bands of 18 to 20 nt were also detected (Fig. 2D, lane vPol, stars), which could correspond to the degradation of the template during purification. To determine the specificity, polarity, and amount of the potential viral RNA associated with the purified polymerase, samples of the purified RNA were spotted onto nylon filters along with known amounts of synthetic oligonucleotides containing either a sequence identical to the model vRNA coexpressed intracellularly or the corresponding complementary sequence. The filters were hybridized with two synthetic oligonucleotide probes after 5′-terminal labeling (Table 1). The results obtained from six independently purified preparations are shown in Fig. 2E. These results indicated that the RNA associated with the polymerase is virus specific and corresponds mostly to negative polarity, although a small proportion was positive-polarity cRNA.
FIG. 2.

Purification and characterization of polymerase-vRNA complexes. (A) Analysis of the purification process by Western blotting with anti-PA antibodies. Aliquots of the purification fractions are presented (I, input extract; NB7m, not bound to 7mGTP-Sepharose; e7m, elutions of the 7mGTP-Sepharose column; R7m, retained in the 7mGTP-Sepharose column; en, elutions of the Ni2+-agarose column; Rn, retained in Ni2+-agarose). The positions of the PA bands are indicated at the right, and the mobilities of molecular mass marker are shown at the left. (B) Western blot analyses of the purified polymerase-RNA preparation (en) using extracts of virus-infected cells (Flu) as a control. The specificities of the antibodies used are indicated at the bottom. (C) The protein composition of two steps in the polymerase purification (e7m, 7mGTP-Sepharose eluates; en, Ni2+-agarose eluates) is shown after polyacrylamide gel electrophoresis and silver staining. The mobilities of molecular weight markers (in thousands) are shown at the left, and the positions or relevant bands are indicated at the right. (D) Analysis of the RNA associated with the polymerase. The RNA present in purified preparations of polymerase (Pol) (Fig. 1) or polymerase-RNA (vPol) was isolated, dephosphorylated, terminally labeled with [γ-32P]ATP, and analyzed by denaturing polyacrylamide gel electrophoresis. The autoradiograph is presented, as are the mobilities of labeled oligonucleotides of 40 and 18 nt used as molecular weight markers. The arrow indicates the major RNA band, and the stars indicate two potential degradation products. (E) The RNA associated with purified polymerase-RNA preparations indicated in C (en) was extracted and analyzed by dot blot hybridization using specific probes of either polarity. Known amounts of homologous synthetic oligonucleotides corresponding to the vRNA template and its complement were included as controls. The averages and standard deviations of data from six independent experiments are presented.
To ascertain the apparent molecular mass of the purified polymerase-vRNA complexes, they were analyzed by gel filtration on Sephacryl S300. The results are presented in Fig. 3 A, which shows a Western blot with PA-specific antibodies and shows that these complexes were not in an aggregated form (compare Fig. 1E and 3A) but, rather, eluted as expected for a monomeric heterotrimer. Since the resolution of the gel filtration chromatography was low, the size of the polymerase-vRNA complexes was determined by native polyacrylamide gel electrophoresis and Western blotting with subunit-specific antibodies. In agreement with the results presented in Fig. 1E, the purified RNA-free polymerase showed a mobility consistent with the presence of aggregates (n > 3, slower than tyroglobulin) (Fig. 3B, lane Pol). On the contrary, the polymerase-vRNA preparation revealed a main species moving close to the catalase marker and a minor species moving as the ferritin marker, whose abundance was variable among preparations (Fig. 3B, lane vPol). These results indicated that the polymerase-vRNA preparations contain mainly polymerase heterotrimers. A minor, variable band that corresponded to a slower-migrating complex was also detected. This could correspond to the dimeric complexes of the polymerase, in agreement with previous results (25). The observation that essentially no aggregation was detected in the polymerase-RNA preparation supports the idea that most of the polymerase present was associated with the template. This contention is also supported by the three-dimensional reconstruction of the monomeric polymerase-vRNA complexes (see below).
FIG. 3.

Size analyses of polymerase-vRNA complexes. Polymerase-vRNA complexes were prepared and purified as indicated in Materials and Methods. (A) Gel filtration on Sephacryl S300. A Western blot analysis of the elution fractions using anti-PA antibodies is presented. Numbers at the top indicate elution fractions. The position of the PA-specific band is shown at the right (Flu). (B) Separation by native blue gel electrophoresis of purified polymerase (Pol) or polymerase-vRNA complexes (vPol). The mobility of the polymerase was determined by Western blotting with subunit-specific antibodies, as indicated at the bottom of each panel. The positions of molecular mass markers are indicated (T, tyroglobulin; F, ferritin; C, catalase).
Functional analyses of the polymerase-vRNA complexes.
The polymerase-vRNA complexes might represent the result of a direct interaction of the polymerase expressed in the transfected cells with the RNA Pol I-vRNA model transcript or, alternatively, might be the product of in vivo replication of the cell-derived vRNA model by the activity of the expressed polymerase. To ascertain which of these possibilities is more likely, the generation of the polymerase-vRNA complexes was tested by the mutation of the polymerase active site. Wild-type (wt) polymerase or a mutant polymerase containing the PB1 D445E mutant (3) was expressed in the presence or absence of the plasmid expressing the 46-nt-long model vRNA template. The polymerase was purified as indicated above, and the purified preparations were analyzed by native polyacrylamide gel electrophoresis. The polymerase complexes expressed in the absence of a template donor behaved as expected; i.e., they aggregated and moved slower than the tyroglobulin marker, close to the gel origin (Fig. 4 lanes Pol and 445). On the other hand, the wt polymerase-vRNA complexes moved as shown in Fig. 3B. Surprisingly, however, most of the material purified from cells expressing PB1 D445E mutant polymerase in combination with the template donor moved as aggregates, in a way similar to that of the polymerase devoid of template vRNA (Fig. 4, lane v445). To ascertain whether the material purified from cells coexpressing PB1 D445E mutant polymerase and the template donor did contain the model vRNA, total RNA present in the preparations shown in Fig. 4 was extracted, applied onto a nylon filter, and analyzed by hybridization with a negative-polarity probe. The results demonstrated that no association with the template vRNA was detected in the v445 preparation (data not shown). Altogether, these results indicate that the polymerization activity of the viral RNA polymerase is required for the generation of the polymerase-vRNA complexes described above.
FIG. 4.

Requirements for the generation of monomeric polymerase-vRNA complexes. Viral polymerase was expressed in the presence (vPol) or absence (Pol) of a template-generating plasmid. Two alternative PB1 alleles were employed, either the wt or the D445E mutant (445). Wild-type or mutant 445 polymerase was expressed in the presence or absence of template-generating plasmid, purified, and analyzed by native blue gel electrophoresis and Western blotting with anti-PA antibodies. The positions of molecular mass markers are indicated (T, tyroglobulin; F, ferritin; C, catalase).
Generation of viral RNA polymerase-cRNA complexes.
If the polymerase-vRNA complexes described above were indeed the result of the intracellular amplification of minimal, NP-free replication complexes, one would expect a similar situation to occur when a positive-polarity template is expressed together with the polymerase. Thus, we coexpressed the polymerase subunits with a 46-nt-long cRNA template analogous to the vRNA used above (Table 1). The polymerase complexes were purified by using a protocol optimized for the purification of polymerase-vRNA complexes, and the purified material was analyzed by Western blotting and silver staining after polyacrylamide gel electrophoresis. The results are presented in Fig. 5 A and B and indicate that highly purified polymerase preparations were obtained. These purified polymerase complexes migrated mostly as a single heterotrimeric complex when analyzed by native gel electrophoresis and Western blotting (Fig. 5C), whereas most of the purified material aggregated and moved slower than the tyroglobulin marker (n > 3) when similar polymerase complexes were prepared by coexpressing the PB1 D445E mutant, PB2, PA, and plasmid pHHcRNA, as described above for mutant polymerase-vRNA complexes (Fig. 4). In agreement with this aggregation, much-reduced levels of virus RNA was associated with mutant D445E polymerase complexes (data not shown).
FIG. 5.

Purification and characterization of polymerase-cRNA complexes. Polymerase-RNA complexes were generated in vivo by the coexpression of the polymerase subunits and a cRNA model template of 46 nt. (A) Western blot analyses of the purified polymerase-RNA preparation (en) using extracts of virus-infected cells (Flu) as a control. The specificities of the antibodies used are indicated at the bottom. (B) The protein composition of two steps in the polymerase purification (e7m, 7mGTP-Sepharose eluates; en, Ni2+-agarose eluates) is shown after polyacrylamide gel electrophoresis and silver staining. The mobilities of molecular weight markers (in thousands) are shown at the left, and the positions or relevant bands are indicated at the right. (C) Wild-type or position 445 mutant polymerase was expressed in the presence of the cRNA template-generating plasmid, purified, and analyzed by native blue gel electrophoresis and Western blotting with anti-PA antibodies. The positions of molecular mass markers are indicated (T, tyroglobulin; F, ferritin; C, catalase).
To analyze the RNA present in the purified polymerase complexes obtained, it was extracted, 5′-terminally labeled, and analyzed by polyacrylamide gel electrophoresis and autoradiography. The results indicated the presence of a band of the expected size (46 nt) (Fig. 6 A, arrow) as well as two additional bands compatible with the degradation of the main band (Fig. 6A, stars). In addition, the specificity and polarity of the polymerase-associated RNA were analyzed by dot blot hybridization with specific probes (Table 1). Although the template RNA provided by the transfecting plasmid was of positive polarity (i.e., cRNA), most of the polymerase-associated RNA was of negative polarity (vRNA) (Fig. 6B), in agreement with the hypothesis that intracellular replication is responsible for the generation of the polymerase-vRNA/cRNA complexes.
FIG. 6.
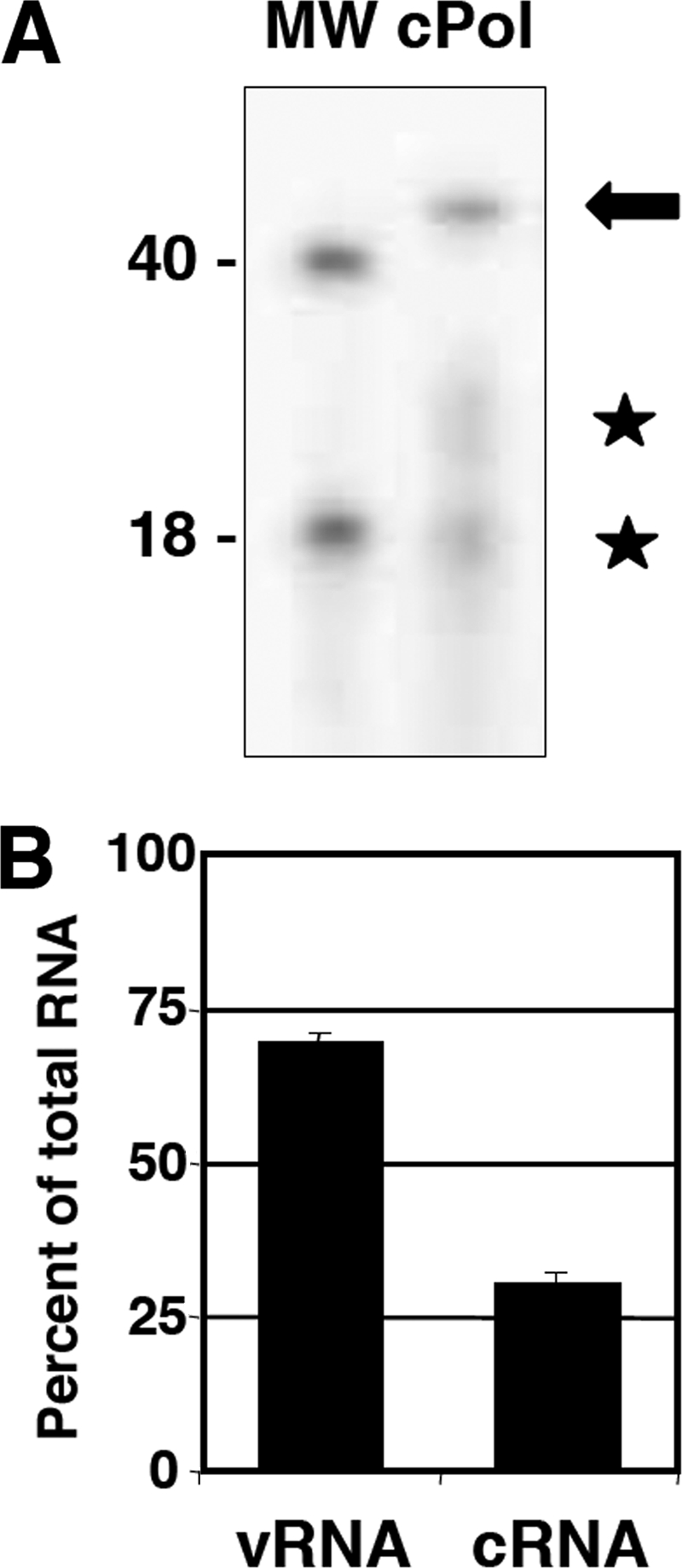
Characterization of the RNA associated with polymerase-cRNA complexes. Polymerase-RNA complexes were generated in vivo by the coexpression of the polymerase subunits and a cRNA model template of 46 nt. (A) The RNA present in purified preparations of polymerase-cRNA complexes (cPol) (Fig. 9) was isolated, dephosphorylated, terminally labeled with [γ-32P]ATP, and analyzed by denaturing polyacrylamide gel electrophoresis. The autoradiograph is presented, as are the mobilities of labeled oligonucleotides of 40 and 18 nt. The arrow indicates the major RNA band, and the stars indicate two potential degradation products. (B) The RNA associated with purified polymerase-RNA preparations indicated in A was extracted and analyzed by dot blot hybridization using specific probes of either polarity. Known amounts of homologous synthetic oligonucleotides corresponding to the cRNA template and its complement were included as controls. The averages and standard deviations of data from four independent experiments are presented.
In vitro analyses of the polymerase-vRNA/cRNA complexes.
Finally, to determine the RNA synthesis activity associated with the purified polymerase-genomic RNA complexes, they were used as an enzyme template for in vitro reactions in the presence of either ApG or β-globin mRNA. These results are presented in Fig. 7. The products obtained using ApG as a primer indicated the synthesis of a full-length copy of the template as well as a truncated copy of about 36 nt (Fig. 7). The proportion of the full-length copy of the template was higher for the preparations generated with a cRNA input than those obtained with a vRNA input (Fig. 7). When β-globin mRNA was used as the primer donor, a major band corresponding in length to the capped oligonucleotide-primed, truncated copy was detected (Fig. 7), which was clearly distinguishable from the ApG-primed full-length product. Only a marginal amount of a product with the length of a cap-primed full copy of the template was found (Fig. 7).
FIG. 7.

In vitro RNA synthesis activities of polymerase-template RNA complexes. (A) Purified polymerase (Pol) or polymerase-template RNA complexes (vPol/cPol) were used for in vitro RNA synthesis using ApG or β-globin mRNA (βg) as a primer. The products were purified and analyzed by polyacrylamide-urea gel electrophoresis and autoradiography. The positions of full-length (FL) or truncated (Tr) products as well as capped oligonucleotide-primed full-length (arrowhead) or capped truncated (star) products are indicated at the right. The mobilities of molecular weight markers (in thousands) are shown at the left.
Three-dimensional reconstruction of monomeric polymerase-vRNA complexes.
The structure of the viral polymerase-vRNA complexes described above was then studied by electron microscopy. The preparations revealed globular particles with a size appropriate for the mass of the polymerase heterotrimers (i.e., monomers) (Fig. 8 A and B) and a minor proportion of dimeric structures (Fig. 8A). Monomeric particles were analyzed by several reference-free classification and averaging methods, revealing a slightly elongated globular protein complex with a narrower end at the bottom and a somewhat wider end at the top, comparable with what was observed previously for the isolated RNA polymerase (65) (Fig. 8C). The comparison of each of these 2D averages with the computational projections from the 3D cryo-EM structure of the polymerase extracted for the RNP after filtering to a resolution similar to those of the present studies (Fig. 8D) revealed the agreement between the two structures (1, 7). Unexpectedly, some 2D averages of the polymerase-vRNA complex showed a roughly tubular density projecting outwards from the core of the structure (Fig. 8E, arrowheads). This density had a significant signal over the background noise of the images and could not be interpreted as a constituent of the core subunits of the polymerase. The most likely explanation is that such a linear density could correspond to a segment of the 46-nt-long RNA that is present in the polymerase-vRNA complex and absent in previous experiments. Even if parts of this RNA are bound and occluded within the structure of the polymerase, some RNA could be projecting outwards, and the length that we observed for this linear density (60 to 70 Å) is within the range expected for the RNA used. Roughly 35 to 40% of all particles were classified into classes containing the putative RNA density. Given that only certain orientations of the polymerase-RNA complex could generate a view where the RNA would project outwards to be detected visually, we believe that a majority of the molecules in the preparation were bound to RNA.
FIG. 8.

Electron microscopy of the polymerase-vRNA complexes. (A) Selected field from an electron micrograph of the polymerase-vRNA complex. Some monomers (arrows) and putative dimers (within circles) are highlighted. (B) Gallery of individual images extracted from the micrographs. (C) Gallery of selected reference-free 2D averages obtained from the EM data collected for the purified polymerase-vRNA complexes. (D) Reference-free 2D averages were computationally compared and aligned with the computational projections of the previously reported cryo-EM structure of the polymerase extracted from the RNP complex (1, 7) and filtered to the resolution of the present studies. (E) Some 2D averages revealed, after alignment with the structure of the polymerase extracted from the RNP, the presence of some extra density with a tubular appearance and projecting outwards from the polymerase (black arrows). (F) The particles classified within selected reference-free 2D averages with some additional density protruding from the structure (only one class average is shown as an example) were further classified using self-organizing maps (49). A mask was used to focus the classification on the protruding density to obtain new averages for subsets of the data.
To further support that this extra density is a genuine feature of the polymerase-vRNA complex, the images within a homogenous subset of the data were further reclassified by using self-organizing maps (58). A mask was used to focus the classification on the variations within an area containing the extra density (Fig. 8F). Several subclasses were obtained, revealing that material consistently protruded from the same region of the polymerase, although differences among the averages suggested a certain degree of flexibility of this element or differences in the way in which it was stained.
The two-dimensional averages obtained after the 3D refinement of the images from the monomers (Fig. 9 A) were very similar to the reference-free averages obtained for the raw data (Fig. 8C), supporting the consistency of the structure solved. Furthermore, the presence of the same density protruding from the polymerase was again consistently found in these averages despite the use of a different image-processing strategy (Fig. 9A). When the 3D reconstruction of the polymerase-vRNA complex was observed at a threshold sufficient to include the expected mass of the polymerase, the model revealed a narrow region and a wider region at opposite ends of the molecule (Fig. 9B). One large opening was found in the front of the molecule, whereas the back lacked any large aperture. Interestingly, when the threshold used to render the structure was slightly modified to include surrounding regions of density significantly above the background noise (Fig. 9C), some material was found to project from the front side of the polymerase-vRNA complex. The terminal end of this density appeared as a blob, likely reflecting an average of a certain “smear” of this putative RNA, suggesting some degree of flexibility, in accordance with the observations for the 2D averages (Fig. 8F). The strong density value of this structural feature in the 3D average supports that RNA is present in the large majority of the molecules. As an additional control, we cropped the density of the putative RNA from the reconstruction, and this model was used as a template for the refinement of the data. Despite the bias introduced by this model, the data rapidly refined into a structure where the RNA density was present, supporting its consistency. The structural features of the 3D structure of the polymerase-vRNA complex were fairly similar to the overall conformation displayed by the polymerase within the RNP (7) (Fig. 10), although the present structure has been solved at a lower resolution. Both structures consist of a closed “back” and an open “front,” which contrasts with the structure of the isolated polymerase in the absence of RNA (65).
FIG. 9.

Three-dimensional reconstruction of the polymerase-vRNA complex. (A) Gallery of selected 2D averages obtained after the angular refinement of the EM data collected for the purified polymerase-vRNA complex. White arrows indicate the added density with a tubular appearance. (B and C) Two orthogonal views of the low-resolution EM structure of the polymerase-vRNA complex. (Top) Structure with a threshold sufficient to enclose 100% of the protein. (Bottom) 3D reconstruction rendered at a threshold that takes into account the surrounding areas of density above the background noise. Arrows indicate the extra density with a tubular appearance.
FIG. 10.

Comparison between the polymerase-vRNA complex and the polymerase present in an RNP. Four views of the 25-Å resolution structure of the polymerase-vRNA complex (blue) are presented in comparison with the cryo-EM structure of the polymerase extracted from the RNP (7) (yellow). F, front view; B, back view; S1 and S2, side views.
DISCUSSION
Interaction with viral RNA abolishes the oligomerization of influenza virus RNA polymerase. Our attempts to improve the expression and purification of the influenza virus RNA polymerase complex for structural studies led to preparations with the appropriate yield and purity, but the purified polymerase showed an apparent molecular mass corresponding to protein aggregates in the MDa range (Fig. 1). Since similarly purified recombinant RNPs behaved as monomers by gel filtration (7), one could presume that the interaction of the polymerase with the template RNA and/or the NP should eliminate its capacity to oligomerize. Indeed, when the polymerase was coexpressed in vivo with a short model vRNA template, we could optimize the purification of polymerase-vRNA complexes that did not oligomerize and could be studied structurally (Fig. 2, 3). These results suggest that the simple interaction of the polymerase with the vRNA template is sufficient to generate a complex that emulates the polymerase-promoter complex in the RNP structure, in line with previous results (5). Indeed, the three-dimensional structure obtained for such a monomeric complex is similar to the structure previously reported for the RNP-associated polymerase by cryo-EM (7) (Fig. 9), although the resolution obtained here was much lower (around 25 Å). In addition, the two-dimensional analysis of the images and the comparison of two-dimensional average images with the projections of the cryo-EM model denoted the presence of a bona fide tubular additional density that could represent the template RNA not covered by the polymerase complex (Fig. 8). The length of such a naked RNA would be around 20 nt, i.e., around 70 Å, which is compatible with the dimensions of the tubular density observed. Furthermore, the location of the RNA loop in the EM structure of the polymerase-vRNA complex provides information on the sites of entry/exit of the template RNA into the polymerase heterotrimer complex (Fig. 9C and 10F).
Generation in vivo of polymerase-RNA complexes requires the polymerization activity of viral polymerase. Attempts to generate polymerase-vRNA complexes using a polymerase with a lethal mutation in PB1 led to complete failures. The purified materials were indistinguishable from the oligomerized polymerase preparations (Fig. 4). These results strongly suggested that the formation of the polymerase-vRNA complexes in vivo is the result of the replication of some initial template provided by the transcription of the genomic plasmid by RNA polymerase I and does not represent a direct complex formation of polymerase and plasmid-driven template RNA. In other words, the polymerase-vRNA complexes obtained could be considered “progeny” of the input template or “micro-RNPs” devoid of NP. This interpretation would be feasible. Thus, although it is well known that NP is essential for the replication of full-length viral templates (7, 21, 23, 38, 60), it has been shown that the polymerase complex can replicate in vitro short RNA templates in the absence of NP (21, 30, 43). Should this interpretation be correct, it follows that a polymerase-cRNA complex should also be generated in vivo, and indeed, a small fraction of the purified polymerase-vRNA complexes does contain cRNA (Fig. 2E). Furthermore, when the polymerase was coexpressed in vivo with a short model cRNA template, polymerase complexes similar to those obtained by providing vRNA were obtained (Fig. 5). Thus, a mixture of monomeric polymerase-RNA complexes could be purified only when a wt PB1 subunit was included but not when the PB1 D445E mutant was employed (Fig. 5C). Importantly, most of the viral RNA associated with these polymerase-RNA complexes was vRNA (Fig. 6B), further implying that they are the final products of in vivo replication. To the best of our knowledge, this is the first description of the in vivo replication of influenza virus RNA in the absence of NP. At this time, it is difficult to compare the efficiencies of RNA replication in the presence and absence of NP. It is expected that, as shown previously in vitro (43), the expression of NP would enhance RNA replication in vivo. In addition, the presence of NP should be relevant for the processivity of the polymerase and/or the stabilization of longer templates in vivo (21, 67). The potential role of NP in the switch from the primary transcription phase to the replication step during infection cannot be ascertained with the experiments presented here, since no primary transcription is required for replication to occur. As newly synthesized polymerase is available in trans, the replication of template RNA can proceed without a previous transcription step. Hence, our results do not distinguish between previous models for the mechanisms of transition from primary transcription to replication (15, 26, 28, 37, 39, 60, 67).
The results obtained when the purified polymerase-template RNA complexes were used for in vitro RNA synthesis indicated that a fraction of the RNA newly generated in vitro using ApG as a primer was a truncated product of about 36 nt. This could correspond to an incomplete transcript primed with ApG and terminated prematurely because of steric hindrance by the polymerase still bound to the template. This product is not polyadenylated because the template lacks the poly(A) signal and could be interpreted as the result of the transcription in cis of the most abundant monomeric complexes (26). In vitro RNA synthesis also led to full-length copies of the template that could correspond to replication products. When the in vitro reaction was primed with β-globin mRNA, most of the products were incomplete transcripts, in this case with an extra length corresponding to the capped oligonucleotide primer, and only a faint band of artifactual, capped full-length product was detected.
In summary, we have generated, purified, and analyzed biochemically, structurally, and functionally polymerase-template RNA complexes that represent final in vivo replication products. The results obtained demonstrate that the NP is not essential for RNA replication of short RNA templates in vivo and provide structural support for the role of the vRNA template in the stabilization and functionality of the polymerase. Furthermore, our results suggest that interaction with the template RNA is sufficient to induce a considerable conformation switch in the polymerase and provide structural information about the potential entry/exit sites of the template RNA in the polymerase complex.
Acknowledgments
We thank D. Hart for the generous donation of the purified C-terminal domain of the polymerase PB1 protein. We gratefully acknowledge the technical assistance of Y. Fernández, C. Terrón, and M. Benavides.
This work has been supported by grants BFU2007-60046 (J.O.) and SAF2008-00451 (O.L.) from the Spanish Ministry of Science; project RD06/0020/1001 of the Red Temática de Investigación Cooperativa en Cáncer (RTICC) (O.L.); CIBER de Enfermedades Respiratorias (J.O.), financed by the Instituto de Salud Carlos III; and the VIRHOST Program, financed by Comunidad de Madrid (J.O.). M.A.R.-C. was supported by an FPI fellowship from the Spanish Ministry of Science, and P.R.-I. was supported by a predoctoral fellowship of the Spanish Research Council. O.L.'s group is additionally supported by the Human Frontiers Science Program Organization (grant RGP39/2008), and J.O.'s group is additionally supported by the Fundación Marcelino Botin.
Footnotes
Published ahead of print on 11 August 2010.
REFERENCES
- 1.Area, E., J. Martín-Benito, P. Gastaminza, E. Torreira, J. M. Valpuesta, J. L. Carrascosa, and J. Ortín. 2004. Three-dimensional structure of the influenza virus RNA polymerase: localization of subunit domains. Proc. Natl. Acad. Sci. U. S. A. 101:308-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bárcena, J., M. Ochoa, S. de la Luna, J. A. Melero, A. Nieto, J. Ortín, and A. Portela. 1994. Monoclonal antibodies against influenza virus PB2 and NP polypeptides interfere with the initiation step of viral mRNA synthesis in vitro. J. Virol. 68:6900-6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biswas, S. K., and D. P. Nayak. 1994. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J. Virol. 68:1819-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaas, D., E. Patzelt, and E. Keuchler. 1982. Identification of the cap binding protein of influenza virus. Nucleic Acids Res. 10:4803-4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brownlee, G. G., and J. L. Sharps. 2002. The RNA polymerase of influenza A virus is stabilized by interaction with its viral RNA promoter. J. Virol. 76:7103-7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cianci, C., L. Tiley, and M. Krystal. 1995. Differential activation of the influenza virus polymerase via template RNA binding. J. Virol. 69:3995-3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coloma, R., J. M. Valpuesta, R. Arranz, J. L. Carrascosa, J. Ortin, and J. Martin-Benito. 2009. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 5:e1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng, T., F. T. Vreede, and G. G. Brownlee. 2006. Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. J. Virol. 80:2337-2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dias, A., D. Bouvier, T. Crepin, A. A. McCarthy, D. J. Hart, F. Baudin, S. Cusack, and R. W. Ruigrok. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914-918. [DOI] [PubMed] [Google Scholar]
- 10.Digard, P., V. C. Blok, and S. C. Inglis. 1989. Complex formation between influenza virus polymerase proteins expressed in Xenopus oocytes. Virology 171:162-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DuBridge, R. B., P. Tang, H. C. Hsia, P. M. Leong, J. H. Miller, and M. P. Calos. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7:379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elton, D., P. Digard, L. Tiley, and J. Ortín. 2006. Structure and function of the influenza virus RNP, p. 1-36. In Y. Kawaoka (ed.), Influenza virology: current topics. Caister Academic Press, Norfolk, United Kingdom.
- 13.Fechter, P., L. Mingay, J. Sharps, A. Chambers, E. Fodor, and G. G. Brownlee. 2003. Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J. Biol. Chem. 278:20381-20388. [DOI] [PubMed] [Google Scholar]
- 14.Fodor, E., M. Crow, L. J. Mingay, T. Deng, J. Sharps, P. Fechter, and G. G. Brownlee. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 76:8989-9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fodor, E., D. C. Pritlove, and G. G. Brownlee. 1994. The influenza virus panhandle is involved in the initiation of transcription. J. Virol. 68:4092-4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gastaminza, P., B. Perales, A. M. Falcón, and J. Ortín. 2003. Influenza virus mutants in the N-terminal region of PB2 protein are affected in virus RNA replication but not transcription. J. Virol. 76:5098-5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guilligay, D., F. Tarendeau, P. Resa-Infante, R. Coloma, T. Crepin, P. Sehr, J. Lewis, R. W. Ruigrok, J. Ortin, D. J. Hart, and S. Cusack. 2008. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 15:500-506. [DOI] [PubMed] [Google Scholar]
- 18.Hay, A. J., J. J. Skehel, and J. McCauley. 1982. Characterization of influenza virus RNA complete transcripts. Virology 116:517-522. [DOI] [PubMed] [Google Scholar]
- 19.He, X., J. Zhou, M. Bartlam, R. Zhang, J. Ma, Z. Lou, X. Li, J. Li, A. Joachimiak, Z. Zeng, R. Ge, Z. Rao, and Y. Liu. 2008. Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus. Nature 454:1123-1126. [DOI] [PubMed] [Google Scholar]
- 20.Hemerka, J. N., D. Wang, Y. Weng, W. Lu, R. S. Kaushik, J. Jin, A. F. Harmon, and F. Li. 2009. Detection and characterization of influenza A virus PA-PB2 interaction through a bimolecular fluorescence complementation assay. J. Virol. 83:3944-3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honda, A., K. Ueda, K. Nagata, and A. Ishihama. 1988. RNA polymerase of influenza virus: role of NP in RNA chain elongation. J. Biochem. 104:1021-1026. [DOI] [PubMed] [Google Scholar]
- 22.Horimoto, T., and Y. Kawaoka. 2005. Influenza: lessons from past pandemics, warnings from current incidents. Nat. Rev. Microbiol. 3:591-600. [DOI] [PubMed] [Google Scholar]
- 23.Huang, T. S., P. Palese, and M. Krystal. 1990. Determination of influenza virus proteins required for genome replication. J. Virol. 64:5669-5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huet, S., S. Avilov, L. Ferbitz, N. Daigle, S. Cusack, and J. Ellenberg. 2010. Nuclear import and assembly of the influenza A virus RNA polymerase studied in live cells by fluorescence cross correlation spectroscopy. J. Virol. 84:1254-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jorba, N., E. Area, and J. Ortin. 2008. Oligomerization of the influenza virus polymerase complex in vivo. J. Gen. Virol. 89:520-524. [DOI] [PubMed] [Google Scholar]
- 26.Jorba, N., R. Coloma, and J. Ortin. 2009. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog. 5:e1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorba, N., S. Juarez, E. Torreira, P. Gastaminza, N. Zamarreno, J. P. Albar, and J. Ortin. 2008. Analysis of the interaction of influenza virus polymerase complex with human cell factors. Proteomics 8:2077-2088. [DOI] [PubMed] [Google Scholar]
- 28.Klumpp, K., R. W. Ruigrok, and F. Baudin. 1997. Roles of the influenza virus polymerase and nucleoprotein in forming a functional RNP structure. EMBO J. 16:1248-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuzuhara, T., D. Kise, H. Yoshida, T. Horita, Y. Murazaki, A. Nishimura, N. Echigo, H. Utsunomiya, and H. Tsuge. 2009. Structural basis of the influenza A virus RNA polymerase PB2 RNA-binding domain containing the pathogenicity-determinant lysine 627 residue. J. Biol. Chem. 284:6855-6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, M. T., K. Bishop, L. Medcalf, D. Elton, P. Digard, and L. Tiley. 2002. Definition of the minimal viral components required for the initiation of unprimed RNA synthesis by influenza virus RNA polymerase. Nucleic Acids Res. 30:429-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee, M. T., K. Klumpp, P. Digard, and L. Tiley. 2003. Activation of influenza virus RNA polymerase by the 5′ and 3′ terminal duplex of genomic RNA. Nucleic Acids Res. 31:1624-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ludtke, S. J., P. R. Baldwin, and W. Chiu. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82-97. [DOI] [PubMed] [Google Scholar]
- 33.Mahy, B. W. J. 1983. Mutants of influenza virus, p. 192-253. In P. Palese and D. W. Kingsbury (ed.), Genetics of influenza viruses. Springer-Verlag, Wien, Germany.
- 34.Marabini, R., I. M. Masegosa, C. San Martín, S. Marco, J. J. Fernández, L. G. de la Fragra, C. Vaquerizo, and J. M. Carazo. 1996. X-mipp: an image processing package for electron microscopy. J. Struct. Biol. 116:237-240. [DOI] [PubMed] [Google Scholar]
- 35.Marión, R. M., T. Zürcher, S. de la Luna, and J. Ortín. 1997. Influenza virus NS1 protein interacts with viral transcription-replication complexes in vivo. J. Gen. Virol. 78:2447-2451. [DOI] [PubMed] [Google Scholar]
- 36.Martín-Benito, J., E. Area, J. Ortega, O. Llorca, J. M. Valpuesta, J. L. Carrascosa, and J. Ortín. 2001. Three dimensional reconstruction of a recombinant influenza virus ribonucleoprotein particle. EMBO Rep. 2:313-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medcalf, L., E. Poole, D. Elton, and P. Digard. 1999. Temperature-sensitive lesions in two influenza A viruses defective for replicative transcription disrupt RNA binding by the nucleoprotein. J. Virol. 73:7349-7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mena, I., S. de la Luna, C. Albo, J. Martín, A. Nieto, J. Ortín, and A. Portela. 1994. Synthesis of biologically active influenza virus core proteins using a vaccinia-T7 RNA polymerase expression system. J. Gen. Virol. 75:2109-2114. [DOI] [PubMed] [Google Scholar]
- 39.Mena, I., E. Jambrina, C. Albo, B. Perales, J. Ortin, M. Arrese, D. Vallejo, and A. Portela. 1999. Mutational analysis of influenza A virus nucleoprotein: identification of mutations that affect RNA replication. J. Virol. 73:1186-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neumann, G., G. G. Brownlee, E. Fodor, and Y. Kawaoka. 2004. Orthomyxovirus replication, transcription, and polyadenylation. Curr. Top. Microbiol. Immunol. 283:121-143. [DOI] [PubMed] [Google Scholar]
- 41.Neumann, G., T. Noda, and Y. Kawaoka. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neumann, G., T. Watanabe, H. Ito, S. Watanabe, H. Goto, P. Gao, M. Hughes, D. R. Perez, R. Donis, E. Hoffmann, G. Hobom, and Y. Kawaoka. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. U. S. A. 96:9345-9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newcomb, L. L., R. L. Kuo, Q. Ye, Y. Jiang, Y. J. Tao, and R. M. Krug. 2009. Interaction of the influenza A virus nucleocapsid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J. Virol. 83:29-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niepmann, M., and J. Zheng. 2006. Discontinuous native protein gel electrophoresis. Electrophoresis 27:3949-3951. [DOI] [PubMed] [Google Scholar]
- 45.Obayashi, E., H. Yoshida, F. Kawai, N. Shibayama, A. Kawaguchi, K. Nagata, J. R. Tame, and S. Y. Park. 2008. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature 454:1127-1131. [DOI] [PubMed] [Google Scholar]
- 46.Ochoa, M., J. Bárcena, S. de la Luna, J. A. Melero, A. R. Douglas, A. Nieto, J. Ortín, J. J. Skehel, and A. Portela. 1995. Epitope mapping of cross-reactive monoclonal antibodies specific for the influenza A virus PA and PB2 polypeptides. Virus Res. 37:305-315. [DOI] [PubMed] [Google Scholar]
- 47.Ortín, J. 2008. Structure and function of the influenza A virus ribonucleoprotein: transcription and replication, p. 168-186. In H.-D. Klenk, M. Matrosovish, and J. Stech (ed.), Avian influenza. Monographs in virology, vol. 27. Karger, Basel, Switzerland. [Google Scholar]
- 48.Ortín, J., R. Nájera, C. López, M. Dávila, and E. Domingo. 1980. Genetic variability of Hong Kong (H3N2) influenza viruses: spontaneous mutations and their location in the viral genome. Gene 11:319-331. [DOI] [PubMed] [Google Scholar]
- 49.Pascual-Montano, A., L. E. Donate, M. Valle, M. Barcena, R. D. Pascual-Marqui, and J. M. Carazo. 2001. A novel neural network technique for analysis and classification of EM single-particle images. J. Struct. Biol. 133:233-245. [DOI] [PubMed] [Google Scholar]
- 50.Peiris, J. S., M. D. de Jong, and Y. Guan. 2007. Avian influenza virus (H5N1): a threat to human health. Clin. Microbiol. Rev. 20:243-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perales, B., and J. Ortín. 1997. The influenza A virus PB2 polymerase subunit is required for the replication of viral RNA. J. Virol. 71:1381-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plotch, S. J., M. Bouloy, I. Ulmanen, and R. M. Krug. 1981. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23:847-858. [DOI] [PubMed] [Google Scholar]
- 53.Poon, L. L. M., D. C. Pritlove, E. Fodor, and G. G. Brownlee. 1999. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J. Virol. 73:3473-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rao, P., W. Yuan, and R. M. Krug. 2003. Crucial role of CA cleavage sites in the cap-snatching mechanism for initiating viral mRNA synthesis. EMBO J. 22:1188-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Resa-Infante, P., N. Jorba, N. Zamarreno, Y. Fernandez, S. Juarez, and J. Ortin. 2008. The host-dependent interaction of alpha-importins with influenza PB2 polymerase subunit is required for virus RNA replication. PLoS One 3:e3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robertson, J. S., M. Schubert, and R. A. Lazzarini. 1981. Polyadenylation sites for influenza mRNA. J. Virol. 38:157-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scheres, S. H., H. Gao, M. Valle, G. T. Herman, P. P. Eggermont, J. Frank, and J. M. Carazo. 2007. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat. Methods 4:27-29. [DOI] [PubMed] [Google Scholar]
- 58.Scheres, S. H., R. Nunez-Ramirez, C. O. Sorzano, J. M. Carazo, and R. Marabini. 2008. Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 3:977-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheres, S. H., M. Valle, R. Nunez, C. O. Sorzano, R. Marabini, G. T. Herman, and J. M. Carazo. 2005. Maximum-likelihood multi-reference refinement for electron microscopy images. J. Mol. Biol. 348:139-149. [DOI] [PubMed] [Google Scholar]
- 60.Shapiro, G. I., and R. M. Krug. 1988. Influenza virus RNA replication in vitro: synthesis of viral template RNAs and virion RNAs in the absence of an added primer. J. Virol. 62:2285-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sorzano, C. O., R. Marabini, J. Velazquez-Muriel, J. R. Bilbao-Castro, S. H. Scheres, J. M. Carazo, and A. Pascual-Montano. 2004. XMIPP: a new generation of an open-source image processing package for electron microscopy. J. Struct. Biol. 148:194-204. [DOI] [PubMed] [Google Scholar]
- 62.Sugiyama, K., E. Obayashi, A. Kawaguchi, Y. Suzuki, J. R. Tame, K. Nagata, and S. Y. Park. 2009. Structural insight into the essential PB1-PB2 subunit contact of the influenza virus RNA polymerase. EMBO J. 28:1803-1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tarendeau, F., J. Boudet, D. Guilligay, P. J. Mas, C. M. Bougault, S. Boulo, F. Baudin, R. W. Ruigrok, N. Daigle, J. Ellenberg, S. Cusack, J. P. Simorre, and D. J. Hart. 2007. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol. 14:229-233. [DOI] [PubMed] [Google Scholar]
- 64.Tarendeau, F., T. Crepin, D. Guilligay, R. W. Ruigrok, S. Cusack, and D. J. Hart. 2008. Host determinant residue lysine 627 lies on the surface of a discrete, folded domain of influenza virus polymerase PB2 subunit. PLoS Pathog. 4:e1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Torreira, E., G. Schoehn, Y. Fernández, N. Jorba, R. W. Ruigrok, S. Cusack, J. Ortin, and O. Llorca. 2007. Three-dimensional model for the isolated influenza virus polymerase heterotrimer. Nucleic Acids Res. 35:3774-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ulmanen, I., B. A. Broni, and R. M. Krug. 1981. The role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 78:7355-7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vreede, F. T., T. E. Jung, and G. G. Brownlee. 2004. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J. Virol. 78:9568-9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Webster, R. G., W. J. Bean, O. T. Gorman, T. M. Chambers, and Y. Kawaoka. 1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56:152-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wigler, M., A. Pellicer, S. Silverstein, R. Axel, G. Urlaub, and L. Chasin. 1979. DNA-mediated transfer of the adenine phosphoribosyltransferase locus into mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 76:1373-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yuan, P., M. Bartlam, Z. Lou, S. Chen, J. Zhou, X. He, Z. Lv, R. Ge, X. Li, T. Deng, E. Fodor, Z. Rao, and Y. Liu. 2009. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458:909-913. [DOI] [PubMed] [Google Scholar]
- 71.Zhang, S., L. Weng, L. Geng, J. Wang, J. Zhou, V. Deubel, P. Buchy, and T. Toyoda. 2010. Biochemical and kinetic analysis of the influenza virus RNA polymerase purified from insect cells. Biochem. Biophys. Res. Commun. 391:570-574. [DOI] [PubMed] [Google Scholar]