

Abstract
Polyomavirus JC (JCV) infects ∼60% of the general population, followed by asymptomatic urinary shedding in ∼20%. In patients with pronounced immunodeficiency, including HIV/AIDS, JCV can cause progressive multifocal leukoencephalopathy (PML), a devastating brain disease of high mortality. While JCV in the urine of healthy people has a linear noncoding control region called the archetype NCCR (at-NCCR), JCV in brain and cerebrospinal fluid (CSF) of PML patients bear rearranged NCCRs (rr-NCCRs). Although JCV NCCR rearrangements are deemed pathognomonic for PML, their role as a viral determinant is unclear. We sequenced JCV NCCRs found in CSF of eight HIV/AIDS patients newly diagnosed with PML and analyzed their effect on early and late gene expression using a bidirectional reporter vector recapitulating the circular polyomavirus early and late gene organization. The rr-NCCR sequences were highly diverse, but all increased viral early reporter gene expression in progenitor-derived astrocytes, glia-derived cells, and human kidney compared to the expression levels with the at-NCCR. The expression of simian virus 40 (SV40) large T antigen or HIV Tat expression in trans was associated with a strong increase of at-NCCR-controlled early gene expression, while rr-NCCRs were less responsive. The insertion of rr-NCCRs into the JCV genome backbone revealed higher viral replication rates for rr-NCCR compared to those of the at-NCCR JCV in human progenitor-derived astrocytes or glia cells, which was abrogated in SV40 large T-expressing COS-7 cells. We conclude that naturally occurring JCV rr-NCCR variants from PML patients confer increased early gene expression and higher replication rates compared to those of at-NCCR JCV and thereby increase cytopathology.
Polyomavirus JC (JCV) infects approximately 60% of the general population, followed by asymptomatic urinary shedding in 20% of healthy individuals (20). Although JCV-associated nephropathy may occur in kidney transplant (14, 33) and HIV/AIDS patients (6, 27), the most prominent JCV disease is progressive multifocal leukoencephalopathy (PML) (44, 60). The pathology of PML was first described in 1958 as a rare complication of patients with chronic lymphocytic leukemia or Hodgkin's lymphoma (3). Today, PML is recognized as a rare, virus-mediated demyelinating disease of the white brain matter in highly immunocompromised patients, including HIV/AIDS, transplantation, and chemotherapy patients and those exposed to immunomodulatory or depleting biologicals for the treatment of autoimmune diseases (29, 40). During the human immunodeficiency virus type 1 (HIV-1) pandemic, the incidence of PML rose significantly to rates of 1 to 8% prior to the use of highly active antiretroviral therapy (2, 5, 34). The definitive diagnosis requires brain tissue, but the detection of JCV by PCR in cerebrospinal fluid (CSF) is generally accepted for a laboratory-confirmed diagnosis in immunocompromised patients with (multi-)focal neurological deficits and corresponding radiological findings (8, 26). Due to the lack of effective antiviral therapy (13), the treatment of PML is based on improving overall immune functions. While this is difficult to achieve in cancer, chemotherapy, and transplantation, prompt antiretroviral therapy in HIV/AIDS patients has significantly improved PML survival, with increasing JCV-specific immune responses and declining intracerebral JCV replication (7, 15, 23, 35, 37). In patients diagnosed with PML after treatment with natalizumab for multiple sclerosis or inflammatory bowel disease, the removal of the monoclonal antibody by plasmapheresis has been tried to restore lymphocyte homing to, and the immune surveillance of, JCV replication sites in the central nervous system (38, 40, 52). However, the success of immune reconstitution in HIV/AIDS- and natalizumab-associated PML cases is limited by the fact that PML is typically diagnosed clinically by neurological deficits resulting from significant brain damage, where mounting antiviral immunity often may be too slow to modify the outcome. On the other hand, rapid recovery may cause immune reconstitution inflammatory syndrome with paradoxical clinical worsening and fatal outcomes (9, 16, 25, 38, 46). Although the etiologic role of JCV in PML is well documented, the pathogenesis and, in particular, the role of viral determinants is less clear. Virtually all JCV strains isolated from the brain or CSF of PML patients are characterized by highly variable genomic rearrangements of the noncoding control region (NCCR), which governs viral early and late genes in opposite directions of the circular polyomavirus DNA genome (1, 4, 31, 39, 41, 43, 49, 54, 59). In contrast, JCV detected in the urine of immunocompetent individuals show a consistent linear architecture called the archetype NCCR (at-NCCR). Thus, detecting rearranged NCCRs (rr-NCCRs) JCV in the central nervous system has been viewed as being derived from the archetype and closely linked to PML (4), but the functional consequences of rearrangements are unclear. To address the consequences of the rr-NCCR for JCV gene expression and replication, we characterized the sequences of JCV rr-NCCR from patients with PML and analyzed their effect on viral gene expression and replication with JCV at-NCCR in a bidirectional reporter assay and in recombinant JCV.
MATERIALS AND METHODS
Patients.
We characterized the JCV NCCRs in the CSF of eight PML patients of white ethnicity (Table 1). From one patient, day-matched CSF, plasma, and urine were available, resulting in a total of 11 samples. The NCCR architecture was determined by an independent diagnostic confirmatory assay. All samples had been submitted for JCV testing to the Molecular Diagnostics Laboratory of the Division of Diagnostics, which is an accredited quality-controlled laboratory (EN ISO 17025-2005) of the University of Basel (www.zid.ch).
TABLE 1.
Patient characteristics, NCCR architecture, and replication capacity of recombinant JCV in cell culture
| Patient | Sex | Age | Sample type | Viral load (geq/mla) | NCCR architecture | Replication in cell cultureb |
|---|---|---|---|---|---|---|
| 1 | M | 41 | Urine sample 1c | 6.43 × 105 | Archetype | (+) |
| Plasma sample 1c | 5.53 × 102 | Insertion | + | |||
| CSF-1c | 1.28 × 107 | Insertion | ++ | |||
| CSF-2 | 3.35 × 107 | Insertion | + | |||
| 2 | M | 56 | CSF | 2.34 × 104 | Insertion | ++ |
| 3 | F | 45 | CSF | 6.12 × 102 | Insertion | + |
| 4 | M | 37 | CSF | 1.51 × 105 | Insertion | + |
| 5 | F | 27 | CSF | 2.58 × 105 | Insertion | +++ |
| 6 | M | 39 | CSF | 2.27 × 103 | Deletion | ++ |
| 7 | M | 43 | CSF | 5.62 × 103 | Deletion | ++ |
| 8 | M | 55 | CSF | 3.28 × 104 | Deletion | +++ |
| NAd | NA | NA | NA | NA | Insertion (Mad-4) | +++ |
geq/ml, genome equivalents per milliliter.
Graduation of virus replication in PDA cells, from only occasionally JCV-positive cells [(+)] to many JCV-positive cells (+++) per microscopy field.
Day matched.
NA, not applicable.
JCV viral load and NCCR analysis.
Cell culture supernatants were harvested at indicated time points, and naked DNA was removed by DNase I treatment according to the QIAamp DNA blood kit instructions (51104; Qiagen, Hilden, Germany). JCV DNA was quantified after DNA extraction from 200 μl urine, plasma, cerebrospinal fluid, or cell culture supernatant using the QIAamp DNA blood kit and a real-time PCR amplification using the forward primer 5′-ACAGGAAAGTCTTTAGGGTCTTCTACC-3′, the reverse primer 5′-TAGGTGCCAACCTATGGAACAGA-3′, and the 6-carboxyfluorescein (FAM)-labeled probe 5′-TGTTGGGATCCTGTGTTTTCATCACTGGCA-3′. The quantitative PCR (qPCR) was specific for JCV large T antigen and does not detect simian virus 40 (SV40) sequences (14). NCCR sequences were amplified from patient material by nested PCR using primer pair 1 (5′-AGGCCTAATAAATCCATAAGCTCCA-3′ and 5′-GTTCCACTCCAGGTTTTACTAACT-3′) and primer pair 2 (5′-AAAAACGCGTCATTTTAGCTTTTTGCAGCAAAAAATTA-3′ and 5′-TTTTGCGCGCCTGGCGAAGAACCATGGCCAG-3′). The sensitivity of the nested PCR to detect minority species was ∼5% (24). The PCR products were compared after 2% Tris-acetate-EDTA-agarose gel electrophoresis to at-NCCR controls and excised for sequence analysis. The sequences were aligned to the CY archetype (55) by the program MultiAlign (10) and arbitrarily divided into sequence blocks Ori, A, B, C, D, E, and F, consisting of 117, 36, 23, 55, 66, 18, and 69 bp, respectively (4).
Cells and culture.
HEK293 (ATCC CRL1573), CV-1 (ATCC CCL70), and Hs683 (ATCC HTB-138) cells were grown in Dulbecco's modified Eagle's medium (DMEM; D5671; Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) (S0113; Biochrome AG, Berlin, Germany). COS-7 cells (ATCC CRL1651) were cultured in DMEM high-glucose formulation (DMEM-H) containing 5% FBS. HeLa (ATCC CCL-2) and HeLa SX-CCR5 cells (36) were maintained in DMEM-H containing 10% FBS. Human fetal brain progenitor-derived astrocyte cells (PDA) (42) were propagated in Eagle's minimum essential medium (MEM; M2279; Sigma, St. Loius, MO) supplemented with 10% FBS. All cell cultures were supplemented with 2 mM l-glutamine (K0302; Biochrome AG, Berlin, Germany).
Plasmids.
The generation of plasmid pHRG has been described already (24). In pHRG-NCCR plasmids, the expression of red fluorescent protein (RFP) (early) and green fluorescent protein (GFP) (late) is controlled by the inserted JCV NCCR. The MluI-BssHII-cleaved NCCR amplicon was ligated into pHRG digested with the same enzymes. The plasmid pHRGJCVΔD was generated from at-NCCR by the deletion of nucleotides D1 to 66 using gene splicing by overlap extension (28). Two DNA fragments were generated using primer pair 1 (forward, 5′-AAAAACGCGTCATTTTAGCTTTTTGCAGCAAAAAATTA-3′; reverse, 5′-CCCCTTGTCCTTTGTTTACTGGCTGTCAGCTGGTTG-3′) and primer pair 2 (forward, 5′-CAACCAGCTGACAGCCAGTAAACAAAGCACAAGGGG-3′; reverse, 5′-TTTTGCGCGCCTGGCGAAGAACCATGGCCAG-3′). The two DNA fragments were joined in a PCR mixture containing the forward primer of pair 1 and reverse primer of pair 2. The MluI-BssHII-cleaved amplification product was cloned as described above. Plasmid pGEMMad-4 was cloned by the extraction of JCV DNA from a 200-μl aliquot of JCV (ATCC VR-1583) as described above and the cleavage of eluted DNA by EcoRI followed by ligation into pGEM3Zf(+) (P2271; Promega, Madison, WI) digested with the same enzyme. Recombinant JCV genomes were generated by synthesizing the NcoI-NcoI insert (Eurogentec, Liège, Belgium) of the corresponding NCCR and ligating it into pGEMMad-4 cleaved with the same enzyme to generate pGEMJCV plasmids. The orientation and intactness of all constructs was verified by sequencing.
Transfection and infection.
The transfection of QIAamp DNA kit-purified JCV genomes into COS-7 cells was initiated by the cleavage of the pGEMJCV plasmid DNAs with EcoRI, followed by the religation of the diluted DNA. The transfection of purified religated JCV and reporter plasmid DNA into cells was performed at 60 to 80% confluence in 12- or 24-well plates using Lipofectamine 2000 (11668-019; Invitrogen, Carlsbad, CA) according to the manufacturer's instructions at an optimized DNA-to-Lipofectamine ratio of 0.8 to 1 (24). Hs683 cells were transfected with GeneExpressoPlus (IV-1032; Lab Supply Mall, InnoVita Inc., Gaithersburg, MD) at 90 to 95% confluence in 24-well plates at a DNA-to-GeneExpressoPlus ratio of 1:3 with religated JCV genomes. PDA cells were transfected with GeneExpresso8000 (IV-1074; Lab Supply Mall) at 90 to 95% confluence in 12-well plates at a DNA-to-GeneExpresso8000 ratio of 1:2 with reporter plasmid DNA. For all transfections, medium was replaced after 4 to 6 h. At 12 days posttransfection, COS-7 cells were harvested by scraping off cells in 1/10 of the cell culture supernatant. Virus was released by six cycles of the freezing-thawing of the cells and centrifugation at 800 × g for 5 min. The infection of PDA cells was performed by the overlay of growth-phase monolayers with the virus-containing COS-7 cell lysate at 37°C for 2 h. Following incubation, virus inoculum was replaced by complete MEM-2% FBS.
Immunofluorescence and antibodies.
Cells were fixed at indicated times with 4% paraformaldehyde in phosphate-buffered saline (PBS) at 37°C for 20 min and permeabilized with 0.2% Triton in PBS at 37°C for 10 min (24). After incubation with monoclonal mouse anti-SV40 large T antigen (1:50; DP02; Merck, Darmstadt, Germany) or monoclonal mouse anti-VP1 (1:20; NCL-JCBK; Novocastra Laboratories, United Kingdom) and polyclonal rabbit anti-agno (1:600) (45), diluted in 3% dry milk-PBS at 37°C for 1 h, cells were washed two times with PBS at room temperature for 5 min and incubated with secondary fluorescently labeled antibodies anti-mouse Cy3 (1:2,000; 115-165-146; Jackson ImmunoResearch, West Grove, PA) and anti-rabbit Cy2 (1:500; 111-255-045; Jackson ImmunoResearch) and Hoechst 33342 dye (0.5 μg/ml; H21492; Invitrogen) as described for primary antibodies. After two washes as before, specimens were mounted in 90% glycerol (1.04095; Merck) in PBS containing 1% N-propyl gallate (P-3130; Sigma) as an antifading agent.
Microscopy and confocal laser-scanning microscopy (CLSM).
Microscopy was performed using TE200 or Eclipse E800 (Nikon) epifluorescence microscopes equipped with suitable filters and digital cameras (Hamamatsu or Nikon DS-2MBWc), respectively. The counting of red and green cells and digital image processing has been described already (24). As transfection controls, plasmids expressing RFP or GFP alone in the pHRG background showed less than 10% variance in independent experiments. Confocal images were recorded with a confocal laser-scanning microscope (Zeiss LSM 510 Meta; Carl Zeiss AG, Oberkochen, Germany), using a Plan-Neofluar 10× magnification/0.3-numeric-aperture or Plan-Neofluar 20× magnification/0.5-numeric-aperture objective, and the following lasers were used: enterprise, 405 nm; argon laser with a 488-nm laser line; and He-Ne, 543 nm. To exclude the possibility of channel cross-talk, images were acquired sequentially using the multitrack mode. In an alternative approach, we also recorded pictures using an automated ImageXpressMicro microscope running MetaXpress 3.0 (Molecular Devices, CA). The PDA cell count was determined by the Hoechst staining of the nuclei (approximately 1,000 cells per picture, 20 pictures per culture well), and the number of red and/or green cells was quantified.
Accession numbers.
NCCR sequences have been deposited in GenBank under the accession numbers HM038523 to HM038533.
RESULTS
Patient CSF samples contain unique JCV rr-NCCRs.
We studied eight consecutive patients with PML (median age, 42 years; range, 27 to 56), all of whom were HIV positive and whose CSF samples had been submitted to our laboratory for diagnostic JCV PCR. The median JCV load in the eight CSF samples was 4.52 log10 genome equivalents (geq)/ml (range, 2.79 to 7.35 log10 geq/ml). For patient 1, we also studied a day-matched sample from plasma and urine as well as a second CSF sample taken 3 days after the first sampling, yielding a total of 11 samples. The entire JCV NCCR was amplified, generating a fragment from the start codon of the large T antigen to the start codon of the agnoprotein (Table 1). Computer-assisted analysis showed that the nine NCCRs amplified from the CSF from eight patients differed from each other in sequence and length and were unique in the NCBI database. In contrast, the JCV NCCR sequence detected in the same-day urine sample of patient 1 was identical to that of the archetype strain CY reported by Yogo and colleagues (55), showing a linear architecture from the origin of replication (ori) followed by the sequence blocks A through F, but with two single-nucleotide changes in the O block (T4→A and T13→C) (Fig. 1). The two nucleotide changes also were found in all of the CSF samples studied here (see Fig. S1 in the supplemental material) and had been noted earlier in JCV sequences identified in the urine of healthy Swiss blood donors (20). The analysis of the eight rr-NCCRs from CSF revealed the partial or complete deletion of sequence block D (patient 1, plasma, CSF-1, and CSF-2; patients 3 to 5; and patient 8). Deletions varied from small (patient 6) to more-complex cessations of whole sequence blocks (patient 8). Insertions ranged from simple duplications (patient 2) to rather complex rearrangements (patient 5). The detailed analysis for patient 1 revealed that despite being unique, the day-matched CSF-1 and the plasma sample as well as the CSF-2 sample showed similarities, since all rr-NCCRs had maintained the archetype architecture from the O block to the end of the C block at nucleotide 55, followed by partial deletions of the D block and partial duplications of the blocks B, C, and/or F, respectively. As the CSF samples were taken only 72 h apart, these findings indicate that there are different, but closely related, JCV majority species in patient 1. Taken together, the data suggest that naturally occurring rr-NCCRs arise as unique genetic rearrangements of the archetype JCV genome, which are replaced by other rr-NCCR variants as predominant species during ongoing replication in the course of the disease.
FIG. 1.

JCV NCCR architecture and reporter gene expression in PDA cells. The at-NCCR of JCV is shown with the following arbitrarily denoted blocks (the number of base pairs is in parentheses): Ori(117)-A(36)-B(23)-C(55)-D(66)-E(18)-F(69). Also shown is the architecture of the rr-NCCRs. Pt, patient; source, origin of sample; ins, insertions corresponding to duplications of the numbered base pairs; del, deletions denoted by gaps and nucleotide number; confluence, samples showing phase contrast; early, red fluorescence; late, green fluorescence. Cells were transfected with the indicated bidirectional reporter constructs, and expression was quantified at 2 dpt. Expression level indicates the fold induction of green and red fluorescence-positive cells normalized to GFP- and RFP-positive cells bearing pHRG/urine at the at-NCCR; ratio, the number of RFP-positive cells divided by the number of GFP-positive cells; n.a., not applicable, since the NCCR has been generated by in vitro mutagenesis or represents the NCCR of reference strain Mad-4. Means from three independent experiments are shown. Error bars represent the means ± standard deviations.
Naturally selected JCV rr-NCCRs increase viral early gene expression.
To investigate the functional consequences of the natural rr-NCCR with regard to viral early and late gene expression, the different NCCRs were inserted into the bidirectional reporter vector pHRG (24), which recapitulates the polyomavirus genome organization. Here, the expression of red fluorescent protein (RFP; early) and green fluorescent protein (GFP; late) is controlled by the inserted JCV NCCR. Following the transfection of human brain progenitor-derived astrocyte (PDA) cells (42), we compared the different JCV rr-NCCR pHRG constructs for red and green fluorescence. The expression pattern of RFP and GFP became detectable after 1 day posttransfection (dpt) and remained stable for at least 7 days. Analyzing red and green fluorescence at 2 dpt, we found that the at-NCCR isolated from urine of patient 1 showed a weak early gene expression (red signal), which typically was less than half that of late gene expression (green signal; ratio, 0.49) (Fig. 1). In contrast, all rr-NCCRs demonstrated an increased early gene expression of 3- to 9-fold, whereas the late gene expression remained weak. Compared to that of the archetype, rr-NCCR showed an increased ratio of early to late gene expression of 3.06 (median, 3.3; range, 1.55 to 4.26). Simple deletions (patient 6) or insertions (patient 2) could have effects similar to those of more complex ones (patient 4 or 8), but there was no common sequence alteration. However, several rr-NCCRs had various deletions of the D block, similarly to many rr-NCCRs reported by others (1, 4, 21, 41). Therefore, we deleted the sequence block D from the archetype NCCR and inserted this or the reference strain Mad-4 NCCR into pHRG. Both ΔD and Mad-4 rr-NCCRs increased early gene expression in PDA cells by 8.6- and 3.2-fold, respectively, with red-to-green ratios of 2.73 and 4.82, respectively (Fig. 1). Similar results were obtained when at-NCCR and rr-NCCR constructs were transfected into human glioma cells (Hs683), CV-1 cells (data not shown), and human embryonic kidney cells (HEK293) (see Fig. S2 in the supplemental material). To assess the quantitative distribution, we performed an automated microscopy analysis of transfected PDA cells. Comparing at-NCCR and rr-NCCR (Fig. 2), the ratio of red to green was inverted, and the early gene expression was significantly higher for rr-NCCR constructs (see Fig. S3 in the supplemental material). This technique also permitted measuring the distribution of the relative red and green signal intensities, indicating higher values for rr-NCCR than at-NCCR (Fig. 2). We concluded that naturally occurring rr-NCCRs from patients newly diagnosed with PML as well as the rr-NCCR from the reference strain Mad-4 or the deletion of the D block are characterized by an increased early gene expression compared to that of the archetype NCCR.
FIG. 2.
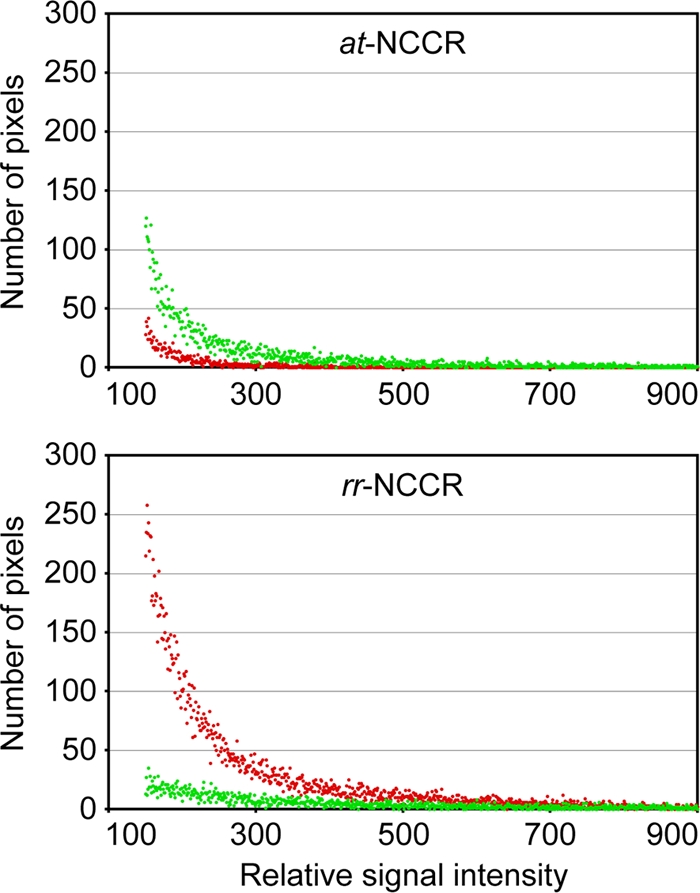
Reporter gene expression in progenitor-derived astrocytes (PDA). The distribution of the red (early gene) and green (late gene) fluorescence of at-NCCR and rearranged CSF-1 (patient 1) rr-NCCR dual-reporter gene expression was examined in PDA cells by automated microscopy and MetaXpress software (see Materials and Methods). The histogram displays pixels carrying red and green signal distributed according to their relative signal intensity along the respective axes.
Naturally occurring rr-NCCRs increase JCV replication.
To directly determine the impact of rr-NCCRs on JCV replication, we generated recombinant JCV strains by inserting the indicated 10 rr-NCCRs, as well as the at-NCCR, into the backbone genome of the JCV Mad-4 strain. The replication of the recombinant JCV was analyzed in the human glioma cell line Hs683 after transfection or infection by determining DNase-protected JCV loads in the culture supernatant. JCV DNA loads increased more rapidly for rr-NCCR JCV than for at-NCCR, reaching more-than 50-fold higher levels after 14 days (Fig. 3 A). Staining for viral early gene large T antigen revealed that significantly higher expression levels at day 7 compared to those of the archetype (Fig. 3B) and the detection of viral late gene agnoprotein confirmed the completion of the viral life cycle (Fig. 3B, inset). The direct sequencing of the NCCR found in the cell culture supernatants at day 14 revealed in all cases identity to the input NCCR. Similar results were obtained in analyses of JCV replication in human PDA cells, which revealed a more rapid replication of rr-NCCR JCV variants and the reference strain Mad-4 compared to that of the at-NCCR JCV (Fig. 3C). The expression of the large T antigen was significantly more abundant at 14 days postinfection (Fig. 3D). Similar results were obtained for the remaining rr-NCCR JCV variants (Table 1). The results indicated that JCV rr-NCCRs from PML patients were sufficient to increase JCV large T antigen expression and replication rates compared to those of the archetype.
FIG. 3.

Replication of rr-NCCR JCV in cell culture. (A) Time course for prototypes CSF (patient 8), CSF-1 (patient 1), Mad-4, and archetype urine (patient 1) in Hs683 cells. DNase-protected JCV DNA was quantified by qPCR from cell supernatants collected at the indicated times after transfection. The data display the means from two independent experiments determined in triplicate. Error bars indicate the means ± standard deviations. (B) Immunofluorescence of JCV prototypes CSF (patient 8) (a), CSF-1 (patient 1) (b), Mad-4 (c), and urine (patient 1) (d) was performed at 7 dpt in Hs683 cells. Large T antigen (red) and agnoprotein (green) were detected using monoclonal mouse anti-large T antigen visualized with anti-mouse Cy3 and polyclonal rabbit anti-agno stained by anti-rabbit Cy2, respectively. Cell nuclei were visualized by Hoechst 33342 dye (blue). (C) Time course for prototypes in PDA cells as described for panel A. DNase-protected JCV DNA was quantified by qPCR from cell supernatants collected at the indicated times after infection. The data display the means from two independent experiments determined in triplicate. Error bars indicate the means ± standard deviations. (D) Immunofluorescence for prototypes was performed at 14 dpi in PDA cells as described for panel B.
SV40 large T antigen or HIV Tat increase JCV rr-NCCR-driven reporter gene expression.
To investigate whether the rr-NCCRs allowed for modulatory effects in trans, we analyzed the replication of the recombinant JCV in COS-7 cells that constitutively express SV40 large T antigen. Similar rates of increasing supernatant JCV loads were observed for the rr-NCCR JCV variants and at-NCCR JCV (Fig. 4 A). Immunofluorescent staining for VP1 indicated comparable expression for rearranged and archetype NCCR JCV (Fig. 4B). These results indicated that the presence of SV40 large T antigen in trans abrogated the difference in replication rates between the archetype and rr-NCCR JCV variants and suggested that the replication advantage of JCV rr-NCCR involves increased large T antigen expression. We therefore compared the reporter gene expression in the COS-7 cells to that of the corresponding precursor CV-1 cells that do not express SV40 large T antigen. In CV-1 cells (Fig. 5 A), we observed a 3.5-fold higher RFP early gene expression of the Mad-4 rr-NCCR compared to that of the at-NCCR, similarly to the expression pattern seen in HEK293 cells (see Fig. S1 in the supplemental material). In the SV40 large T antigen-expressing COS-7, early gene expression was higher for both the archetype and the Mad-4 rr-NCCR reporter (13.3- and 17.3-fold, respectively) (Fig. 5A), but the difference between the archetype and rr-NCCR was reduced (ratio, 1.3). Taken together, the results supported the model that increased early gene expression from JCV rr-NCCR in cis was further increased by the expression of large T antigen in trans, which is key to the higher replication rate of JCV rr-NCCR variants.
FIG. 4.

Replication of recombinant rr-NCCR JCV in COS-7 cells. (A) Time course for prototypes CSF (patient 8), CSF-1 (patient 1), Mad-4, and urine (patient 1). DNase-protected JCV DNA was quantified by qPCR from cell supernatants collected at the indicated times after transfection. The data display the means from two independent experiments determined in triplicate. Error bars indicate the means ± standard deviations. (B) Immunofluorescence of JCV prototypes CSF (patient 8) (a), CSF-1 (patient 1) (b), Mad-4 (c), and urine (patient 1) (d) for VP1 (red) and agnoprotein (green) was performed at 6 dpt using monoclonal mouse anti-VP1 visualized with anti-mouse Cy3 and polyclonal rabbit anti-agno stained by anti-rabbit Cy2, respectively. Cell nuclei were detected by Hoechst 33342 dye (blue).
FIG. 5.
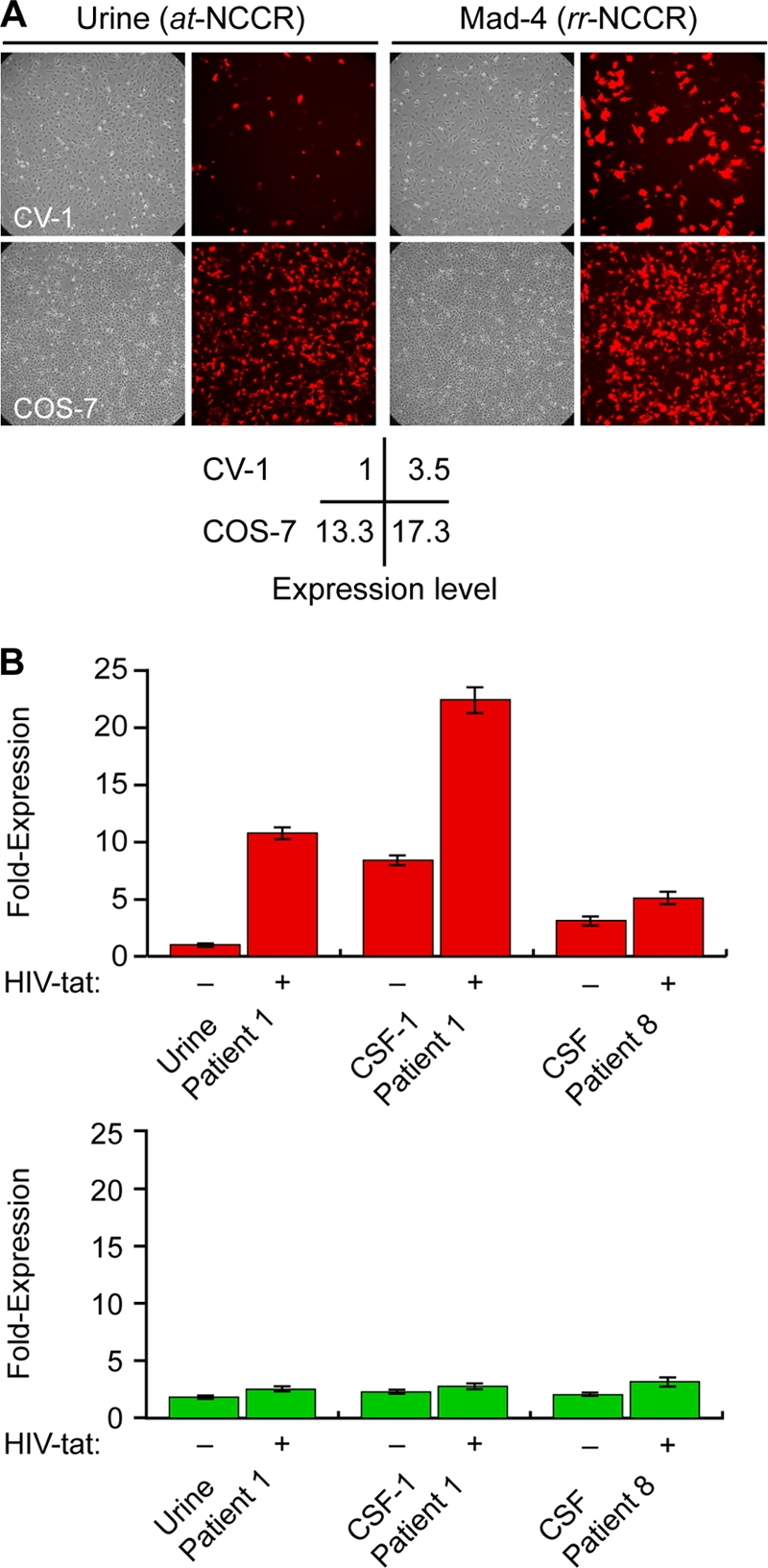
Modulation of NCCR-directed viral early and late gene expression in trans. (A) Effect of SV40 LTag on NCCR-mediated reporter gene expression. RFP-positive cells (red) were analyzed in CV-1 and COS-7 cells at 2 dpt. Early gene expression was normalized for at-NCCR-mediated expression in CV-1 cells. (B) Effect of HIV Tat on NCCR-directed reporter gene expression. At 2 dpt, RFP-positive (early) and GFP-positive (late) cells were analyzed after the transfection of pHRGNCCR reporter constructs into HeLa cells or HeLa SX cells constitutively expressing HIV Tat protein. The number of RFP-positive cells was set to 1 for at-NCCR-mediated reporter gene expression. The data display means from two independent experiments. Error bars indicate the means ± standard deviations.
We next analyzed the effect of HIV Tat as another potentially relevant modulator of JCV replication in HIV-infected patients. The archetype NCCR of patient 1 and the rr-NCCR CSF-1 (patient 1) and CSF (patient 8) pHRG reporter plasmids were transfected into HeLa cells or into HeLa SX-CCR5 cells constitutively expressing HIV Tat (36). As shown in Fig. 5B, at-NCCR-mediated early gene expression was increased by more than 10-fold in HIV Tat-expressing HeLa cells. The baseline activity of the JCV rr-NCCR from CSF of patient 1 and patient 8 in HeLa cells were already 8- and 3-fold higher, respectively, than that of at-NCCR-driven expression, and the increase by HIV Tat expression was less than 2-fold. Of note, the CSF of patient 8 was missing a functional Tat response element due to a block C sequence deletion (see Fig. S1 in the supplemental material). The results indicated that JCV at-NCCR mediated a low basal-level activity that could be increased further by trans-acting regulators, while naturally occurring rr-NCCRs had a constitutively higher activity, which permitted only smaller responses to trans-acting factors.
DISCUSSION
Rearrangements of the JCV NCCR are rare in immunocompetent individuals (20, 57), but they have been identified most consistently in PML and thereby are closely linked to this polyomavirus disease (4, 39). The reported rearrangements of presumed control sequences are highly variable, which has rendered a delineation of their functional role difficult. In this study, we characterized JCV NCCR sequences from the CSF of eight newly diagnosed PML patients and found eight different rearrangements consisting of novel deletions, duplications, and complex combinations thereof. Using a bidirectional reporter gene construct that precisely recapitulated the polyomavirus genome architecture regarding early and late gene organization, we identified increased JCV early gene expression as the functional hallmark of these naturally occurring rr-NCCRs compared to that of the at-NCCR. The generation of recombinant JCV variants confirmed increased early gene expression of rr-NCCR in the context of the JCV genome by immunofluorescence staining for large T antigen. Moreover, JCV rr-NCCR recombinants showed a faster JCV replication rate, with almost 2 log10 geq/ml higher viral loads at 7 dpi than that of the archetype. In COS-7 cells constitutively expressing the homologous SV40 large T antigen, this difference in replication rate between archetype and rearranged variants disappeared, since the at-NCCR JCV now also replicated at similarly high rates. Compared to levels in CV-1 cells, reporter gene expression levels of the archetype NCCR were increased in SV40 large T antigen-expressing COS-7 cells, now reaching levels comparable to those of the rr-NCCR. Taken together, these data indicate that JCV rr-NCCRs in PML patients represent genetic alterations conferring a cis-acting activation of viral early gene expression and, as a result, higher JCV replication rates in vitro. The consistent presence of this functional advantage for different rr-NCCRs suggests that this viral determinant is favored in PML and contributes to disease progression.
We noted that different sequence alterations had a similar potential of upregulating viral early gene expression. Similarly to naturally occurring rr-NCCR of BK virus (BKV) in kidney transplant patients (24), duplications of the JCV NCCR were found more often in the left blocks A to C, close to the origin of genome replication, while deletions were more frequent toward the right end in the vicinity of the late genes (51).
A simple hypothesis is that deletions in the vicinity of the late genes represent a loss of function, e.g., removing a suppressing control sequence, whereas duplications in the vicinity of the early genes represent a gain of function, e.g., increasing activating control sequences that reduce downregulating effects from the late gene area. Daniel et al. reported that deletions and duplications within the at-NCCR are consistent with the loss of negative or the creation of a positive transcriptional control signal and significant effects on the host cell range (11). Several JCV rr-NCCRs identified here were characterized by different deletions of the D block, a feature shared with many JCV rr-NCCRs described in other studies (1, 4, 21, 41). Our experimental deletion of the D block from the JCV at-NCCR was sufficient to confer a strong increase in early gene expression, indicating that important cis-acting negative controls must reside in this sequence. Single-nucleotide differences also were present in the O block in most of the rr-NCCRs (20, 55) but have little, if any, functional impact. Their presence in strains from other areas of the world do not permit the delineation of a specific geographical or racial preponderance (48).
The detailed analysis of the JCV NCCR in same-day urine, plasma, and the CSF of patient 1 (Table 1) and in the follow-up CSF sample, provided some remarkable insights. Regarding the architecture, we noted that the NCCR from the urine JCV was the archetype, while the rr-NCCR from the CSF and plasma samples represented unique but clearly related rearrangements. This triple discordance of JCV NCCR architecture in the same-day samples of a single patient is direct molecular evidence for different virus populations replicating at the same time as majority species in different anatomic sites. One site is the renourinary tract of this PML patient, where the archetype JCV was detected. However, this replication compartment is active in approximately 20% of healthy nonimmunosuppressed persons, but it is without clinical significance (20). The other site is the brain, where the rr-NCCR variant CSF-1 was detected and has diagnostic significance, while the third site giving rise to the plasma rr-NCCR JCV variant is not clear. The latter could come from an unknown replication outside the central nervous system such as leukocytes, bone marrow, or the kidney (14, 39, 52). More likely, given the extensive and multifocal damage of the brain by PML, the plasma rr-NCCR variant comes from another replication site in the central nervous system that is better accessed by, and therefore represented in the blood, as described, e.g., for some PML patients after exposure to natalizumab (52). Indeed, four JCV strains with different NCCRs were isolated from urine, blood, serum, and CSF samples of a PML patient (12), and distinct JCV strains had been reported in different areas of the brain of PML patients (53, 56). Of note, the rapid replacement of CSF-1 and CSF-2 in our patient points to a very dynamic process of this formerly called slow virus disease, suggesting that JCV replication rates significantly drive the PML pathology, similarly to BKV nephropathy (22).
The variability and dynamics of rearrangements affecting only a small area of less than one-tenth of the total JCV genome are intriguing and link the emergence of these variants to specific steps during replication. Polyomaviruses depend on host cell DNA polymerases operating with high-fidelity and proof-reading functions. rr-NCCRs, however, do not carry multiple point mutations but rather deletions and duplications of preexisting archetype sequences of, typically, six or more nucleotides, as reported previously (4, 39), suggesting that other error mechanisms must be operating. Presumably, this could involve imperfect homologous recombination rejoining the viral daughter DNA strands after bidirectional replication from the ori in the NCCR (31, 58).
Given the better replication capacity of JCV with rr-NCCR, why are archetype JCVs not readily replaced but instead persist in the human population? Apparently there are effective barriers to rr-NCCR emergence and persistence. Antiviral immunity seems to be a key barrier for the appearance of PML (15, 17, 18, 35). In immunocompetent individuals, cellular immunity may preferentially eliminate JCV with rr-NCCR that are readily detectable due to constitutive early gene expression, higher replication rate, and impaired ability to hide as latent infections. In contrast, uncontrolled JCV replication in profoundly immunosuppressed patients may permit the emergence of an NCCR quasispecies, from which variants with higher fitness emerge as majority species in the CSF of PML patients.
Do rr-NCCR JCVs emerge from preexisting variants that are suppressed by the immune system or de novo from replication errors of archetype JCV? The current data do not allow the answering of this question. The occasional detection of JCV in blood and bone marrow has supported the notion that rr-NCCR JCV arises outside the central nervous system and travels with mononuclear or precursor cells into the brain, and they generate PML through immunologically uncontrolled replication in oligodendrocytes and possibly other cells (30). Indeed, in HIV/AIDS patients, cellular immune surveillance is systemically compromised. In natalizumab-treated patients, CD34+ precursor cells have been proposed to be activated and mobilized from the bone marrow and might carry the virus to the brain (40). However, natalizumab effectively blocks lymphocyte homing, as evidenced by the clinical improvement of patients with otherwise refractory multiple sclerosis and a vigorous response to JCV (19, 32). Alternatively, JCV may reach the brain during primary infection, since transmission is thought to occur via the oral or respiratory route, followed by subsequent persistence in the renourinary tract, which calls for a viremic phase. This would provide an opportunity for JCV to reach a variety of other tissues, including the kidney and the central nervous system. The predilection of PML in HIV/AIDS patients might then reflect the synergy of the breakdown of immune control, HIV infection of the brain, and local intracerebral activation of archetype JCV replication by HIV Tat (47, 50). Our results demonstrate that HIV Tat is able to increase the viral early gene expression of archetype NCCR JCV to a level comparable to the intrinsic activity of the rearranged NCCR. Further studies are needed to identify details of JCV-activating stimuli in non-HIV patients, which would allow us to build a risk profile beyond mere JCV seropositivity, HIV, and profound immunodeficiency. In summary, our study indicates that JCV rr-NCCR variants arising in HIV patients with PML consistently confer increased early gene expression and higher replication rates compared to those of the archetype JCV and represent a significant viral disease determinant.
Supplementary Material
Acknowledgments
We thank Thomas Klimkait for the HeLa SX-CCR5 cells, Christoph Kasper for help with the automated microscope and data analysis by MetaXpress software, Marion Wernli for excellent technical assistance, and Christine H. Rinaldo for critical comments on the manuscript.
Footnotes
Published ahead of print on 4 August 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Agostini, H. T., C. F. Ryschkewitsch, E. J. Singer, and G. L. Stoner. 1997. JC virus regulatory region rearrangements and genotypes in progressive multifocal leukoencephalopathy: two independent aspects of virus variation. J. Gen. Virol. 78:659-664. [DOI] [PubMed] [Google Scholar]
- 2.Albrecht, H., C. Hoffmann, O. Degen, A. Stoehr, A. Plettenberg, T. Mertenskotter, C. Eggers, and H. J. Stellbrink. 1998. Highly active antiretroviral therapy significantly improves the prognosis of patients with HIV-associated progressive multifocal leukoencephalopathy. AIDS 12:1149-1154. [DOI] [PubMed] [Google Scholar]
- 3.Astrom, K. E., E. L. Mancall, and E. P. Richardson, Jr. 1958. Progressive multifocal leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin's disease. Brain 81:93-111. [DOI] [PubMed] [Google Scholar]
- 4.Ault, G. S., and G. L. Stoner. 1993. Human polyomavirus JC promoter/enhancer rearrangement patterns from progressive multifocal leukoencephalopathy brain are unique derivatives of a single archetypal structure. J. Gen. Virol. 74:1499-1507. [DOI] [PubMed] [Google Scholar]
- 5.Berger, J. R., B. Kaszovitz, M. J. Post, and G. Dickinson. 1987. Progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. A review of the literature with a report of sixteen cases. Ann. Intern. Med. 107:78-87. [DOI] [PubMed] [Google Scholar]
- 6.Boldorini, R., E. Omodeo-Zorini, M. Nebuloni, E. Benigni, L. Vago, A. Ferri, and G. Monga. 2003. Lytic JC virus infection in the kidneys of AIDS subjects. Mod. Pathol. 16:35-42. [DOI] [PubMed] [Google Scholar]
- 7.Bossolasco, S., G. Calori, F. Moretti, A. Boschini, D. Bertelli, M. Mena, S. Gerevini, A. Bestetti, R. Pedale, S. Sala, S. Sala, A. Lazzarin, and P. Cinque. 2005. Prognostic significance of JC virus DNA levels in cerebrospinal fluid of patients with HIV-associated progressive multifocal leukoencephalopathy. Clin. Infect. Dis. 40:738-744. [DOI] [PubMed] [Google Scholar]
- 8.Cinque, P., I. J. Koralnik, and D. B. Clifford. 2003. The evolving face of human immunodeficiency virus-related progressive multifocal leukoencephalopathy: defining a consensus terminology. J. Neurovirol 9(Suppl. 1):88-92. [DOI] [PubMed] [Google Scholar]
- 9.Cinque, P., C. Pierotti, M. G. Vigano, A. Bestetti, C. Fausti, D. Bertelli, and A. Lazzarin. 2001. The good and evil of HAART in HIV-related progressive multifocal leukoencephalopathy. J. Neurovirol. 7:358-363. [DOI] [PubMed] [Google Scholar]
- 10.Corpet, F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16:10881-10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daniel, A. M., J. J. Swenson, R. P. Mayreddy, K. Khalili, and R. J. Frisque. 1996. Sequences within the early and late promoters of archetype JC virus restrict viral DNA replication and infectivity. Virology 216:90-101. [DOI] [PubMed] [Google Scholar]
- 12.Delbue, S., G. Sotgiu, D. Fumagalli, M. Valli, E. Borghi, R. Mancuso, E. Marchioni, R. Maserati, and P. Ferrante. 2005. A case of a progressive multifocal leukoencephalopathy patient with four different JC virus transcriptional control region rearrangements in cerebrospinal fluid, blood, serum, and urine. J. Neurovirol. 11:51-57. [DOI] [PubMed] [Google Scholar]
- 13.De Luca, A., A. Ammassari, P. Pezzotti, P. Cinque, J. Gasnault, J. Berenguer, S. Di Giambenedetto, A. Cingolani, Y. Taoufik, P. Miralles, C. M. Marra, and A. Antinori. 2008. Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis. AIDS 22:1759-1767. [DOI] [PubMed] [Google Scholar]
- 14.Drachenberg, C. B., H. H. Hirsch, J. C. Papadimitriou, R. Gosert, R. K. Wali, R. Munivenkatappa, J. Nogueira, C. B. Cangro, A. Haririan, S. Mendley, and E. Ramos. 2007. Polyomavirus BK versus JC replication and nephropathy in renal transplant recipients: a prospective evaluation. Transplantation 84:323-330. [DOI] [PubMed] [Google Scholar]
- 15.Du Pasquier, R. A., P. Autissier, Y. Zheng, J. Jean-Jacques, and I. J. Koralnik. 2005. Presence of JC virus-specific CTL in the cerebrospinal fluid of PML patients: rationale for immune-based therapeutic strategies. AIDS 19:2069-2076. [DOI] [PubMed] [Google Scholar]
- 16.Du Pasquier, R. A., and I. J. Koralnik. 2003. Inflammatory reaction in progressive multifocal leukoencephalopathy: harmful or beneficial? J. Neurovirol. 9(Suppl. 1):25-31. [DOI] [PubMed] [Google Scholar]
- 17.Du Pasquier, R. A., M. J. Kuroda, Y. Zheng, J. Jean-Jacques, N. L. Letvin, and I. J. Koralnik. 2004. A prospective study demonstrates an association between JC virus-specific cytotoxic T lymphocytes and the early control of progressive multifocal leukoencephalopathy. Brain 127:1970-1978. [DOI] [PubMed] [Google Scholar]
- 18.Du Pasquier, R. A., J. E. Schmitz, J. Jean-Jacques, Y. Zheng, J. Gordon, K. Khalili, N. L. Letvin, and I. J. Koralnik. 2004. Detection of JC virus-specific cytotoxic T lymphocytes in healthy individuals. J. Virol. 78:10206-10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du Pasquier, R. A., M. C. Stein, M. A. Lima, X. Dang, J. Jean-Jacques, Y. Zheng, N. L. Letvin, and I. J. Koralnik. 2006. JC virus induces a vigorous CD8+ cytotoxic T cell response in multiple sclerosis patients. J. Neuroimmunol. 176:181-186. [DOI] [PubMed] [Google Scholar]
- 20.Egli, A., L. Infanti, A. Dumoulin, A. Buser, J. Samaridis, C. Stebler, R. Gosert, and H. H. Hirsch. 2009. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 199:837-846. [DOI] [PubMed] [Google Scholar]
- 21.Frisque, R. J., G. L. Bream, and M. T. Cannella. 1984. Human polyomavirus JC virus genome. J. Virol. 51:458-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Funk, G. A., R. Gosert, P. Comoli, F. Ginevri, and H. H. Hirsch. 2008. Polyomavirus BK replication dynamics in vivo and in silico to predict cytopathology and viral clearance in kidney transplants. Am. J. Transplant. 8:2368-2377. [DOI] [PubMed] [Google Scholar]
- 23.Giudici, B., B. Vaz, S. Bossolasco, S. Casari, A. M. Brambilla, W. Luke, A. Lazzarin, T. Weber, and P. Cinque. 2000. Highly active antiretroviral therapy and progressive multifocal leukoencephalopathy: effects on cerebrospinal fluid markers of JC virus replication and immune response. Clin. Infect. Dis. 30:95-99. [DOI] [PubMed] [Google Scholar]
- 24.Gosert, R., C. H. Rinaldo, G. A. Funk, A. Egli, E. Ramos, C. B. Drachenberg, and H. H. Hirsch. 2008. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J. Exp. Med. 205:841-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirsch, H. H., G. Kaufmann, P. Sendi, and M. Battegay. 2004. Immune reconstitution in HIV-infected patients. Clin. Infect. Dis. 38:1159-1166. [DOI] [PubMed] [Google Scholar]
- 26.Hirsch, H. H., P. R. Meylan, W. Zimmerli, A. Iten, M. Battegay, and P. Erb. 1998. HIV-1-infected patients with focal neurologic signs: diagnostic role of PCR for Toxoplasma gondii, Epstein-Barr virus, and JC virus. Clin. Microbiol. Infect. 4:577-584. [DOI] [PubMed] [Google Scholar]
- 27.Hirsch, H. H., and J. Steiger. 2003. Polyomavirus BK. Lancet Infect. Dis. 3:611-623. [DOI] [PubMed] [Google Scholar]
- 28.Horton, R. M., H. D. Hunt, S. N. Ho, J. K. Pullen, and L. R. Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61-68. [DOI] [PubMed] [Google Scholar]
- 29.Hou, J., and E. Major. 2005. Management of infections by the human polyomavirus JC: past, present and future. Expert Rev. Anti-Infect. Ther. 3:629-640. [DOI] [PubMed] [Google Scholar]
- 30.Houff, S. A., E. O. Major, D. A. Katz, C. V. Kufta, J. L. Sever, S. Pittaluga, J. R. Roberts, J. Gitt, N. Saini, and W. Lux. 1988. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N. Engl. J. Med. 318:301-305. [DOI] [PubMed] [Google Scholar]
- 31.Imperiale, M. J., and E. O. Major. 2007. Polyomaviruses, p. 2263-2298. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 32.Jilek, S., E. Jaquiery, H. H. Hirsch, A. Lysandropoulos, M. Canales, L. Guignard, M. Schluep, G. Pantaleo, and R. A. Du Pasquier. 2010. Immune responses to JC virus in patients with multiple sclerosis treated with natalizumab: a cross-sectional and longitudinal study. Lancet Neurol. 9:264-272. [DOI] [PubMed] [Google Scholar]
- 33.Kazory, A., D. Ducloux, J. M. Chalopin, R. Angonin, B. Fontaniere, and H. Moret. 2003. The first case of JC virus allograft nephropathy. Transplantation 76:1653-1655. [DOI] [PubMed] [Google Scholar]
- 34.Khanna, N., L. Elzi, N. J. Mueller, C. Garzoni, M. Cavassini, C. A. Fux, P. Vernazza, E. Bernasconi, M. Battegay, H. H. Hirsch, et al. 2009. Incidence and outcome of progressive multifocal leukoencephalopathy in 20 years of the Swiss HIV cohort study. Clin. Infect. Dis. 48:1459-1466. [DOI] [PubMed] [Google Scholar]
- 35.Khanna, N., M. Wolbers, N. J. Mueller, C. Garzoni, R. A. Du Pasquier, C. A. Fux, P. Vernazza, E. Bernasconi, R. Viscidi, M. Battegay, and H. H. Hirsch. 2009. JC virus-specific immune responses in human immunodeficiency virus type 1 patients with progressive multifocal leukoencephalopathy. J. Virol. 83:4404-4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klimkait, T., F. Stauffer, E. Lupo, and C. Sonderegger-Rubli. 1998. Dissecting the mode of action of various HIV-inhibitor classes in a stable cellular system. Arch. Virol. 143:2109-2131. [DOI] [PubMed] [Google Scholar]
- 37.Koralnik, I. J., R. A. Du Pasquier, and N. L. Letvin. 2001. JC virus-specific cytotoxic T lymphocytes in individuals with progressive multifocal leukoencephalopathy. J. Virol. 75:3483-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindå, H., A. von Heijne, E. O. Major, C. Ryschkewitsch, J. Berg, T. Olsson, and C. Martin. 2009. Progressive multifocal leukoencephalopathy after natalizumab monotherapy. N. Engl. J. Med. 361:1081-1087. [DOI] [PubMed] [Google Scholar]
- 39.Loeber, G., and K. Dorries. 1988. DNA rearrangements in organ-specific variants of polyomavirus JC strain GS. J. Virol. 62:1730-1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Major, E. O. 2009. Reemergence of PML in natalizumab-treated patients-new cases, same concerns. N. Engl. J. Med. 361:1041-1043. [DOI] [PubMed] [Google Scholar]
- 41.Martin, J. D., D. M. King, J. M. Slauch, and R. J. Frisque. 1985. Differences in regulatory sequences of naturally occurring JC virus variants. J. Virol. 53:306-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Messam, C. A., J. Hou, R. M. Gronostajski, and E. O. Major. 2003. Lineage pathway of human brain progenitor cells identified by JC virus susceptibility. Ann. Neurol. 53:636-646. [DOI] [PubMed] [Google Scholar]
- 43.Newman, J. T., and R. J. Frisque. 1997. Detection of archetype and rearranged variants of JC virus in multiple tissues from a pediatric PML patient. J. Med. Virol. 52:243-252. [DOI] [PubMed] [Google Scholar]
- 44.Padgett, B. L., D. L. Walker, G. M. ZuRhein, R. J. Eckroade, and B. H. Dessel. 1971. Cultivation of papova-like virus from human brain with progressive multifocal leukoencephalopathy. Lancet i:1257-1260. [DOI] [PubMed] [Google Scholar]
- 45.Rinaldo, C. H., T. Traavik, and A. Hey. 1998. The agnogene of the human polyomavirus BK is expressed. J. Virol. 72:6233-6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Safdar, A., R. J. Rubocki, J. A. Horvath, K. K. Narayan, and R. L. Waldron. 2002. Fatal immune restoration disease in human immunodeficiency virus type 1-infected patients with progressive multifocal leukoencephalopathy: impact of antiretroviral therapy-associated immune reconstitution. Clin. Infect. Dis. 35:1250-1257. [DOI] [PubMed] [Google Scholar]
- 47.Stettner, M. R., J. A. Nance, C. A. Wright, Y. Kinoshita, W. K. Kim, S. Morgello, J. Rappaport, K. Khalili, J. Gordon, and E. M. Johnson. 2009. SMAD proteins of oligodendroglial cells regulate transcription of JC virus early and late genes coordinately with the Tat protein of human immunodeficiency virus type 1. J. Gen. Virol. 90:2005-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugimoto, C., M. Hasegawa, A. Kato, H. Y. Zheng, H. Ebihara, F. Taguchi, T. Kitamura, and Y. Yogo. 2002. Evolution of human polyomavirus JC: implications for the population history of humans. J. Mol. Evol. 54:285-297. [DOI] [PubMed] [Google Scholar]
- 49.Sugimoto, C., D. Ito, K. Tanaka, H. Matsuda, H. Saito, H. Sakai, K. Fujihara, Y. Itoyama, T. Yamada, J. Kira, R. Matsumoto, M. Mori, K. Nagashima, and Y. Yogo. 1998. Amplification of JC virus regulatory DNA sequences from cerebrospinal fluid: diagnostic value for progressive multifocal leukoencephalopathy. Arch. Virol. 143:249-262. [DOI] [PubMed] [Google Scholar]
- 50.Tada, H., J. Rappaport, M. Lashgari, S. Amini, F. Wong-Staal, and K. Khalili. 1990. Trans-activation of the JC virus late promoter by the tat protein of type 1 human immunodeficiency virus in glial cells. Proc. Natl. Acad. Sci. U. S. A. 87:3479-3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vacante, D. A., R. Traub, and E. O. Major. 1989. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology 170:353-361. [DOI] [PubMed] [Google Scholar]
- 52.Van Assche, G., M. Van Ranst, R. Sciot, B. Dubois, S. Vermeire, M. Noman, J. Verbeeck, K. Geboes, W. Robberecht, and P. Rutgeerts. 2005. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N. Engl. J. Med. 353:362-368. [DOI] [PubMed] [Google Scholar]
- 53.Yasuda, Y., H. Yabe, H. Inoue, T. Shimizu, M. Yabe, Y. Yogo, and S. Kato. 2003. Comparison of PCR-amplified JC virus control region sequences from multiple brain regions in PML. Neurology 61:1617-1619. [DOI] [PubMed] [Google Scholar]
- 54.Yogo, Y., J. Guo, T. Iida, K. Satoh, F. Taguchi, H. Takahashi, W. W. Hall, and K. Nagashima. 1994. Occurrence of multiple JC virus variants with distinctive regulatory sequences in the brain of a single patient with progressive multifocal leukoencephalopathy. Virus Genes 8:99-105. [DOI] [PubMed] [Google Scholar]
- 55.Yogo, Y., T. Kitamura, C. Sugimoto, T. Ueki, Y. Aso, K. Hara, and F. Taguchi. 1990. Isolation of a possible archetypal JC virus DNA sequence from nonimmunocompromised individuals. J. Virol. 64:3139-3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yogo, Y., T. Matsushima-Ohno, T. Hayashi, C. Sugimoto, M. Sakurai, and I. Kanazawa. 2001. JC virus regulatory region rearrangements in the brain of a long surviving patient with progressive multifocal leukoencephalopathy. J. Neurol. Neurosurg. Psychiatry 71:397-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yogo, Y., S. Zhong, A. Shibuya, T. Kitamura, and Y. Homma. 2008. Transcriptional control region rearrangements associated with the evolution of JC polyomavirus. Virology 380:118-123. [DOI] [PubMed] [Google Scholar]
- 58.Yoshiike, K., and K. K. Takemoto. 1986. Studies with BK virus and monkey lymphotropic papovavirus, p. 295-326. In N. P. Salzman (ed.), The Polyomaviridae, vol. 1. Plenum Press, New York, NY. [Google Scholar]
- 59.Zheng, H. Y., Y. Yasuda, S. Kato, T. Kitamura, and Y. Yogo. 2004. Stability of JC virus coding sequences in a case of progressive multifocal leukoencephalopathy in which the viral control region was rearranged markedly. Arch. Pathol. Lab. Med. 128:275-278. [DOI] [PubMed] [Google Scholar]
- 60.Zurhein, G., and S. M. Chou. 1965. Particles resembling papova viruses in human cerebral demyelinating disease. Science 148:1477-1479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.