

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiologic agent of primary effusion lymphoma (PEL). All PEL cell lines are infected with KSHV, and 70% are coinfected with Epstein-Barr virus (EBV). KSHV reactivation from latency requires promoter-specific transactivation by the KSHV Rta protein through interactions with RBP-Jk (CSL), the cellular DNA-binding component of the Notch signal transduction pathway. EBV transformation of primary B cells requires EBV nuclear antigen 2 (EBNA-2) to interact with RBP-Jk to direct the latent viral and cellular gene expression program. Although KSHV Rta and EBV EBNA-2 both require RBP-Jk for transactivation, previous studies have suggested that RBP-Jk-dependent transactivators do not function identically. We have found that the EBV latent protein LMP-1 is expressed in less than 5% of KSHV+/EBV+ PEL cells but is induced in an Rta-dependent fashion when KSHV reactivates. KSHV Rta transactivates the EBV latency promoters in an RBP-Jk-dependent fashion and forms a ternary complex with RBP-Jk on the promoters. In B cells that are conditionally transformed by EBV alone, we show that KSHV Rta complements a short-term EBNA-2 growth deficiency in an autocrine/paracrine manner. Complementation of EBNA-2 deficiency by Rta depends on RBP-Jk and LMP-1, and Rta transactivation is required for optimal growth of KSHV+/EBV+ PEL lines. Our data suggest that Rta can contribute to EBV-driven cellular growth by transactivating RBP-Jk-dependent EBV latency genes. However, our data also suggest that EBNA-2 and Rta induce distinct alterations in the cellular proteomes that contribute to the growth of infected cells.
Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiologic agent of Kaposi's sarcoma (KS) and primary effusion lymphoma (PEL). PEL is a body cavity based lymphoma that is rapidly fatal (15, 24, 37, 56, 63). Multiple, continuous PEL cell lines were established by culturing clinical samples from PEL patients (13, 54, 60). These cell lines were the first tissue culture models for KSHV infection (53, 60). Approximately 70% of PEL cell lines are coinfected with KSHV and Epstein-Barr virus (EBV) (17).
The KSHV lytic switch protein, replication and transcriptional activator (Rta), encoded by open reading frame 50 (ORF50), is both necessary and sufficient for viral reactivation in PEL cells (47, 48, 65, 70). Lytic reactivation requires the formation of a ternary complex between Rta, delayed early promoter DNA, and the host cell's recombination signal binding protein (RBP)-Jk (also known as CSL-1 and CBF1) (11, 44, 45). RBP-Jk is a sequence-specific DNA-binding protein that is the nuclear effector of the canonical Notch signal transduction pathway (23).
Whereas RBP-Jk is required for productive KSHV reactivation (45), it is also required for latent (nonproductive) transformation of primary B cells by EBV (39). In this program, termed latency III, EBV nuclear antigen 2 (EBNA-2) transactivates two EBV promoters by interacting with RBP-Jk. The two promoters express transcripts that encode seven proteins that promote viral persistence by stabilizing the EBV genome, stimulating B-cell growth and expansion, and blocking B-cell apoptosis (39). One of the transforming EBV latency proteins is latent membrane protein 1 (LMP-1). LMP-1 is a constitutively active ortholog of the cellular tumor necrosis factor (TNF) receptor CD40 (55, 68). LMP-1 induces cell proliferation and transformation by engaging multiple signaling pathways including NF-κB, TNF receptor-associated factors (TRAFs) 1 to 3, Akt kinase, Jun kinase, c-Rel, and p38 (16, 18-20, 27, 31, 40, 43, 46, 51, 52, 55). EBV transformation also requires transactivation of cellular genes by EBNA-2 in an RBP-Jk-dependent fashion (22, 34, 38, 62, 64, 71).
RBP-Jk's principal role in KSHV and EBV infection is to specify transcriptional targets of Rta and EBNA-2. RBP-Jk also specifies transcriptional targets for the activated form of the cellular Notch receptor (Notch intracellular domain 1 [NICD-1]); despite the apparent mechanistic similarity of NICD-1 transactivation to that of Rta and EBNA-2, these proteins are not always phenotypically interchangeable. For example, NICD-1 and EBNA-2 do not productively reactivate KSHV from latency (11, 14, 45), and NICD-1 does not fully complement EBNA-2 deficiency in long-term outgrowth of lymphoblastoid cell lines (LCLs) (25, 28). Furthermore, the KSHV genome contains 177 RBP-Jk sites and yet, in the absence of de novo protein expression, Rta only transactivates eight KSHV genes in infected cells (5). These data and studies from other systems (32, 57) suggest that a binding site for RBP-Jk is often not sufficient to specify a promoter as a target of an RBP-Jk-dependent transactivator. Moreover, KSHV+/EBV+ PEL cell lines express little to no EBNA-2 and LMP-1, a finding consistent with the most restricted program of EBV latent gene expression (type I latency) (6, 7, 30, 42, 50, 66, 67). Since there is currently no evidence in the literature that RBP-Jk is required for type I latency, the significance of RBP-Jk in PEL cell growth is questionable.
The goal of the present study was to determine whether the KSHV lytic switch protein, Rta, could function in an EBNA-2-like fashion and cross talk to EBV in coinfected cells. We demonstrate here that Rta transactivates EBV latency promoters in an RBP-Jk-dependent fashion and forms ternary complexes with RBP-Jk on those promoters. We demonstrate that Rta complements an EBNA-2 growth deficiency in EBV-infected LCLs that conditionally express EBNA-2 in a paracrine manner. This complementation requires Rta transactivation, RBP-Jk DNA binding, and LMP-1. Our data suggest that KSHV and EBV cooperate to stimulate cell growth in PEL cells by a mechanism in which Rta induces LMP-1 and cellular gene expression in a background of EBV type I latency. Nonetheless, cellular proteomic analyses reveal that Rta rescues the growth defect in a manner distinct from EBNA-2.
MATERIALS AND METHODS
Cell culture, transfections, and inductions.
BC-1 and BL-41 B cells were grown in 15 and 10% fetal calf serum, respectively. BC-1 viral inductions were performed as previously described, with 20 ng of tetradecanoyl phorbol acetate (TPA)/ml to induce EBV reactivation and 3 mM n-Butyrate to induce KSHV reactivation (50). Cells were harvested after 18 h for immunofluorescence. BC-1 cells were electroporated with 20 to 40 μg of DNA at 200V, 975uF; BL-41 cells were electroporated as previously described (12). EREB2-5 cells (gift of Bettina Kempkes) were propagated as previously described (38). 107 cells were electroporated with 30 μg of DNA at 210V, 975uF. The cells were grown in the presence or absence of β-estradiol for 7 days postelectroporation and then stained for mitochondrial function, described below. OT11 (RBP-Jk−/−) Mouse Embryonic Fibroblasts were propagated and transfected as previously described (12). All transfections were performed in triplicate, in at least two experiments. In all transfections, total DNA was normalized with empty expression vector.
Paracrine effects of conditioned media.
EREB2-5 cells were transfected as described for the Mitotracker assay. Seven days posttransfection, cells were collected by centrifugation at 250g, and the clarified media was filtered through a sterile, 0.22 μM membrane. This conditioned media replaced the media of untransfected EREB2-5 cells that were deprived of β-estradiol for 7 days. The cells grown in the conditioned media were incubated for an additional 7 days and then harvested for the Mitotracker assay as described.
Luciferase and β-galactosidase assays.
Luciferase and β-galactosidase assays were performed as described previously (11).
Plasmids.
The following plasmid constructs were described previously: pV5-FLc50, pV5-50ΔSTAD, pcDNA3-FLg50, and pcDNA3.1-HisLacZ (47), pSG5-EBNA2, pCMX-VP16-RBP-Jk, and 2x EBV RBP-hsp-luc (11), pV5-ORF50-ILL140AAA (12), and pV5-50ΔSTADΔLR (4). EBV promoter-reporter vectors were constructed by PCR amplifying the clarified supernatant of boiled, concentrated B95-8 cells (American Type Culture Collection). The promoters of LMP-1 (B95-8 nucleotide [nt] positions 169871 to 169521) and Cp (B95-8 nt positions 10755 to 11394) were independently amplified with oligonucleotide primers that introduced 5′ XhoI and 3′ HindIII sites, respectively, and cloned into pGL3-Basic (Promega) at those sites. pEF-BOS Neo/RBP-J (R218H)-myc (35) was a gift from T. Honjo (Kyoto University). pSG5-LMP-1-AAAG (DN1) was a gift from M. Rowe (New York University). pSV-HA-LMP-1-DN (2) (DN2) was a gift from E. Adriaenssens (Université des Sciences et Technologies de Lille).
Immunofluorescence.
Immunofluorescence analysis was performed as described previously (48) with rabbit anti-Rta serum at 1:5,000, rabbit anti-V5 serum at 1:1,280 (Sigma), and LMP-1 S12 hybridoma supernatant at 1:20 (a gift from G. Bishop [University of Iowa] with permission of D. Thorley-Lawson [Tufts University]). A minimum of 500 V5 or Rta-positive cells, per transfection, was counted by visual inspection using fluorescence microscopy.
RNA isolation and quantitative reverse transcriptase PCR (qRT-PCR).
BC-1 cells were either treated with sodium butyrate to reactivate KSHV or left uninduced and transfected with empty vector or expression plasmids for Rta, RtaΔSTAD, or RtaΔSTADΔLR. RNA was purified from the cells 18 h posttransfection as described previously (5). The RNA was resuspended in RNase-free water, and cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen) as recommended by the manufacturer. LMP-1 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) transcripts were quantitated in 1 μg of cDNA from each cell population by using a QuantiTect SYBR green PCR kit (Qiagen) and a Corbett RotorGene instrument. Primer sequences were as follows: LMP-1 (forward, GGCCTTCTTCCTAGCCTTCT; reverse, GAGTCATCGTGGTGGTGTTC) and GAPDH (forward, CCAGCAAGAGCACAAGAGGA; reverse, GGAGATTCAGTGTGGTGGG). PCR conditions were an initial incubation at 95°C for 5 min, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. PCR products were confirmed by using a terminal melting curve and agarose gel electrophoresis. Control reactions omitting RT demonstrated the absence of contaminating genomic DNA. The fold change in expression was determined by using the ΔΔCT method (5).
Generation of proteins in E. coli.
Proteins were generated as described previously (4).
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed as described previously (11). Two probes for the LMP-1 promoter were used (top strand is shown with RBP-Jk sites underlined): probe 1 (CCTCTTTGTGCAGATTACACTGCCGCTTCCCACAAC) and probe 2 (ATTTCCTGTTGCACTTGGCCACCGCATTCCCACAGC).
Mitochondrial function.
Mitochondria were stained by MitoTracker Red CMX-Ros as recommended by the manufacturer (Invitrogen). Briefly, 2 × 106 EREB2-5 cells were pelleted at 1,000 rpm for 10 min and washed with 1× phosphate-buffered saline (PBS). The cells were then resuspended in 2 ml of prewarmed RPMI 1640 complete medium and 100 nM MitoTracker-Red CMX-Ros dye. Cells were incubated for 45 min at 37°C and then pelleted and washed with 1× PBS. Cells were fixed with 3.7% formaldehyde for 10 min and washed with 1× PBS. Cells were adhered to poly-l-lysine-coated slides at 2.5 × 105 cells/200 μl and incubated for 30 min. At least 500 Bright red cells were quantitated, per transfection, by visual inspection with fluorescence microscopy.
Cytotoxicity.
EREB2-5 cells were resuspended in fresh medium at 104 cells per 100 μl and plated in duplicate in a black 96-well plate. Cytotoxicity was quantitated by using a MultiTox-Fluor multiplex cytotoxicity assay (Promega), as recommended by the manufacturer (58). Dead cells were detected by fluorescence at 485-nm excitation and 520-nm emission by using a SpectraMax plate reader (Molecular Devices).
Antibody array.
EREB2-5 cells were transfected and grown in the presence or absence of β-estradiol, as described elsewhere. Seven days posttransfection, protein extracts were prepared, and 200 μg of each was analyzed by using a RayBio human apoptosis antibody array kit (RayBiotech) according to the manufacturer's suggestions. The membranes were exposed to autoradiography film for different times to detect the chemiluminescent signals. Images with signals in linear range were quantitated by using the program ImageJ (1). For each membrane, signals from the negative control spots were averaged and then subtracted from each of the other spots. A signal was considered valid if its value exceeded both its average local background and the average of all valid negative control values. Valid signals were normalized by using the positive control spots (for cellular BID protein). The fold changes in signals for each spot were quantitated by dividing by the valid signals for each corresponding spot on the minus β-estradiol membrane. The average fold change, and standard deviation, were calculated for each protein. Raw data have been submitted to the Gene Expression Omnibus under accession number GSE21375.
RESULTS
KSHV reactivation induces EBV LMP-1 expression in KSHV+/EBV+ PEL cells.
Successful reactivation of KSHV, and EBV transformation, both require the cellular protein RBP-Jk (39, 45). To test whether KSHV could induce expression of EBV latency genes in the context of a coinfection, we utilized the KSHV+/EBV+ PEL cell line BC-1. We chose BC-1 cells because they express little or undetectable EBNA-2 and LMP-1 (type I latency), and each virus can be selectively induced with different chemicals (6, 7, 30, 42, 50, 66, 67). We treated the cells independently with TPA, the EBV-selective inducer, or sodium butyrate, the KSHV-selective inducer. We quantitated the percentage of cells expressing Rta and/or LMP-1 by indirect immunofluorescence and normalized them to the control cells that were left untreated. Induction of KSHV reactivation was accompanied by ∼11-fold induction of Rta and 8-fold induction of LMP-1, and ca. 70% of Rta-expressing cells also expressed LMP-1 (Fig. 1 A). A representative image is shown in Fig. 1B. We were also surprised to find that a small percentage of untreated cells also expressed LMP-1 spontaneously, despite the classification of the EBV infection in PELs as type I latency (Fig. 1). Remarkably, spontaneous LMP-1 expression corresponded to 94% of cells in which Rta was also expressed. Treatment of the cells with TPA, the EBV inducer, also resulted in a slight increase in the percentage of cells expressing both Rta and LMP-1 (Fig. 1A). This single-cell relationship between Rta and LMP-1 expression was reminiscent of the relationship between Rta and KSHV DE proteins in KSHV+/EBV− cells (5, 12, 48, 59).
FIG. 1.

KSHV reactivation induces LMP-1 expression in KSHV+/EBV+ cells. (A) BC-1 cells were treated with 20 ng of TPA/ml or 3 mM sodium butyrate, as indicated, to stimulate reactivation of EBV and KSHV, respectively. At 18 h postinduction, LMP1 and Rta were detected by immunofluorescence. A minimum of 500 immunopositive cells were counted, and the fold induction of either protein was calculated by comparison to untreated cells. The numbers above each bar graph represent the percentage of Rta-expressing cells that also expressed LMP1. (B) Representative image demonstrating correspondence between Rta and LMP-1 expression.
KSHV Rta transactivates EBV LMP-1 in KSHV+/EBV+ PEL cells.
To verify that LMP-1 was induced by Rta expression, and not by n-butyrate directly, we ectopically expressed Rta in BC-1 cells from an electroporated vector in the absence of any chemical inducer. Using indirect immunofluorescence, we quantitated the percentage of ectopic Rta-expressing cells (identified by V5 epitope tag) that also expressed LMP-1. Similar to n-butyrate induction, ca. 70% of Rta-expressing cells induced LMP-1 expression (Fig. 2 A).
FIG. 2.

Rta transactivates LMP-1 in KSHV+/EBV+ cells. BC-1 cells were electroporated with 10 μg of V5-Rta plasmid and 10 and 20 μg of either empty expression vector (vector), RtaΔSTAD vector (ΔSTAD), or RtaΔSTADΔLR (ΔSTADΔLR) vector. At 42 h postelectroporation, LMP1 and V5-Rta were detected by immunofluorescence. (A) Graph representing the percentages of V5-Rta-positive (transfected) cells, in which LMP1 was induced. (B) Representative images demonstrating induction of LMP-1 in cells transfected with the V5-Rta expression vector with either empty vector, RtaΔSTAD vector, RtaΔSTADΔLR vector. (C) Total RNA was purified from BC-1 cells transfected with empty vector and treated with sodium butyrate (n-BA) or transfected with the indicated plasmids alone. The fold activation of LMP-1 was quantitated by qRT-PCR measuring LMP-1 and GAPDH RNA and comparing each to cells transfected with empty vector (Vec).
To confirm that LMP-1 was induced by Rta transcriptional transactivation, we coelectroporated the Rta plasmid together with a vector that expressed the Rta-specific dominant-negative transcriptional mutant protein RtaΔSTAD (also known as ORF50ΔSTAD and dominant-negative [DN] Rta) (4, 47). Electroporation of wild-type Rta plasmid with two different amounts of RtaΔSTAD expression vector resulted in >50% reduction in the percentage of electroporated cells in which LMP-1 was induced, in a dose-dependent fashion (Fig. 2A). When we repeated the coelectroporation using RtaΔSTADΔLR, an inactive variant of RtaΔSTAD (also known as nonfunctional DN Rta) (4), Rta-mediated induction of LMP-1 returned to levels equivalent to Rta alone at both amounts of the mutant (Fig. 2A). An example of the indirect immunofluorescence is shown in Fig. 2B; the merged color image is shown in Fig. S1 in the supplemental material. Quantitation of LMP-1 RNA by qRT-PCR also demonstrated that Rta activated LMP-1 transcription in coinfected cells (Fig. 2C). These data demonstrate that induction of LMP-1 expression during KSHV reactivation depends on bona fide Rta transcriptional transactivation.
Rta transactivates the EBV Cp and LMP-1 promoters.
In EBV type III latency, EBNA-2 transactivates the Cp and LMP-1 promoters by piggy-backing onto the cellular DNA-binding protein RBP-Jk (39). Rta transactivates delayed early KSHV promoters in an RBP-Jk-dependent fashion also but uses a distinct mechanism (11, 12, 44). To determine whether Rta could directly transactivate the EBV type III latency promoters, we coelectroporated increasing amounts of Rta expression vector with reporter plasmids for the EBV promoters in uninfected cells. In the Burkitt's lymphoma cell line BL-41, Rta transactivated both the Cp (Fig. 3 A) and LMP-1 promoters (Fig. 3B). In fact, Rta transactivated the Cp promoter with a magnitude greater than EBV EBNA-2 (Fig. 3A).
FIG. 3.

KSHV Rta transactivates EBV latency III promoters in an RBP-Jk-dependent manner. Uninfected BL-41 cells were coelectroporated with 2 μg of a luciferase reporter construct of the EBV Cp (A) or the LMP-1 promoter (B) alone and increasing amounts of either KSHV Rta or EBNA-2 expression plasmids, as indicated. The fold activation was calculated for each amount of Rta or EBNA-2 plasmid by comparison to the luciferase expressed by the reporter vectors coelectroporated with empty expression vector (“0”). OT-11 (RBP-Jk−/−) cells were transfected with 0.25 μg of the EBV Cp (C) or the LMP-1 promoter (D) reporters alone, or together with Rta expression vector with increasing amounts of either RBP-Jk expression vector or empty vector (Vec) (as indicated). The fold activation was calculated as in panels A and B.
Rta transactivation of the Cp and LMP-1 promoter depends on RBP-Jk.
As outlined in the introduction, an RBP-Jk binding site is not always sufficient to specify a promoter as a target of RBP-Jk-dependent activators. RBP-Jk-dependent activators also display heterogeneous phenotypes. We previously demonstrated that Rta transactivation of several KSHV DE promoters was dependent on RBP-Jk by performing promoter-reporter assays in the RBP-Jk-null mouse fibroblast line OT-11 (11, 12, 44). To determine whether Rta required RBP-Jk to transactivate the Cp promoter, we cotransfected OT-11 cells with the Rta expression and the Cp reporter plasmids, in the presence or absence of an RBP-Jk expression plasmid. In OT-11 cells, Rta did not transactivate the Cp promoter in the absence of the RBP-Jk plasmid (Fig. 3C). However, when we cotransfected increasing amounts of the RBP-Jk plasmid, Rta transactivation of Cp was rescued in a dose-dependent fashion. Similarly, Rta failed to transactivate the LMP-1 promoter in OT-11 cells unless ectopic RBP-Jk was coexpressed (Fig. 3D). Therefore, similar to many KSHV DE promoters, Rta transactivation of the EBV latency Cp and LMP-1 promoters was RBP-Jk dependent.
Rta forms a ternary complex with RBP-Jk and EBV latency promoter DNAs.
Both the KSHV ORF57/Mta and the thymidine kinase (TK) promoters contain RBP-Jk binding sites. However, Rta forms ternary complexes with RBP-Jk only on the ORF57 promoter, corresponding to strong transactivation of ORF57 but not TK (11). To determine whether Rta forms a ternary complex with RBP-Jk and the EBV latency promoters in a manner similar to KSHV lytic promoters, we performed EMSAs. In this approach, we utilize 35S-labeled RBP-Jk in rabbit reticulocyte lysates, unlabeled promoter DNA, and unlabeled RtaΔSTAD fused to the maltose-binding protein epitope (MBP). RBP-Jk formed a complex with the LMP-1 DNA containing an RBP-Jk element but did not form a complex with nonspecific DNA (Fig. 4 A, compare lanes 1 and 3). When we added MBP-RtaΔSTAD to RBP-Jk and the LMP-1 DNA, a new Rta/RBP-Jk/DNA (ternary) complex with slower migration formed in addition to the faster-migrating RBP-Jk/DNA complex (Fig. 4A, lanes 4 to 8). Confirming that the supershifted complex contained MBP-RtaΔSTAD, anti-MBP serum created an additional supershift of the Rta/RBP-Jk/DNA complex (Fig. 4A, lanes 12 to 14). As a negative control, the anti-MBP serum did not affect migration of the RBP-Jk/DNA complex (Fig. 4A, lanes 15 to 17).
FIG. 4.

Rta forms a ternary complex with RBP-Jk and the EBV Cp and LMP1 promoter DNAs. (A) EMSA was performed by incubating [35S]methionine (35S-Met)-labeled RBP-Jk alone or together with LMP-1 promoter DNA, MBP-Rta protein, and/or anti-MBP serum, as indicated. (B) EMSA was performed by incubating 35S-Met-labeled RBP-Jk alone or together with Cp promoter DNA and MBP-Rta or MBP proteins, as indicated. (C) 35S-Met-labeled RBP-Jk as visualized by SDS-PAGE/autoradiogram. The position of migration of molecular mass standards are indicated at left in kilodaltons. Rta stimulates DNA binding of RBP-Jk to the EBV Cp RBP-Jk element (D) and the LMP-1 promoter (E). BL-41 cells were electroporated with the indicated plasmids, as in Fig. 3 and reference 11, and the reporter plasmid 2× EBV RBP-hsp-luc (D) and pLMP-1-GL3basic (E). In panel E, RtaΔSTAD weakly repressed the LMP-1 promoter alone, so the values for RBP-Jk/VP16+RtaΔSTAD were normalized to RtaΔSTAD alone, which was set to 1-fold. *, P = 0.0011 (compared to the RBP-Jk/VP16, alone, column).
We performed a similar analysis using Cp DNA containing an RBP-Jk element. As expected, RBP-Jk bound to the Cp, but not nonspecific, DNA (Fig. 4B, compare lanes 1 and 2). Similar to the LMP-1 promoter, RtaΔSTAD formed a ternary complex with RBP-Jk and DNA (Fig. 4B, lanes 3 and 4). However, the negative control MBP moiety did not form a ternary complex with RBP-Jk and Cp DNA (Fig. 4B, lanes 5 and 6), proving that the ternary complex formed specifically with the Rta portion of the MBP fusion protein. An image of the RBP-Jk protein used in these experiments is shown in Fig. 4C. These data demonstrate that Rta and RBP-Jk form ternary complexes on the RBP-Jk elements in the Cp and LMP-1 promoters, similar to complexes formed on KSHV DE promoters.
Rta stimulates RBP-Jk binding to the RBP-Jk element from the EBV promoters.
We previously demonstrated that Rta stimulates RBP-Jk DNA binding to multiple KSHV and cellular promoters to reactivate KSHV from latency (11), a mechanism distinct from that of EBNA-2. Stimulation of RBP-Jk DNA binding corresponded with ternary Rta/RBP-Jk/DNA complex formation. To determine whether this novel mechanism also mediated cross talk between KSHV and EBV, we sought to determine whether Rta stimulated RBP-Jk DNA binding to a promoter containing two tandem copies of a 25-bp EBV Cp promoter element containing the RBP-Jk site. Constitutively active RBP-Jk (“RBP-Jk/VP16”; fused to the herpes simplex virus VP16 transcriptional activation domain) activated the Cp reporter ∼4-fold in BL-41 cells, confirming that RBP-Jk can bind to Cp in vivo (Fig. 4D). Transfecting higher amounts of RBP-Jk/VP16 resulted in higher transactivation (not shown). Coelectroporation of the RBP-Jk/VP16 vector with two different amounts of RtaΔSTAD expression vector increased transactivation of the reporter to nearly 20-fold (Fig. 4D). RtaΔSTAD retains the ability to bind DNA and RBP-Jk but lacks a transcriptional activation domain. Therefore, all transactivation in this experiment is attributable to RBP-Jk/VP16, suggesting that RtaΔSTAD stimulates DNA binding of RBP-Jk to the Cp promoter in vivo. Rta's interaction with RBP-Jk at the Cp promoter is thus mechanistically different than that of EBNA-2. A similar analysis of the LMP-1 promoter shows that RBP-Jk/VP16 and RtaΔSTAD alone failed to transactivate the promoter, but the two proteins together demonstrate a weak but statistically significant 2.4-fold transactivation (Fig. 4E).
EBNA-2 is required for mitochondrial function in EBV-transformed B cells.
EBNA-2 is required for EBV transformation of primary B cells (39). EREB2-5 cells are EBV-infected B cells that express a recombinant EBNA-2 protein fused to the hormone-binding domain of the estrogen receptor (38). In the presence of β-estradiol, EBNA-2 localizes to the nucleus and maintains immortalization of the cells. When β-estradiol is withdrawn from the growth medium, EBNA-2 is sequestered in the cytoplasm, and the cells die via apoptosis.
An early event in programmed cell death is abrogation of mitochondrial function by the loss of mitochondrial membrane potential (41). Positively charged, lipophilic dyes, such as CMX-Ros, exclusively label functioning mitochondria and distinguish healthy from apoptotic cells in a highly quantitative manner (49). We stained EREB2-5 cells with CMX-Ros and found that mitochondrial function was significantly reduced between 6 and 7 days after the removal of β-estradiol (Fig. 5 A). Quantitation of functioning mitochondria by fluorescence-activated cell sorting and trypan blue staining also confirmed a difference in viability at day 7 (not shown). Cell nuclei were intact regardless of CMX-Ros staining (compare panels in Fig. 5B), confirming that loss of mitochondrial function is an early event in the apoptotic death of β-estradiol-deprived EREB2-5 cells. However, cells that were CMX-Ros negative contained condensed nuclei with hyperintense DAPI (4′,6′-diamidino-2-phenylindole) staining, characteristics of apoptosis (Fig. 5B, arrows; merged color image is shown in Fig. S2 in the supplemental material). Therefore, EBNA-2 is required to maintain mitochondrial function and inhibit apoptosis in EBV-transformed B cells.
FIG. 5.

EBNA-2 is required for mitochondrial function in EBV-transformed B cells. EREB2-5 cells were grown in the presence or absence of β-estradiol and stained with MitoTracker CMX-Ros dye at the indicated time points. Cells were fixed and quantitated by visual inspection using fluorescence microscopy. (A) The percentages of total viable cells are plotted over time with the indicated treatments. (B) Representative image demonstrating healthy and apoptotic cells (arrows).
Rta complements the short-term growth defect of EBNA-2 deficiency in EBV-infected cells.
Our data showed that Rta transactivated the EBV LMP-1 and Cp latency promoters, similar to EBNA-2 in type III latency. To determine whether Rta could complement the short-term growth deficiency in an EBNA-2-like fashion in EREB2-5 cells, we quantitated the number of cells that were labeled by CMX-Ros in the presence or absence of β-estradiol, 7 days after electroporation of various plasmids. We set the baseline number of CMX-Ros-positive cells electroporated with empty vector, cultured in the absence of β-estradiol, at 1-fold (Fig. 6 A). Maintenance of β-estradiol in the growth media of empty-vector electroporated cells resulted in a 40-fold increase in the number of CMX-Ros-positive cells (i.e., functional mitochondria) (Fig. 6A). As a positive control, electroporation of an EBNA-2 expression vector completely rescued the EREB2-5 cells from apoptosis when grown in the absence of β-estradiol.
FIG. 6.

Rta complements the short-term growth defect of EBNA-2 deficiency in EBV-infected cells. (A) EREB2-5 cells were electroporated with plasmids expressing the indicated proteins (1, 2.5, 5, 10, and 20 μg of Rta [pcDNA3-FLg50], or 20 μg of EBNA-2 positive control). β-Estradiol (β-e'diol) was either maintained or removed from the growth media at the time of electroporation. Mitochondrial function was determined 7 days postelectroporation by MitoTracker dye. Quantitation is expressed as the fold relative to cells electroporated with empty vector in the absence of β-estradiol. (B) EREB2-5 were electroporated with 20 μg of the indicated plasmids, and β-estradiol (β-e'diol) was either maintained or removed from the growth medium at the time of electroporation. Cell death was measured 7 days posttransfection by fluorogenic cytotoxicity assay. (C) Representative images of cells. Functioning mitochondria are red, and DNA is blue (DAPI). (D) Relationship of Rta-transfected cells to functioning mitochondria. EREB2-5 cells were transfected with plasmids expressing Rta and histone H2b-GFP. Green, transfected cells; red, functioning mitochondria; blue, DNA. Green/Blue/Red overlap appears white in color.
Electroporation of increasing amounts of Rta expression vector revealed that Rta also protected the cells from apoptosis, in a dose-responsive fashion, when EBNA-2 was inactivated by removing β-estradiol (Fig. 6A).
To confirm these data, we utilized a second assay for cell viability that measures protease activity released from dead cells by cleaving a cell-impermeable fluorogenic peptide substrate (58). The dead cell protease activity from EREB2-5 cells grown in the absence of β-estradiol for 7 days was set to 100%. Maintenance of β-estradiol in the media reduced relative dead cell protease activity to ca. 60% (Fig. 6B). A similar reduction of cell death protease activity was observed when cells were transfected with EBNA-2 or Rta expression vectors and cultured in the absence of β-estradiol (Fig. 6B). Thus, this cell death assay confirmed that Rta could rescue the short-term growth defect in EREB2-5 cells.
Representative fields of cells from the CMX-Ros experiments are shown in Fig. 6C. In these experiments, 40 to 60% of the cell population was protected from apoptosis even though Rta or EBNA-2 were expressed in only ca. 3 to 5% of cells. These data suggested that Rta- or EBNA-2-expressing cells supported short-term growth of the remaining cell population in a paracrine fashion. A typical field containing a transfected cell is shown in Fig. 6D. When we measured mitochondrial function only in transfected cells, 100% of the cells expressing Rta or EBNA-2 were rescued from apoptosis (not shown).
To ensure that the Rta rescue was not due to a spontaneous change in the cell population after a prolonged time without β-estradiol, we altered our experimental strategy. In contrast to the previous experiment in which β-estradiol was removed from the cells immediately on the day of transfection, we withdrew β-estradiol 4 days prior to transfection. We analyzed the cells at 48 h posttransfection. As shown in Fig. S3 in the supplemental material, the cells were rescued from the EBNA-2 deficiency as effectively as in Fig. 6.
Media conditioned by Rta-transfected cells complement the short-term growth defect of EBNA-2 deficiency in EBV-infected cells.
The data in Fig. 6 suggested that mitochondrial function was rescued by a paracrine effect of Rta or EBNA-2 expression on the cell population as a whole. To formally test this hypothesis, we repeated the experimental approach shown in Fig. 6A but transferred the media conditioned by the cells to new, nontransfected EREB2-5 cells that had been deprived of β-estradiol for 7 days. Positive control media conditioned by addition of β-estradiol to vector electroporated cells increased the percentage of viable cells in a paracrine fashion from 20 to 52% (Fig. 7). Expression of either KSHV Rta, or EBNA-2, conditioned the electroporated-cell media in a quantitatively similar fashion as adding β-estradiol (Fig. 7). Therefore, media conditioned by Rta expression can complement the growth defect in EBNA-2-deficient cells in a paracrine fashion.
FIG. 7.
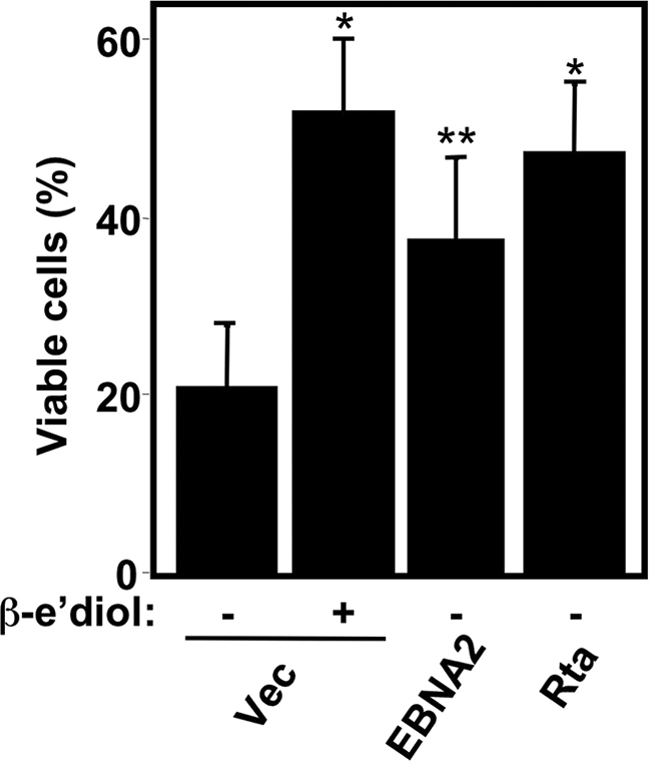
Medium conditioned by Rta-transfected cells complements the short-term growth defect of EBNA2 deficiency in EBV-infected cells. EREB2-5 cells were grown in the absence of β-estradiol for 7 days, after which their media were replaced with the conditioned medium from EREB2-5 cells that had been electroporated with the indicated plasmids and cultured in the presence or absence of β-estradiol (β-e'diol). After growth for an additional 7 days, the mitochondrial function of the cells was determined by MitoTracker dye. *, P < 0.0001; **, P < 0.0357 (compared to the first column).
LMP-1 signaling is required for Rta to complement the short-term growth defect of EBNA-2 deficiency.
In type III EBV latency, EBNA-2 transactivates the LMP-1 promoter, and EBNA-2 and LMP-1 cooperate to maintain growth of the infected cells (39, 71). To determine whether LMP-1 was required for Rta complementation of EBNA-2 deficiency, we coexpressed Rta with two dominant-negative signaling mutants of LMP-1 (LMP1-DM-AAAG and LMP1-DN) (2, 3). Rta expressed alone restored ca. 60% of cell viability when β-estradiol was removed from the culture media, a phenotype similar to expression of EBNA-2 alone (Fig. 8). However, coelectroporation of the Rta vector with either of the vectors for the dominant-negative LMP-1 alleles inhibited Rta-mediated complementation of the EBNA-2 deficiency (Fig. 8). Therefore, LMP-1 signaling is required for Rta to complement the EBNA-2 defect in EBV-infected cells.
FIG. 8.
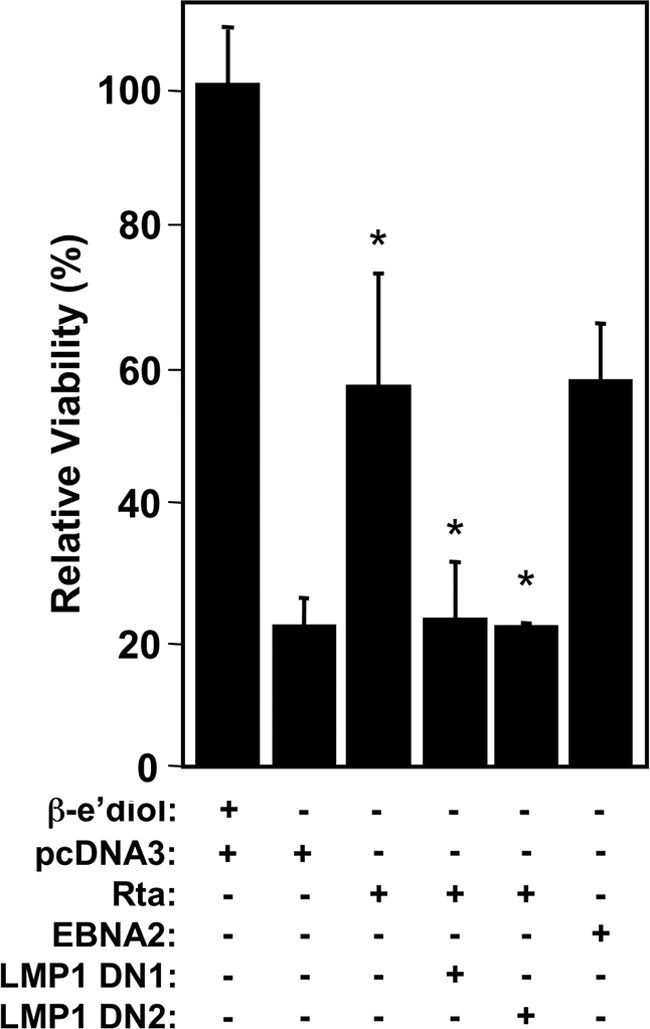
LMP-1 signaling is required for Rta to complement the short-term growth defect of EBNA2 deficiency. EREB2-5 cells were electroporated with plasmids expressing the indicated proteins. Relative viabilities were calculated by dividing MitoTracker-positive (Red) cells by total cells. “LMP1 DN1” is pSG5-LMP1-AAAG, and “LMP1 DN2” is psVHA-LMP1 DN. *, P < 0.0001 (Rta alone compared to the first column, Rta with DNs compared to Rta alone).
DNA binding of Rta and RBP-Jk are required for Rta to complement the short-term growth defect of EBNA-2 deficiency.
Rta reactivates KSHV from latency in part by transactivating DE promoters to which it binds in combination with RBP-Jk (4, 5, 11, 12, 44, 47, 48). Our data showed that Rta transactivated the EBV latency promoters in a mechanistically similar fashion to some KSHV DE promoters. To determine whether this mechanism was required for Rta to complement short-term EBNA-2 deficiency, we electroporated plasmids expressing Rta and RBP-Jk DNA binding mutants. The Rta DNA-binding mutant (Rta-ILL mt) (12) expressed alone was unable to reverse the affect of removing β-estradiol from the media of EREB2-5 cells (Fig. 9). Moreover, while expression of wild-type Rta alone restored ca. 40% viability of EREB2-5 when we removed β-estradiol in this experiment, coexpression of Rta with a dominant-negative, DNA-binding mutant of RBP-Jk (RBP-J [R218H]-myc [35]) eliminated Rta complementation (Fig. 9). Thus, DNA binding of Rta and RBP-Jk is required for Rta to complement the short-term growth defect of EBNA-2 deficiency.
FIG. 9.

DNA binding of Rta and RBP-Jk are required for Rta to complement the short-term growth defect of EBNA2 deficiency. EREB2-5 cells were electroporated with plasmids expressing the indicated proteins. Mitochondrial function was determined 7 days postelectroporation by MitoTracker dye. Relative viabilities were calculated by dividing MitoTracker-positive cells (Red) by total cells. “RBP-Jk DN” is pEF-BOS Neo/RBP-J R218H-myc. “Rta-ILL” is pV5-ORF50-ILL140AAA. *, P = 0.0264 (Rta compared to the first column, Rta plus DN compared to Rta alone).
The partial cellular proteomes associated with rescue of the EREB2-5 short-term growth defect by EBNA-2 and Rta are distinct.
Our data suggested that KSHV Rta phenocopied EBV EBNA-2 in short-term inhibition of LCL apoptosis and maintenance of growth. It has been clearly established that EBNA-2's contribution to LCL growth is not simply to transactivate LMP-1 but also to transactivate cellular promoters (22, 34, 38, 62, 64, 71). However, RBP-Jk-dependent activators can be functionally heterogeneous. To gain a broader perspective on the cellular changes engendered by short-term Rta or EBNA-2 expression in EREB2-5 cells, we analyzed the partial cellular proteomes. We used an antibody array capable of detecting 43 intracellular and secreted proteins implicated in the regulation of apoptosis. We prepared whole-cell protein extracts from EREB2-5 cells transfected with empty, Rta, or EBNA-2 vectors, grown in the presence or absence of β-estradiol and harvested 7 days posttransfection. We calculated fold changes for each protein relative to vector-transfected cells grown in the absence of β-estradiol. A total of 31 proteins displayed at least a 1.5-fold difference in response to at least one of the three experimental conditions (Fig. 10; additional unchanged proteins are shown in Fig. S4 in the supplemental material). All three conditions resulted in induction of cellular protein expression; however, the most striking result was that β-estradiol also broadly repressed cellular proteins, while ectopic Rta and EBNA-2 had little repressive effect on cellular protein expression. β-Estradiol altered the expression of the greatest number of proteins, with 7 induced and 20 repressed (Fig. 10). Transfection of the EBNA-2 vector induced 6 proteins, and Rta induced 14 and repressed 2. The overall range of values was a maximum of 7.4-fold for β-estradiol activation of insulin-like growth factor 2 (IGF-II), to a minimum of −15.6-fold for β-estradiol repression of Bad (albeit with a high standard error) (Fig. 10). Although there was no obvious progrowth or antiapoptotic pattern of protein expression associated with all three conditions, the greatest correspondence was activation of caspases 8 and 3, IGF-II, and Bcl-w. The effect of Rta expression showed a greater correspondence with β-estradiol treatment than did EBNA-2 expression, sharing five induced proteins and two repressed proteins; among these shared effects were induction of IGFs I and II, and repression of tumor necrosis factor alpha (TNF-α) and TNF-receptor 2 (TNF-R2). The largest divergence occurred for DR-6 and Bad, which were strongly repressed by β-estradiol, but induced by EBNA-2 or Rta expression (Fig. 10). Overall, these proteomic analyses support the notion that rescue of the EREB2-5 cells from apoptosis is associated with cellular protein changes that are largely distinct for β-estradiol treatment, or expression of Rta or EBNA-2 in less than 5% of cells.
FIG. 10.

The partial cellular proteomes associated with rescue of the short-term EREB2-5 growth defect by EBNA2 and Rta are distinct. EREB2-5 cells were electroporated with the indicated plasmids and cultured in the absence of β-estradiol (EBNA2 and Rta) or were transfected with empty vector and cultured in the presence of β-estradiol (+ β-e'diol). Equal amounts of protein extracts from each condition were analyzed by using a RayBio human apoptosis antibody array kit (RayBiotech). The figure shows the fold changes and standard deviations (sd) determined by comparison to results from vector-transfected EREB2-5 cells that were cultured in the absence of β-estradiol (data not shown). Fold changes greater than or equal to 1.5 are indicated by boxes, and fold changes less than or equal to −1.5 are indicated by gray shading.
Rta transactivation is required for optimal growth of KSHV+/EBV+ PEL cells.
Our data suggested that Rta rescued a short-term growth defect of EBNA-2 deficiency in EREB2-5 cells, in part by inducing LMP-1 expression. In Fig. 1 and 2 we showed that Rta induced expression of LMP-1 in KSHV+/EBV+ PEL cells. To determine whether this mechanism contributed to the growth of these dually infected cells, we quantitated growth of BC-1 cells that were transfected with vectors expressing cognate DN Rta, nonfunctional DN Rta, DN LMP-1, or negative control empty vector. Both DN Rta (ΔSTAD) and DN LMP-1 reduced BC-1 cell growth (Fig. 11 A). However, the nonfunctional DN Rta (ΔSTADΔLR) and the empty vector had no effect on cell growth (Fig. 11A). These data support a contribution of spontaneously expressed Rta to growth of KSHV+/EBV+ cells by transactivating LMP-1.
FIG. 11.

Rta transactivation is required for optimal growth of KSHV+/EBV+ PEL cells. BC-1 cells (A) and BL-41 cells (B) were electroporated with equal amounts of the indicated expression vectors, and total live cells were counted at the indicated times postelectroporation. The fold cell number equals total live cells at the indicated times, divided by total live cells 1 h postelectroporation. “LMP-1 DN” is pSG5-LMP-1-AAAG. (A) *, P < 0.0231; **, P < 0.0633 (both compared to the pcDNA3 column). (B) *, P = 0.3508 (not significantly different than empty vector [pcDNA3]).
To verify that the vectors tested in the above experiment did not have a nonspecific effect on B-cell growth, we transfected uninfected B cells, BL-41, in the same manner as Fig. 11A. The data show there was no significant difference in growth between the indicated expression plasmids compared to empty vector (Fig. 11B). Thus, the expression plasmids did not have a nonspecific toxic effect on the B cells, and we conclude that the RtaΔSTAD suppressive effect on growth required KSHV and/or EBV infection.
DISCUSSION
Reactivation of KSHV from latency requires the cellular protein RBP-Jk to specify transcriptional targets for the KSHV Rta lytic switch protein (45). The KSHV genome includes 177 predicted binding sites for RBP-Jk. RBP-Jk also specifies transcriptional targets for EBV EBNA-2 and the activated form of the cellular Notch receptor NICD1. Despite the apparent mechanistic similarity of all three RBP-Jk-dependent transactivators, neither EBNA-2 nor NICD productively reactivates KSHV from latency (11, 14, 45). These observations support the inference that these proteins are not always phenotypically interchangeable, and a noncanonical mechanism for regulating RBP-Jk promoter specification may operate in KSHV-infected cells. Since KSHV and EBV both maintain latency in B cells and coinfect 70% of the established PEL cell lines, we wanted to determine whether the KSHV lytic switch protein, Rta, could function in an EBNA-2-like fashion and cross talk to EBV in coinfected cells.
In the present study, we demonstrate that Rta transactivates the EBV LMP-1 and latency C promoters (Cp) in uninfected cells and in dually infected PEL cells (Fig. 3). RBP-Jk was required for Rta to transactivate both EBV latency promoters (Fig. 3C and D), and Rta formed a ternary complex with RBP-Jk and both the Cp and the LMP-1 promoter DNAs (Fig. 4A and B). We showed that dominant-negative Rta (RtaΔSTAD) potentiated transactivation of Cp and LMP-1 by a constitutively active RBP-Jk allele (Fig. 4D and E). We have previously shown that all three mechanisms are used by Rta to transactivate the promoter of the essential delayed-early (DE) KSHV gene ORF57/Mta and correspond with the stimulation of RBP-Jk DNA binding by Rta (11). Therefore, Rta regulates the EBV latency promoters as if they were KSHV DE promoters. Although RBP-Jk specifies these EBV promoters as Rta targets, it is likely that Rta stimulates RBP-Jk binding to them, a mechanism distinct from that of EBNA-2 (11).
Since Rta and EBNA-2 shared similar promoter specificities, we tested their biological significance in EBV-infected cells that are conditionally immortalized by EBNA-2 (EREB2-5 cells). We showed that Rta maintains short-term growth of the cells when EBNA-2 is inactivated (Fig. 6) and that this effect required DNA binding of Rta and RBP-Jk (Fig. 9). Moreover, our data reveal that Rta's ability to maintain short-term viability of the cells in the absence of EBNA-2 is attributable to a paracrine effect (Fig. 7). Therefore, the minor population of Rta-expressing cells supported the growth of the remaining cells that were not expressing Rta. Rta's complementation of the EBNA-2 deficiency also required LMP-1 signaling (Fig. 8). Since LMP-1 expression induced paracrine signaling in other studies (19, 36), we hypothesize that LMP-1 is playing the same role in EREB2-5 cells transiently expressing Rta.
Although our short-term cell growth assays demonstrated an EBNA-2-like function for Rta, our array data suggest that short-term rescue of LCL growth by Rta is associated with a pattern of cellular proteome modulation distinct from that of EBNA-2. Although all of the EBNA-2 targets were shared with Rta, Rta uniquely activated the antiapoptotic proteins survivin, livin, IGF-1 receptor, and the cellular inhibitor of apoptosis protein (cIAP2) (Fig. 10). Although we have not determined whether Rta and EBNA-2 differentially regulate the LCL transcriptome, we hypothesize that Rta's broader set of induced targets is attributable to its unique ability to stimulate RBP-Jk DNA binding (11).
One question raised by our data is whether the three treatments rescue short-term LCL growth by unique effects on the cell proteome or whether the rescue is attributable to proteomic changes common to the three treatments. The data suggest that paracrine effects of expressing EBNA-2 and Rta are sufficient to rescue short-term LCL growth, without significantly repressing cellular protein expression within the subset of the proteome that we screened. It has long been recognized that LCL growth is dependent on paracrine/autocrine mechanisms (19, 26, 61). The obvious cellular candidates for rescuing short-term growth are those secreted proteins induced by all three treatments. In particular, Rta and β-estradiol both repressed TNF-α, which suppresses LCL growth (21) (Fig. 10) (unfortunately, EBNA-2's effect on TNF-α is unknown since it was invalidated in our quality control procedure for the assay). If instead, the short-term growth of the LCLs is rescued by inducing unique proteomes, then another important question is whether the effect of Rta is sufficient for long-term outgrowth of LCLs. Partial rescue of EBNA-2 deficiency, and initialization, but not complete LCL transformation, have been previously reported for NICD1 (25, 29) and truncated LMP-1 (36), respectively. In this scenario, the unique proteomic changes associated with re-addition of β-estradiol to EREB2-5 cells in our study would provide the most likely candidates for supporting long-term outgrowth of LCLs. Finally, it is currently unclear how induction of proapoptotic proteins in our experiments is consistent with maintaining short-term growth of the EREB2-5 cells (Fig. 10).
While the EREB2-5 experiments were a key to investigating the similarities between Rta and EBNA-2, the biologically relevant experiments used PEL cells coinfected by KSHV and EBV. We showed that the dominant-negative Rta allele, RtaΔSTAD, but not a nonfunctional RtaΔSTADΔLR allele, inhibited growth of coinfected PEL cells (Fig. 11A). Importantly, the growth-inhibitory effect of RtaΔSTAD required cells to be infected by EBV and KSHV, since the dominant-negative had no effect on growth of uninfected BL-41 cells (Fig. 11B). Similar to EREB2-5 cells, we achieved only ca. 5% transfection efficiency in BC-1 PEL cells. Therefore, the growth-suppressing effect of RtaΔSTAD in BC-1 PELs supports two conclusions: (i) RtaΔSTAD is likely inhibiting a paracrine effect of Rta on cell growth of PELs, and (ii) a large proportion of the BC-1 cell population spontaneously expressed endogenous Rta at some point during the 7-day duration of the experiment. In the latter situation, spontaneous Rta expression must have been asynchronous and unsustained, since spontaneous Rta is typically only detected in ca. 5% of cells when assayed by immunofluorescence (48). We recognize that immunofluorescence may underestimate the actual percentage of cells expressing Rta; however, regardless of the total number of Rta-expressing cells, the growth rescue of untransfected EREB2-5 cells by media conditioned by a small percentage of Rta-expressing cells (Fig. 7) strongly supports a paracrine mechanism for Rta's effect. Further, we hypothesize that the growth-suppressing effect of the dominant-negative RtaΔSTAD in coinfected cells (Fig. 11A) might be due to a “feed-forward” mechanism in which RtaΔSTAD transfected cells inhibit spontaneous Rta expression in the cell population in a paracrine fashion, also. We note that we have previously shown that RtaΔSTAD completely inhibits spontaneous KSHV reactivation in singly infected cells (47).
We propose a model for convergence of KSHV reactivation with EBV latency in coinfected B cells through their common cellular target, the Notch effector RBP-Jk (Fig. 12). Rta activates the EBV type III latency promoters as if they were KSHV DE promoters (Fig. 12), in a background of type I latency. We propose that the small amount of LMP-1 detected in PEL cell lines and primary tumors (6, 7, 9, 10, 30, 67) may be dependent on spontaneous Rta expression. The set of cellular proteins induced by EBNA-2 or Rta only partially overlap (Fig. 12). Previous publications have revealed negative, reciprocal feedback between KSHV and EBV in dually infected cells (33, 50, 69). Most notably, LMP-1 expression inhibits KSHV Rta expression and reactivation (Fig. 12) (69). We surmise that LMP-1 provides a negative-feedback loop to promote a cellular environment in which Rta indirectly contributes to PEL growth without productively reactivating KSHV and lysing the infected cell. Although we have not investigated the role of KSHV oncogenes in our studies, we point out that a similar negative-feedback relationship has been demonstrated for Rta and its transcriptional target, the KSHV oncogene ORF74/vG-protein-coupled receptor (vGPCR) (8).
FIG. 12.

Model of convergence of KSHV reactivation with EBV latency in KSHV+/EBV+ B cells. Rta transactivates EBV latency promoters as if they were KSHV delayed early promoters. Rta activates EBV latency type III promoters in a background of latency type I. In accordance with previous published data suggesting a negative, reciprocal feedback between KSHV and EBV (see the text), the subsequent expression of LMP1 would inhibit Rta. Sets of cellular proteins induced by Rta and EBNA2 partially overlap.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes of Health (AI 078138) and the American Heart Association (0855879D).
We thank Nikhat Parveen, Michael B. Mathews, Vivian Bellofatto, and Hua Zhu for helpful discussions. We gratefully acknowledge Bettina Kempkes, Tasuku Honjo, Martin Rowe, Eric Adriaenssens, David Thorley-Lawson, and Gail Bishop for gifts of plasmids, cell lines, and reagents.
Footnotes
Published ahead of print on 4 August 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Abramoff, M., P. Magelhaes, and S. Ram. 2004. Image processing with ImageJ. Biophotonics Int. 11:36-42. [Google Scholar]
- 2.Adriaenssens, E., A. Mougel, G. Goormachtigh, E. Loing, V. Fafeur, C. Auriault, and J. Coll. 2004. A novel dominant-negative mutant form of Epstein-Barr virus latent membrane protein-1 (LMP1) selectively and differentially impairs LMP1 and TNF signaling pathways. Oncogene 23:2681-2693. [DOI] [PubMed] [Google Scholar]
- 3.Brennan, P., J. E. Floettmann, A. Mehl, M. Jones, and M. Rowe. 2001. Mechanism of action of a novel latent membrane protein-1 dominant negative. J. Biol. Chem. 276:1195-1203. [DOI] [PubMed] [Google Scholar]
- 4.Bu, W., K. D. Carroll, D. Palmeri, and D. M. Lukac. 2007. The Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 81:5788-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bu, W., D. Palmeri, R. Krishnan, R. Marin, V. M. Aris, P. Soteropoulous, and D. M. Lukac. 2008. Identification of direct transcriptional targets of the KSHV Rta lytic switch protein by conditional nuclear localization. J. Virol. 82:10709-10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callahan, J., S. Pai, M. Cotter, and E. S. Robertson. 1999. Distinct patterns of viral antigen expression in Epstein-Barr virus and Kaposi's sarcoma-associated herpesvirus coinfected body-cavity-based lymphoma cell lines: potential switches in latent gene expression due to coinfection. Virology 262:18-30. [DOI] [PubMed] [Google Scholar]
- 7.Cannon, J. S., D. Ciufo, A. L. Hawkins, C. A. Griffin, M. J. Borowitz, G. S. Hayward, and R. F. Ambinder. 2000. A new primary effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma herpesvirus-containing supernatant. J. Virol. 74:10187-10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cannon, M., E. Cesarman, and C. Boshoff. 2006. KSHV G protein-coupled receptor inhibits lytic gene transcription in primary-effusion lymphoma cells via p21-mediated inhibition of Cdk2. Blood 107:277-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carbone, A., G. Gaidano, A. Gloghini, L. M. Larocca, D. Capello, V. Canzonieri, A. Antinori, U. Tirelli, B. Falini, and R. Dalla-Favera. 1998. Differential expression of BCL-6, CD138/syndecan-1, and Epstein-Barr virus-encoded latent membrane protein-1 identifies distinct histogenetic subsets of acquired immunodeficiency syndrome-related non-Hodgkin's lymphomas. Blood 91:747-755. [PubMed] [Google Scholar]
- 10.Carbone, A., A. Gloghini, E. Vaccher, V. Zagonel, C. Pastore, P. Dalla Palma, F. Branz, G. Saglio, R. Volpe, U. Tirelli, and G. Gaidano. 1996. Kaposi's sarcoma-associated herpesvirus DNA sequences in AIDS-related and AIDS-unrelated lymphomatous effusions. Br. J. Hematol. 94:533-543. [DOI] [PubMed] [Google Scholar]
- 11.Carroll, K. D., W. Bu, D. Palmeri, S. Spadavecchia, S. J. Lynch, S. A. Marras, S. Tyagi, and D. M. Lukac. 2006. Kaposi's sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 80:9697-9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carroll, K. D., F. Khadim, S. Spadavecchia, D. Palmeri, and D. M. Lukac. 2007. Direct interactions of KSHV/HHV-8 ORF50/Rta protein with the cellular protein Octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 81:8451-8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cesarman, E., P. S. Moore, P. H. Rao, G. Inghirami, D. M. Knowles, and Y. Chang. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708-2714. [PubMed] [Google Scholar]
- 14.Chang, H., D. P. Dittmer, S. Y. Chul, Y. Hong, and J. U. Jung. 2005. Role of Notch signal transduction in Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 79:14371-14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 16.Dawson, C. W., G. Tramountanis, A. G. Eliopoulos, and L. S. Young. 2003. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J. Biol. Chem. 278:3694-3704. [DOI] [PubMed] [Google Scholar]
- 17.Drexler, H., C. Uphoff, G. Gaidano, and A. Carbone. 1998. Lymphoma cell lines: in vitro models for the study of HHV-8+ primary effusion lymphomas (body cavity-based lymphomas). Leukemia 12:1507-1517. [DOI] [PubMed] [Google Scholar]
- 18.Eliopoulos, A. G., C. W. Dawson, G. Mosialos, J. E. Floettmann, M. Rowe, R. J. Armitage, J. Dawson, J. M. Zapata, D. J. Kerr, M. J. Wakelam, J. C. Reed, E. Kieff, and L. S. Young. 1996. CD40-induced growth inhibition in epithelial cells is mimicked by Epstein-Barr virus-encoded LMP1: involvement of TRAF3 as a common mediator. Oncogene 13:2243-2254. [PubMed] [Google Scholar]
- 19.Eliopoulos, A. G., N. J. Gallagher, S. M. Blake, C. W. Dawson, and L. S. Young. 1999. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J. Biol. Chem. 274:16085-16096. [DOI] [PubMed] [Google Scholar]
- 20.Eliopoulos, A. G., and L. S. Young. 1998. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene 16:1731-1742. [DOI] [PubMed] [Google Scholar]
- 21.Estrov, Z., R. Kurzrock, E. Pocsik, S. Pathak, H. M. Kantarjian, T. F. Zipf, D. Harris, M. Talpaz, and B. B. Aggarwal. 1993. Lymphotoxin is an autocrine growth factor for Epstein-Barr virus-infected B-cell lines. J. Exp. Med. 177:763-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faumont, N., S. Durand-Panteix, M. Schlee, S. Gromminger, M. Schuhmacher, M. Holzel, G. Laux, R. Mailhammer, A. Rosenwald, L. M. Staudt, G. W. Bornkamm, and J. Feuillard. 2009. c-Myc and Rel/NF-κB are the two master transcriptional systems activated in the latency III program of Epstein-Barr virus-immortalized B cells. J. Virol. 83:5014-5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fortini, M. E. 2009. Notch signaling: the core pathway and its posttranslational regulation. Dev. Cell 16:633-647. [DOI] [PubMed] [Google Scholar]
- 24.Gao, S. J., L. Kingsley, M. Li, W. Zheng, C. Parravicini, J. Ziegler, R. Newton, C. R. Rinaldo, A. Saah, J. Phair, R. Detels, Y. Chang, and P. S. Moore. 1996. KSHV antibodies among Americans, Italians, and Ugandans with and without Kaposi's sarcoma. Nat. Med. 2:925-928. [DOI] [PubMed] [Google Scholar]
- 25.Gordadze, A. V., R. Peng, J. Tan, G. Liu, R. Sutton, B. Kempkes, G. W. Bornkamm, and P. D. Ling. 2001. Notch1 IC partially replaces EBNA2 function in B cells immortalized by Epstein-Barr virus. J. Virol. 75:5899-5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon, J., S. C. Ley, M. D. Melamed, P. Aman, and N. C. Hughes-Jones. 1984. Soluble factor requirements for the autostimulatory growth of B lymphoblasts immortalized by Epstein-Barr virus. J. Exp. Med. 159:1554-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammarskjold, M. L., and M. C. Simurda. 1992. Epstein-Barr virus latent membrane protein transactivates the human immunodeficiency virus type 1 long terminal repeat through induction of NF-κB activity. J. Virol. 66:6496-6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofelmayr, H., L. J. Strobl, G. Marschall, G. W. Bornkamm, and U. Zimber-Strobl. 2001. Activated Notch1 can transiently substitute for EBNA2 in the maintenance of proliferation of LMP1-expressing immortalized B cells. J. Virol. 75:2033-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofelmayr, H., L. J. Strobl, C. Stein, G. Laux, G. Marschall, G. W. Bornkamm, and U. Zimber-Strobl. 1999. Activated mouse Notch1 transactivates Epstein-Barr virus nuclear antigen 2-regulated viral promoters. J. Virol. 73:2770-2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horenstein, M. G., R. G. Nador, A. Chadburn, E. M. Hyjek, G. Inghirami, D. M. Knowles, and E. Cesarman. 1997. Epstein-Barr virus latent gene expression in primary effusion lymphomas containing Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8. Blood 90:1186-1191. [PubMed] [Google Scholar]
- 31.Huen, D. S., S. A. Henderson, D. Croom-Carter, and M. Rowe. 1995. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-κB and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 10:549-560. [PubMed] [Google Scholar]
- 32.Jarriault, S., C. Brou, F. Logeat, E. H. Schroeter, R. Kopan, and A. Israel. 1995. Signaling downstream of activated mammalian Notch. Nature 377:355-358. [DOI] [PubMed] [Google Scholar]
- 33.Jiang, Y., D. Xu, Y. Zhao, and L. Zhang. 2008. Mutual inhibition between Kaposi's sarcoma-associated herpesvirus and Epstein-Barr virus lytic replication initiators in dually-infected primary effusion lymphoma. PLoS One 3:e1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jochner, N., D. Eick, U. Zimber-Strobl, M. Pawlita, G. W. Bornkamm, and B. Kempkes. 1996. Epstein-Barr virus nuclear antigen 2 is a transcriptional suppressor of the immunoglobulin mu gene: implications for the expression of the translocated c-myc gene in Burkitt's lymphoma cells. EMBO J. 15:375-382. [PMC free article] [PubMed] [Google Scholar]
- 35.Kato, H., Y. Taniguchi, H. Kurooka, S. Minoguchi, T. Sakai, S. Nomura-Okazaki, K. Tamura, and T. Honjo. 1997. Involvement of RBP-J. in biological functions of mouse Notch1 and its derivatives. Development 124:4133-4141. [DOI] [PubMed] [Google Scholar]
- 36.Kaye, K. M., K. M. Izumi, H. Li, E. Johannsen, D. Davidson, R. Longnecker, and E. Kieff. 1999. An Epstein-Barr virus that expresses only the first 231 LMP1 amino acids efficiently initiates primary B-lymphocyte growth transformation. J. Virol. 73:10525-10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kedes, D., E. Operskalski, M. Busch, R. Kohn, J. Flood, and D. Ganem. 1996. The seroepidemiology of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nat. Med. 2:918-924. [DOI] [PubMed] [Google Scholar]
- 38.Kempkes, B., D. Spitkovsky, P. Jansen-Durr, J. W. Ellwart, E. Kremmer, H. J. Delecluse, C. Rottenberger, G. W. Bornkamm, and W. Hammerschmidt. 1995. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J. 14:88-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kieff, E., and A. Rickinson. 2007. Epstein-Barr virus and its replication, p. 2603-2654. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 2. Lippincott/The Williams & Wilkins Co., Philadelphia, PA. [Google Scholar]
- 40.Kieser, A., E. Kilger, O. Gires, M. Ueffing, W. Kolch, and W. Hammerschmidt. 1997. Epstein-Barr virus latent membrane protein-1 triggers AP-1 activity via the c-Jun N-terminal kinase cascade. EMBO J. 16:6478-6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kroemer, G., L. Galluzzi, and C. Brenner. 2007. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87:99-163. [DOI] [PubMed] [Google Scholar]
- 42.Lacoste, V., J. G. Judde, G. Bestett, J. Cadranel, M. Antoine, F. Valensi, E. Delabesse, E. Macintyre, and A. Gessain. 2000. Virological and molecular characterisation of a new B lymphoid cell line, established from an AIDS patient with primary effusion lymphoma, harbouring both KSHV/HHV8 and EBV viruses. Leuk. Lymphoma 38:401-409. [DOI] [PubMed] [Google Scholar]
- 43.Laherty, C. D., H. M. Hu, A. W. Opipari, F. Wang, and V. M. Dixit. 1992. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor κB. J. Biol. Chem. 267:24157-24160. [PubMed] [Google Scholar]
- 44.Liang, Y., J. Chang, S. Lynch, D. M. Lukac, and D. Ganem. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jk, the target of the Notch signaling pathway. Genes Dev. 16:1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang, Y., and D. Ganem. 2003. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. U. S. A. 100:8490-8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luftig, M., T. Yasui, V. Soni, M. S. Kang, N. Jacobson, E. Cahir-McFarland, B. Seed, and E. Kieff. 2004. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-κB activation. Proc. Natl. Acad. Sci. U. S. A. 101:141-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 49.Macho, A., D. Decaudin, M. Castedo, T. Hirsch, S. A. Susin, N. Zamzami, and G. Kroemer. 1996. Chloromethyl-X-Rosamine is an aldehyde-fixable potential-sensitive fluorochrome for the detection of early apoptosis. Cytometry 25:333-340. [DOI] [PubMed] [Google Scholar]
- 50.Miller, G., L. Heston, E. Grogan, L. Gradoville, M. Rigsby, R. Sun, D. Shedd, V. M. Kushnaryov, S. Grossberg, and Y. Chang. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller, W. E., J. L. Cheshire, and N. Raab-Traub. 1998. Interaction of tumor necrosis factor receptor-associated factor signaling proteins with the latent membrane protein 1 PXQXT motif is essential for induction of epidermal growth factor receptor expression. Mol. Cell. Biol. 18:2835-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mitchell, T., and B. Sugden. 1995. Stimulation of NF-κB-mediated transcription by mutant derivatives of the latent membrane protein of Epstein-Barr virus. J. Virol. 69:2968-2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moore, P. S., C. Boshoff, R. A. Weiss, and Y. Chang. 1996. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274:1739-1744. [DOI] [PubMed] [Google Scholar]
- 54.Moore, P. S., S. J. Gao, G. Dominguez, E. Cesarman, O. Lungu, D. M. Knowles, R. Garber, P. E. Pellett, D. J. McGeoch, and Y. Chang. 1996. Primary characterization of a herpesvirus agent associated with Kaposi's sarcomae. J. Virol. 70:549-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mosialos, G., M. Birkenbach, R. Yalamanchili, T. VanArsdale, C. Ware, and E. Kieff. 1995. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 80:389-399. [DOI] [PubMed] [Google Scholar]
- 56.Nador, R. G., E. Cesarman, A. Chadburn, D. B. Dawson, M. Q. Ansari, J. Sald, and D. M. Knowles. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpesvirus. Blood 88:645-656. [PubMed] [Google Scholar]
- 57.Nellesen, D. T., E. C. Lai, and J. W. Posakony. 1999. Discrete enhancer elements mediate selective responsiveness of enhancer of split complex genes to common transcriptional activators. Dev. Biol. 213:33-53. [DOI] [PubMed] [Google Scholar]
- 58.Niles, A. L., R. A. Moravec, P. Eric Hesselberth, M. A. Scurria, W. J. Daily, and T. L. Riss. 2007. A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal. Biochem. 366:197-206. [DOI] [PubMed] [Google Scholar]
- 59.Palmeri, D., S. Spadavecchia, K. Carroll, and D. M. Lukac. 2007. Promoter and cell-specific transcriptional activation by the Kaposi's sarcoma-associated herpesvirus ORF57/Mta protein. J. Virol. 81:13299-13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Renne, R., W. Zhong, B. Herndier, M. McGrath, N. Abbey, D. Kedes, and D. Ganem. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342-346. [DOI] [PubMed] [Google Scholar]
- 61.Scala, G., I. Quinto, M. R. Ruocco, A. Arcucci, M. Mallardo, P. Caretto, G. Forni, and S. Venuta. 1990. Expression of an exogenous interleukin 6 gene in human Epstein Barr virus B cells confers growth advantage and in vivo tumorigenicity. J. Exp. Med. 172:61-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schlee, M., T. Krug, O. Gires, R. Zeidler, W. Hammerschmidt, R. Mailhammer, G. Laux, G. Sauer, J. Lovric, and G. W. Bornkamm. 2004. Identification of Epstein-Barr virus (EBV) nuclear antigen 2 (EBNA2) target proteins by proteome analysis: activation of EBNA2 in conditionally immortalized B cells reflects early events after infection of primary B cells by EBV. J. Virol. 78:3941-3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simpson, G. R., T. F. Schulz, D. Whitby, P. M. Cook, C. Boshoff, L. Rainbow, M. R. Howard, S. J. Gao, R. A. Bohenzky, P. Simmonds, C. Lee, A. de Ruiter, A. Hatzakis, R. S. Tedder, I. V. Weller, R. A. Weiss, and P. S. Moore. 1996. Prevalence of Kaposi's sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 348:1133-1138. [DOI] [PubMed] [Google Scholar]
- 64.Spender, L. C., G. H. Cornish, B. Rowland, B. Kempkes, and P. J. Farrell. 2001. Direct and indirect regulation of cytokine and cell cycle proteins by EBNA-2 during Epstein-Barr virus infection. J. Virol. 75:3537-3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun, R., S. F. LIn, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szekely, L., F. Chen, N. Teramoto, B. Ehlin-Henriksson, K. Pokrovskaja, A. Szeles, A. Manneborg-Sandlund, M. Lowbeer, E. T. Lennette, and G. Klein. 1998. Restricted expression of Epstein-Barr virus (EBV)-encoded, growth transformation-associated antigens in an EBV- and human herpesvirus type 8-carrying body cavity lymphoma line. J. Gen. Virol. 79:1445-1452. [DOI] [PubMed] [Google Scholar]
- 67.Trivedi, P., K. Takazawa, C. Zompetta, L. Cuomo, E. Anastasiadou, A. Carbone, S. Uccini, F. Belardelli, K. Takada, L. Frati, and A. Faggioni. 2004. Infection of HHV-8+ primary effusion lymphoma cells with a recombinant Epstein-Barr virus leads to restricted EBV latency, altered phenotype, and increased tumorigenicity without affecting TCL1 expression. Blood 103:313-316. [DOI] [PubMed] [Google Scholar]
- 68.Uchida, J., T. Yasui, Y. Takaoka-Shichijo, M. Muraoka, W. Kulwichit, N. Raab-Traub, and H. Kikutani. 1999. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 286:300-303. [DOI] [PubMed] [Google Scholar]
- 69.Xu, D., T. Coleman, J. Zhang, A. Fagot, C. Kotalik, L. Zhao, P. Trivedi, C. Jones, and L. Zhang. 2007. Epstein-Barr virus inhibits Kaposi's sarcoma-associated herpesvirus lytic replication in primary effusion lymphomas. J. Virol. 81:6068-6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu, Y., D. P. AuCoin, A. R. Huete, S. A. Cei, L. J. Hanson, and G. S. Pari. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79:3479-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zimber-Strobl, U., B. Kempkes, G. Marschall, R. Zeidler, C. Van Kooten, J. Banchereau, G. W. Bornkamm, and W. Hammerschmidt. 1996. Epstein-Barr virus latent membrane protein (LMP1) is not sufficient to maintain proliferation of B cells but both it and activated CD40 can prolong their survival. EMBO J. 15:7070-7078. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.