

Abstract
The mechanisms underlying HIV-1 control by protective HLA class I alleles are not fully understood and could involve selection of escape mutations in functionally important Gag epitopes resulting in fitness costs. This study was undertaken to investigate, at the population level, the impact of HLA-mediated immune pressure in Gag on viral fitness and its influence on HIV-1 pathogenesis. Replication capacities of 406 recombinant viruses encoding plasma-derived Gag-protease from patients chronically infected with HIV-1 subtype C were assayed in an HIV-1-inducible green fluorescent protein reporter cell line. Viral replication capacities varied significantly with respect to the specific HLA-B alleles expressed by the patient, and protective HLA-B alleles, most notably HLA-B*81, were associated with lower replication capacities. HLA-associated mutations at low-entropy sites, especially the HLA-B*81-associated 186S mutation in the TL9 epitope, were associated with lower replication capacities. Most mutations linked to alterations in replication capacity in the conserved p24 region decreased replication capacity, while most in the highly variable p17 region increased replication capacity. Replication capacity also correlated positively with baseline viral load and negatively with baseline CD4 count but did not correlate with the subsequent rate of CD4 decline. In conclusion, there is evidence that protective HLA alleles, in particular HLA-B*81, significantly influence Gag-protease function by driving sequence changes in Gag and that conserved regions of Gag should be included in a vaccine aiming to drive HIV-1 toward a less fit state. However, the long-term clinical benefit of immune-driven fitness costs is uncertain given the lack of correlation with longitudinal markers of disease progression.
There is broad heterogeneity in the ability of HIV-infected individuals to control virus replication, ranging from elite controllers, who maintain undetectable viral loads without treatment, to rapid progressors, who progress to AIDS within 2 years of infection (9, 22, 32). Many interrelated factors, including host and viral genetic factors involved in antiviral immunity and the viral life cycle, may partially account for the differences in the course of disease progression (10, 11, 30, 41). The complex interplay between host genetic factors and viral factors is exemplified by human leukocyte antigen (HLA) class I-restricted cytotoxic T-lymphocyte (CTL) responses, which exert considerable immune pressure on the virus, resulting in escape mutations that affect the interaction of viral and host proteins, thereby influencing infection outcome.
The exact mechanisms by which some HLA class I alleles, such as HLA-B*57 and HLA-B*27, are associated with slower progression to AIDS, while others, such as B*5802 and B*18, are associated with accelerated disease progression (6, 20, 42), are unclear. The magnitude and/or breadth of HLA-restricted CTL responses to the conserved Gag protein has been correlated inversely with disease progression or markers of disease progression in several studies (12, 21, 28, 31, 35, 43, 46), although there are some exceptions (4, 16, 37), while preferential targeting of the highly variable envelope protein (as occurs in HLA-B*5802-positive individuals) correlates with higher viral loads (21, 29). Protective HLA alleles restrict CTL responses that impose a strong selection pressure on a few specific Gag p24 epitopes, resulting in escape mutations (14) for which fitness costs have been demonstrated either through site-directed mutations introduced into a reference strain background (2, 8, 25, 38) or through in vivo reversion of these mutations after transmission to an HLA-mismatched individual (8, 24). Recent evidence suggests that Gag escape mutations with a fitness cost, particularly those in p24, are a significant determinant of disease progression: the transmitted number of HLA-B-associated polymorphisms in Gag was found to significantly impact the viral set point in recipients (although an associated fitness cost was not shown) (7, 15), and in a small number of infants, decreased fitness of the transmitted virus with HLA-B*5703/5801-selected mutations in Gag p24 epitopes resulted in slower disease progression (33, 39). Also, the number of reverting Gag mutations (thought to revert as a consequence of fitness costs) associated with individual HLA-B alleles was strongly correlated with the HLA-linked viral set point in chronically infected patients (26). A recent in vitro study showed that HLA-associated variation in Gag-protease, with resulting reduced replication capacity, may contribute to viral control in HIV-1 subtype B-infected elite controllers (27). Taken together, these studies suggest that CTL responses restricted by favorable HLA alleles select for escape mutations in conserved epitopes, particularly those in Gag, resulting in a fitness cost to HIV and therefore at least partly explaining the slower disease progression in individuals carrying these alleles.
To date, many of the studies investigating the fitness cost of Gag escape mutations and their clinical relevance have concentrated on escape mutations associated with protective HLA alleles, have not assessed fitness consequences in the natural sequence background (in the presence of other escape and compensatory mutations), and/or have focused on a limited number of patients. Most importantly, the majority of studies have focused on HIV-1 subtype B. The present study is the first to use a large population-based approach and clinically derived Gag-protease sequences to investigate comprehensively the relationships between immune-driven sequence variation in Gag, viral replication capacity, and markers of disease progression in chronic infection with HIV-1 subtype C, the most predominant subtype in the epidemic. We assayed the replication capacity of recombinant viruses encoding patient Gag-protease in an HIV-1-inducible green fluorescent protein (GFP) reporter cell line and found associations between lower replication capacities, protective HLA alleles, protective HLA-associated mutations, lower baseline viral loads, and higher baseline CD4 counts. However, Gag-protease replication capacity did not correlate with the subsequent rate of CD4 decline.
MATERIALS AND METHODS
Study subjects.
The study subjects included 406 antiretroviral-naïve individuals chronically infected with HIV-1 subtype C from the Sinikithemba cohort in Durban, South Africa. These individuals were HLA typed to 4-digit resolution by molecular methods (20). Viral load (Roche Amplicor assay, version 1.5) and CD4 count (Trucount technology) measurements were obtained at study entry (baseline) for all participants and at 3-month intervals thereafter for 339 of the participants (20). At baseline, the median viral load of the cohort was 4.77 log10 HIV RNA copies/ml (interquartile range [IQR], 4.15 to 5.27 log10 HIV RNA copies/ml), and the median CD4 count was 340 cells/mm3 (IQR, 238 to 477 cells/mm3). Over the subsequent course of study follow-up (the mean follow-up time was 2.28 years per individual; IQR, 1.21 to 3.02 years), the median rate of CD4 decline was −30 cells/mm3 per year (IQR, −73 to −3 cells/mm3 per year). The median age of the study subjects at baseline was 31 years (IQR, 27 to 36 years), and 322 (79%) patients were female. Among the study participants, there was no significant association between age, gender, and baseline viral load or CD4 count as was reported previously (42), and therefore we did not control for these variables in analyses. Written informed consent was obtained from all study subjects, and the study protocol was approved by the Biomedical Research Ethics Committee of the University of KwaZulu-Natal.
Generation of Gag-protease NL4-3 recombinant virus stocks.
Patient Gag-protease was isolated and inserted into an NL4-3 backbone to generate recombinant viruses. Protease was included to maintain the important interaction between Gag and protease, namely, cleavage of the Gag polyprotein by protease. Viral RNA was isolated from plasma by use of a QIAamp Viral RNA Mini kit from Qiagen (Valencia). Reverse transcription-PCR (RT-PCR) was performed as previously described (27), using a Superscript III One-Step RT-PCR kit (Invitrogen, Carlsbad, CA) and the following Gag-protease-specific primers: 5′ CAC TGC TTA AGC CTC AAT AAA GCT TGC C 3′ (HXB2 nucleotides 512 to 539) and 5′ TTT AAC CCT GCT GGG TGT GGT ATY CCT 3′ (nucleotides 2851 to 2825). A second round of PCR was performed with 100-mer forward and reverse primers that were exactly complementary to NL4-3 on either side of Gag-protease, using a TaKaRa Ex Taq HS enzyme kit (Takara, Shiga, Japan). Two 50-μl PCR mixtures were prepared for each sample, comprising 37 μl diethyl pyrocarbonate (DEPC)-treated water, 5 μl 10× Ex Taq buffer, 4 μl of deoxynucleoside triphosphates (dNTPs), 0.8 μl forward primer (10 μM), 0.8 μl reverse primer (10 μM), 0.25 μl Ex Taq, and 2 μl RT-PCR product. Thermocycler conditions were as follows: 94°C for 2 min; 40 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 2 min; and 72°C for 7 min. PCR products from two 50-μl reaction mixtures were pooled, and 10 μl was set aside for sequencing. The remainder was used in the generation of recombinant viruses. Gag-protease-deleted pNL4-3 plasmid was prepared as previously described (27), and large stocks of the plasmid were generated using a Plasmid Maxi kit (Qiagen, Valencia, CA). The plasmid was digested for 2 h at 60°C immediately prior to cotransfection of 2 × 106 Tat-inducible GFP reporter GXR T cells (3) in R10 medium (800 μl RPMI-1640 [Sigma, St. Louis, MO] supplemented with 10% fetal bovine serum [Gibco, NY], 2 mM l-glutamine [Sigma], 10 mM HEPES [Gibco], and 50 U/ml penicillin-streptomycin [Gibco]) with 10 μg digested plasmid and ≈85 μl Gag-protease PCR product via electroporation at 300 V and 500 μF (27). Following a 1-h incubation at room temperature, GXR cells were transferred to T25 flasks containing 4 ml of medium each. Five days later, 5 ml R10 medium was added to each flask. The percentage of infected cells was monitored from day 12 onwards by flow cytometry on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Culture supernatants were harvested when approximately 30% of the GXR cells were infected and were stored in 1-ml aliquots at −80°C for use in subsequent titration and replication assays.
Titration and replication assays.
Titration of virus stocks and replication assays were performed as previously described (2, 27, 38), using a multiplicity of infection (MOI) of 0.003. The mean slope of exponential growth from days 3 to 6 was calculated using the semilog method in Excel. This was divided by the slope of growth of the wild-type NL4-3 control included in each assay to generate a normalized measure of replication capacity. Replication assays were performed at least in duplicate, and results were averaged.
Sequencing of Gag-protease gene.
The Gag-protease PCR product was diluted 1:15 in DEPC-treated water and population sequenced using Big Dye Terminator ready reaction mix V3 (Applied Biosystems, Foster City, CA) and the following sequencing primers: 5′ CTT GTC TAG GGC TTC CTT GGT 3′ (nucleotides 1098 to 1078), 5′ CTT CAG ACA GGA ACA GAG GA 3′ (nucleotides 991 to 1010), 5′ GGT TCT CTC ATC TGG CCT GG 3′ (nucleotides 1481 to 1462), 5′ CAA CAA GGT TTC TGT CAT CC 3′ (nucleotides 1755 to 1736), 5′ CCT TGC CAC AGT TGA AAC ATT T 3′ (nucleotides 1981 to 1960), 5′ TAG AAG AAA TGA TGA CAG 3′ (nucleotides 1817 to 1834), 5′ CAG CCA AGC TGA GTC AA 3′ (nucleotides 2536 to 2520), and 5′ GGA GCA GAT GAT ACA GTA TT 3′ (nucleotides 2331 to 2350). Sequences were analyzed on an ABI 3130xl genetic analyzer (Applied Biosystems) and were visualized and edited in Sequencher 4.8. Sequence data were aligned with the sequence of HIV-1 subtype B reference strain HXB2 (GenBank accession no. K03455), using a modified NAP algorithm (18), and insertions with respect to the HXB2 sequence were stripped from the sequences. A neighbor-joining tree was constructed from nucleotide sequences by using Paup 4.0 and was edited in Figtree (http://tree.bio.ed.ac.uk/software/figtree/). Nucleotide differences between plasma and recombinant virus Gag-protease sequences were quantified using Highlighter (http://www.hiv.lanl.gov/content/sequence/HIGHLIGHT/highlighter.html). BioEdit 7.0 (http://www.mbio.ncsu.edu/BioEdit/bioedit.html) was used to calculate the similarity of each sequence to the consensus subtype C Gag sequence from 2004.
Identification of HLA-associated polymorphisms.
Statistical methods (described in detail in reference 5) that correct for phylogenetic relatedness between HIV sequences, amino acid covariation, and HLA linkage disequilibrium effects were used to identify HLA-associated polymorphisms. Briefly, a maximum likelihood phylogenetic tree was constructed for each gene, and a model of conditional adaptation was inferred for each observed amino acid at each codon. In this model, the amino acid is assumed to evolve independently down the phylogeny, until it reaches the observed sequences at the tree tips. In each host, the selection pressure arising from HLA-restricted CTL and covariation between HIV codons is modeled directly by a stochastic additive process. To identify which factors contribute to the observed sequences, a forward selection procedure is employed, in which the most significant association is iteratively added to the model, with P values computed using the likelihood ratio test. Each observed amino acid variant at each codon is evaluated in a binary fashion (presence versus absence thereof). Multiple tests are addressed using q values, the P value analogue of the false discovery rate (FDR), for each P value threshold (40). The FDR is the expected proportion of false-positive results among results deemed significant at a given threshold. For example, at a q value of ≤0.2, we would expect a false-positive proportion of 20% among identified associations.
Data analysis.
Viral replication capacities were grouped according to the HLA class I alleles expressed by the host. Analysis of variance (ANOVA) was used to assess whether significant differences in replication capacities were observed within expressed HLA-A, -B, and -C alleles. Then, for each individual allele (n ≥ 5), Student's t test (or the Mann-Whitney U test in cases where the assumptions of Student's t test were not met) was used to compare replication capacities of viruses generated from persons expressing versus not expressing the allele in question. The relationships between replication capacity and log viral load, CD4 count, rate of CD4 decline, number of HLA-associated polymorphisms, and sequence similarity to the HIV-1 subtype C Gag consensus were assessed using Pearson's correlation (for normally distributed variables) or Spearman's rank correlation (for non-normally distributed variables). Viruses were also categorized according to the 10th and 90th percentiles of the replication capacity data, into low- and high-replication-capacity groups, respectively. Clinical and sequence parameters were compared between these groups, using Student's t test (or the Mann-Whitney U test) or Fisher's exact test (in the case of proportion comparisons). The association between single amino acid residues in Gag-protease and the replication capacity was analyzed by Mann-Whitney U tests (univariate method) and linear regression with a forward selection process (multivariate method). q values were calculated in both cases to account for multiple comparisons (40). The significance cutoff for all analyses, unless indicated otherwise, was a P value of <0.05.
Nucleotide sequence accession numbers.
Gag-protease sequences obtained in this study are available in the GenBank database under accession numbers HM593106 to HM593510.
RESULTS
Validation of generated Gag-protease NL4-3 recombinant viruses.
Gag-protease NL4-3 recombinant virus stocks from 406 subjects were generated in a median of 27 days (IQR, 23 to 32 days) following cotransfection of HIV-inducible, GFP-expressing T cells with Gag-protease-deleted NL4-3 plasmid and clinically derived Gag-protease amplicons. To test whether recombinant viruses were representative of the original plasma quasispecies, Gag-protease was resequenced from 40 randomly selected recombinant viruses and compared with the original plasma HIV RNA sequences. The median number of total nucleotide differences between the recombinant virus sequence and the original plasma HIV RNA sequence (when mixtures were not included as differences) was 0 (IQR, 0 to 1.5), resulting in an average nucleotide similarity of 0.99% between pairs. The average number of nucleotide mixtures in recombinant virus sequences was 21 (standard deviation [SD] = 17), indicating somewhat reduced diversity (Student's t test; P = 0.0002) compared to that of the original plasma sequences (mean = 35; SD = 36). Recombinant virus Gag sequences closely clustered with respective plasma Gag sequences in a phylogenetic tree (see Fig. SA1 in the supplemental material). These data indicate that the Gag-protease recombinant viruses were representative of the original plasma quasispecies. All further analyses were based on the original plasma HIV sequences.
Assay variability.
The replication capacities of Gag-protease NL4-3 recombinant viruses were assayed in duplicate, independently, in an HIV-1-inducible GFP reporter cell line. Replication capacity was defined as the slope of the increase in the percentage of infected cells from days 3 to 6 following infection, normalized to wild-type NL4-3. Duplicate measurements were highly concordant (Pearson's correlation; r = 0.88 and P < 0.0001). Accuracy of recombinant viral titers was achieved: on day 3 of the assay, the mean % GFP-expressing cells was 0.65% (SD = 0.28%). Importantly, the observed variability in day 3 readings did not influence viral replication capacity measurements (Pearson's correlation; r = 0.04 and P = 0.44).
Distribution of replication capacities of recombinant viruses.
The NL4-3-normalized replication capacities of the recombinant viruses generated from the 406 cohort participants approximated a normal distribution (mean = 0.62; SD = 0.1) (Fig. 1). The replication capacities of the recombinant HIV-1 subtype C Gag-protease sequences inserted into the NL4-3 backbone were considerably lower than those of the wild-type NL4-3 control and 25 subtype B Gag-protease NL4-3 recombinant viruses, whose mean replication capacity normalized to wild-type NL4-3 was 0.95 (SD = 0.13) (data not shown).
FIG. 1.

Distribution of Gag-protease NL4-3 recombinant virus replication capacities, normalized to the growth of wild-type NL4-3.
Association of recombinant virus replication capacities with specific HLA types.
Replication capacities of recombinant viruses were grouped according to HLA alleles expressed by the host (Fig. 2). Overall, replication capacities varied significantly between the different HLA-B alleles (ANOVA; P = 0.01) but not between HLA-A or HLA-C alleles, suggesting that HLA-B alleles have the greatest impact on Gag-protease-mediated replication capacity. Relationships between specific HLA alleles and replication capacity were also observed, the strongest of which was the association of HLA-B*81 with lower replication capacities (Student's t test; P < 0.0001). P values presented are uncorrected for multiple comparisons. Only the association of HLA-B*81 with lower replication capacities would remain statistically significant following Bonferroni adjustment for multiple comparisons.
FIG. 2.

Associations between HLA alleles and replication capacities of Gag-protease NL4-3 recombinant viruses. (A to C) Graphs show the mean (dot), median (vertical line), interquartile range (edges of boxes), and most extreme values (edges of whiskers) of replication capacities for each different HLA allele for which n was ≥5. *, individual significant (P < 0.05) associations (Student's t test); **, associations that survive Bonferroni correction for multiple comparisons. Overall P values (ANOVA) indicate the significance of differences in replication capacity between alleles of each group. (D) Graph showing a significantly greater proportion (Fisher's exact test) of individuals with protective HLA alleles (HLA-B*57, HLA-B*5801, and HLA-B*81) in the group with the least-fit viruses (replication capacity of ≤0.5; n = 41) than in the group with the fittest viruses (replication capacity of ≥0.74; n = 41).
Besides HLA-B*81, other alleles that were associated with low-replication-capacity recombinant viruses were HLA-B*5801, HLA-A*0205 (Mann-Whitney U test; P = 0.05 and P = 0.04, respectively), HLA-A*3009, and HLA-A*3001 (Student's t test; P = 0.02 and P = 0.05, respectively). Due to tight linkage between HLA-B*5801 and HLA-A*0205 (D′ = 0.56 [23]), the allele driving the effect could not be identified. Among 5 individuals with HLA-B*3009, 4 possessed HLA-B*81, which likely explains the association of HLA-B*3009 with lower replication capacities.
Alleles associated with higher replication capacities were HLA-Cw*0702 (Student's t test; P = 0.05) and HLA-Cw*0501 (Mann-Whitney U test; P = 0.001). Only 6 individuals in this study possessed HLA-Cw*0501, and all were linked to replication capacities above the 80th percentile of the data set (>0.7). HLA-Cw*0501 is in linkage disequilibrium with HLA-B*1801—5 of 6 individuals with HLA-Cw*0501 also carried HLA-B*1801—and could therefore partly contribute to the disadvantage associated with HLA-B*1801 in subtype C infection (20).
In an additional analysis, the HLA types of the individuals corresponding to the fittest recombinant viruses (≥90th percentile of the data set, i.e., ≥0.74; n = 41) were compared to the HLA types of individuals with the least-fit recombinant viruses (10th percentile of the data set, i.e., ≤0.5; n = 41). Protective alleles were defined as those that were most strongly associated with lower viral loads in HIV-1 subtype C-infected individuals, namely, HLA-B*57, HLA-B*5801, and HLA-B*8101 (20), and were also found later to be the most strongly associated with lower viral loads or higher CD4 counts in a cohort of over 1,000 HIV-1 subtype C-infected individuals (43). The proportion of individuals possessing a protective allele was significantly greater in the low-replication-capacity group than in the high-replication-capacity group (Fisher's exact test; P = 0.003) (Fig. 2D).
When HLA-A, -B, and -C alleles were ranked according to viral load and then according to replication capacity, the ranks correlated positively with one another for each group of HLA alleles, although not significantly for HLA-C alleles (Spearman's rank correlation; r = 0.43 and P = 0.03, r = 0.42 and P = 0.04, and r = 0.47 and P = 0.06, respectively), which indicates a relationship between viral load and Gag-protease replication capacity (data not shown).
Correlation between replication capacity and baseline log viral load or CD4 count.
Replication capacities of recombinant viruses correlated positively with baseline log viral loads (Spearman's rank correlation; r = 0.24 and P < 0.0001) and negatively with baseline CD4 counts (Spearman's rank correlation; r = −0.17 and P = 0.0004) (Fig. 3A and B). These effects remained after removal of the protective alleles HLA-B*57, HLA-B*5801, and HLA-B*81 from analysis (Spearman's rank correlation; r = 0.18 and P = 0.001 for baseline log viral load and r = −0.14 and P = 0.01 for baseline CD4 count). Interestingly, analysis of the relationship between viral load or CD4 count and replication capacity among individuals expressing these protective alleles also revealed a significant positive correlation (Pearson's correlation; r = 0.33 and P = 0.001 for viral load and r = −0.33 and P = 0.001 for CD4 count).
FIG. 3.
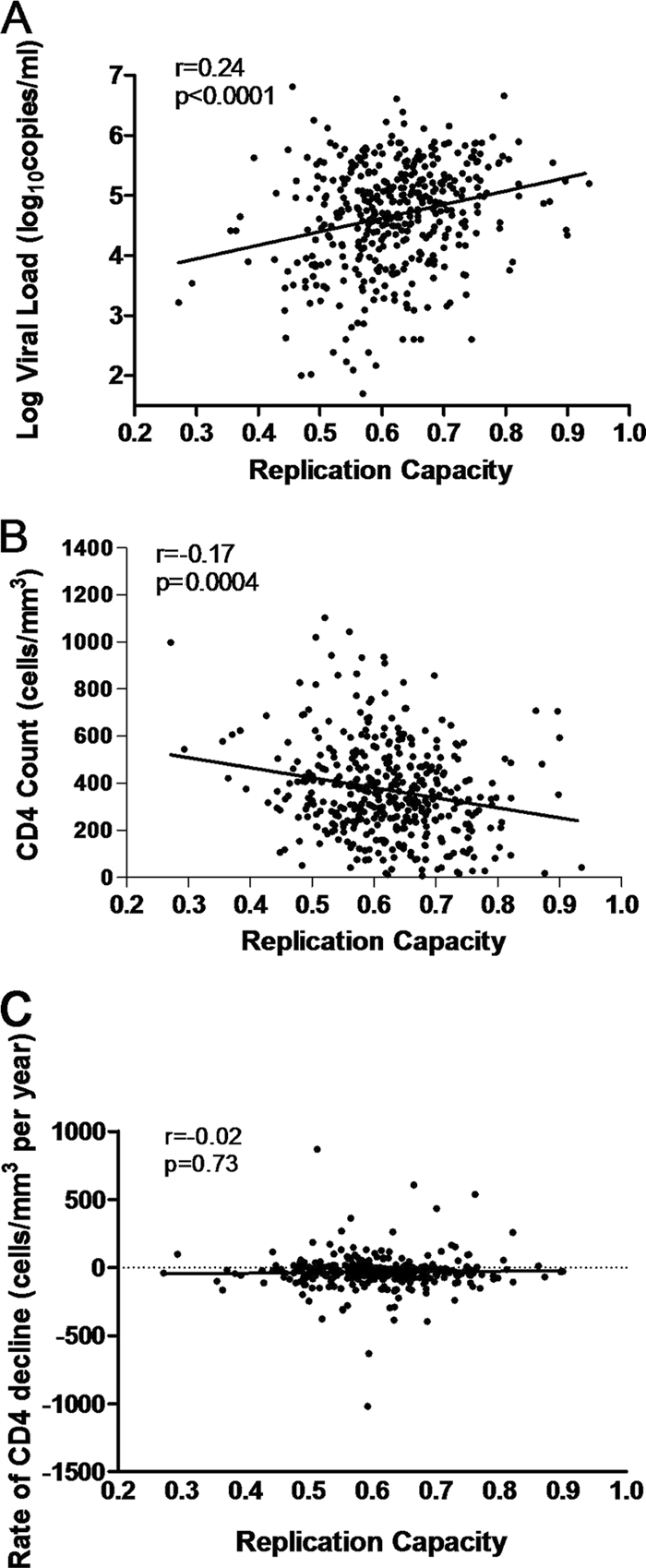
Correlations between replication capacities of recombinant viruses encoding patient Gag-protease and markers of disease progression. The graphs show replication capacity versus baseline log viral load (A), baseline CD4 count (B), and rate of CD4 decline (C). Correlations were calculated with Spearman's rank correlation test.
Relationship between replication capacity and subsequent longitudinal rate of CD4 decline.
An average of 2.28 years (SD = 1.3 years) of untreated follow-up was available for 339 Sinikithemba patients. For each study subject, linear regression was used to compute the rate of CD4 decline for the duration of untreated clinical follow-up. Spearman's correlation was then used to investigate the relationship between viral replication capacity and subsequent rate of CD4 decline. We observed no statistically significant relationship overall between replication capacity and CD4 decline (Spearman's rank correlation; r = −0.01 and P = 0.79). Stratification of the analysis by baseline CD4 counts (≤200, ≥200, ≤350, and ≥350 cells/mm3) also failed to reveal any significant correlations between replication capacity and the rate of CD4 decline (not shown). Figure 3C shows a lack of correlation between CD4 decline and Gag-protease-mediated replication capacity at baseline CD4 counts of ≥200 cells/mm3 (Spearman's rank correlation; r = −0.02 and P = 0.73).
Association of sequence variability in Gag with replication capacity. (i) Overall variability.
To investigate whether an increasing number of polymorphisms in Gag would tend to reduce replication capacity, the percent amino acid similarities of Gag sequences to the 2004 consensus subtype C Gag sequence were calculated using the sequence identity matrix function in BioEdit 7.0 and correlated with replication capacity. Unexpectedly, the calculated Gag percent similarity correlated negatively, although weakly, with replication capacity (Pearson's correlation; r = −0.18 and P = 0.0004), i.e., the fittest viruses were generally least like the consensus sequence (Fig. 4A). This analysis was repeated separately for each region of Gag, namely, p17, p24, p7, and p6, to see whether this relationship differed between regions. There remained an inverse relationship between percent similarity to consensus and replication capacity for every region of Gag except p24, although this was statistically significant only for p17 and p7 (Fig. 4B). There was no correlation between percent similarity to the subtype C Gag p24 consensus and replication capacity. In contrast, the majority of nonconsensus residues in p17/p7 increased replication capacity. It should be noted that divergence from the consensus subtype C sequence did not represent convergence to the consensus subtype B sequence, which would have indicated that divergence from the consensus subtype C sequence resulted in better compatibility with the subtype B NL4-3 backbone, and therefore in fitter viruses.
FIG. 4.

Associations between sequence variability in Gag-protease and replication capacities of recombinant viruses encoding Gag-protease. (A) Significant negative correlation (Pearson's correlation; n = 405) between percent similarity of Gag sequences to the consensus subtype C Gag sequence and replication capacity. (B) Gag p17, p24, p6, and p7 percent similarity to consensus subtype C sequence versus replication capacity (Spearman's rank correlation; n = 405). (C) Significant positive correlation (Pearson's correlation; n = 17) between entropy of HLA-associated sites in or within five amino acids of Gag epitopes and the average replication capacity of viruses with mutations at these sites. (D and E) Significant differences (Fisher's exact test) in proportions of p24 variant epitopes and p24 consensus epitopes (D) and of TL9 variant epitopes and TL9 consensus epitopes (E) between the least-fit viruses (replication capacity of ≤0.5; n = 41) and the fittest viruses (replication capacity of ≥0.74; n = 41). (F and G) Significant differences (Fisher's exact test) in proportions of nonconsensus amino acids associated with decreased replication capacity (RC) in p24 versus p17 (n = 12 and n = 23) (F) and in HLA-A- versus HLA-B-restricted epitopes (n = 13 and n = 17) (G).
(ii) HLA-associated variability.
HLA-associated polymorphisms—amino acids that are significantly more likely to occur in the presence of a particular HLA allele—were identified in the current data set by use of methods that take into account the phylogenetic relatedness of sequences, amino acid covariation, and HLA linkage disequilibrium effects (5). Each sequence was then analyzed in the context of the patient's HLA class I profile, and the number of HLA-associated polymorphisms was computed. To further analyze the influence of HLA alleles on Gag-protease replication capacity, the computed polymorphisms were correlated with replication capacity. The numbers of HLA-A-, -B-, and -C-associated polymorphisms in each sequence did not correlate significantly with replication capacity overall. Likewise, no dose-dependent effects of polymorphisms on replication capacity were observed among polymorphisms associated with protective HLA types. Similarly, when the relationship between the number of HLA-associated polymorphisms and replication capacity was investigated irrespective of patient HLA class I profile, i.e., also taking into account inherited polymorphisms, no significant associations were found. Therefore, while some HLA-associated polymorphisms significantly impact replication capacity (8, 45), the sum of HLA-selected polymorphisms, irrespective of location in Gag, was not associated with replication capacity in this chronic infection cohort. There was, however, a weak trend (Spearman's rank correlation; r = −0.09 and P = 0.08) toward lower replication capacities with increasing numbers of HLA-associated polymorphisms in epitopes or within five amino acids of epitopes restricted by the selecting HLA allele (these polymorphisms are more likely to represent escape mutations, not secondarily arising compensatory mutations [15]). Previously, increasing numbers of HLA-B-associated polymorphisms in or within five amino acids of Gag epitopes were strongly associated with lower viral loads in early infection, and this was attributed to lower fitness levels of these viruses (15). The number of HLA-B-associated polymorphisms in or within five amino acids of Gag epitopes was negatively correlated with fitness (Spearman's rank correlation; r = −0.11 and P = 0.03), although not strongly so. The relatively weak relationship between the number of HLA-associated polymorphisms in Gag and replication capacity in the present chronic infection cohort might be explained by the accumulation of compensatory mutations during the course of infection. In fact, evidence has been found for a strong effect of HLA-mediated selection pressure in Gag on replication capacity in early infection and no such significant relationship in the very late chronic stage of infection, suggesting that this effect wanes over time, presumably due to the development of compensatory mutations (M. A. Brockman et al., submitted for publication).
Since there is some evidence that HLA-associated escape mutations occurring in conserved sites of HIV carry a greater fitness cost than those occurring in regions of high variability (45), we compared the average replication capacity of viruses possessing each HLA-associated polymorphism with the corresponding entropy at that position. A trend toward a significant correlation between these two parameters was found (Pearson's correlation; r = 0.24 and P = 0.06). When the analysis was restricted to those polymorphisms in epitopes or within five amino acids of epitopes restricted by the selecting HLA allele, the correlation was much stronger (Pearson's correlation; r = 0.68 and P = 0.003) (Fig. 4C). Thus, HLA-associated escape mutations at more conserved sites (with lower entropy) in Gag were associated with greater fitness costs.
(iii) Epitope variability: association of TL9 variant with lower replication capacity.
Next, the relationship between sequence variability in specific HLA-restricted epitopes and replication capacity was examined. The proportion of variant Gag epitopes (i.e., nonconsensus) versus consensus epitopes was compared between the least-fit and fittest virus groups by Fisher's exact test, and no significant difference was found. However, there were marginally more variant Gag p24 epitopes in the sequences from the least-fit group (Fisher's exact test; P = 0.04) (Fig. 4D). This significant result was driven mainly by the greater proportion of variant HLA-B*81-restricted TL9 epitopes in the least-fit group (Fisher's exact test; P = 0.007) (Fig. 4E), although there were also significantly more variant HLA-B*57-restricted QW9 epitopes in viruses of lower fitness (Fisher's exact test; P = 0.04) (data not shown).
(iv) Single amino acid associations with replication capacity.
In an exploratory analysis, the Mann-Whitney U test was used to identify specific amino acids in Gag-protease associated with increased or decreased replication capacity. Although none of the comparisons yielded Q values of ≤0.2, 58 associations with P values of <0.05 were found for Gag, and 9 were found for protease, when consensus-nonconsensus pairs were counted as a single association (n ≥ 5 for both groups compared [see Table SA1 in the supplemental material]). Of the 58 associations in Gag, 23 occurred in p17, 12 in p24, 3 in the p2 linker peptide, 9 in p7, and 11 in p6.
Considering amino acids in Gag associated with alterations in viral replication capacity, most of the nonconsensus amino acids in p24 were associated with lower replication capacity (10/12 residues), while most of the nonconsensus residues in p17 were associated with increased replication capacity (15/23 residues). This difference was statistically significant (Fisher's exact test; P = 0.01) (Fig. 4F). Only 17 of 58 amino acids associated with replication capacity alterations corresponded to an HLA association at that position (not necessarily with the same amino acid), and 11 of these were HLA-B associated. Twenty-six associations occurred in published or previously predicted epitopes (13, 36), with 13 in HLA-A-restricted epitopes, 17 in HLA-B-restricted epitopes, and 6 in HLA-C-restricted epitopes (10 of these occurred in epitopes that were restricted by more than one HLA allele class and were thus considered under more than one category). Within HLA-B-restricted epitopes, 13 (8 of these were in p24) of 17 nonconsensus amino acids were associated with decreased replication capacity, while in the HLA-A-restricted epitopes, 8 of 13 nonconsensus amino acids (10 of these were in p17) were linked with increased replication capacity (Fisher's exact test; P = 0.06) (Fig. 4G). These results are suggestive of HLA-B-mediated selective pressure on Gag p24, with resulting lower replication capacity. It should also be noted that half of the amino acids associated with changes in Gag-protease-mediated replication capacity were neither HLA class I associated nor within known or predicted epitopes.
Multivariate analysis (linear regression with forward selection) was also undertaken. Seventeen of the 58 associations in Gag and 4 of the 9 associations in protease identified by univariate analysis also had P values of <0.05 (but Q values of >0.2) in the multivariate model, and the strongest of these associations was the consensus T at position 186, with increased replication capacity (see Table SA1 in the supplemental material).
(v) HLA-B*81-associated T186S mutation is linked to lower replication capacity.
Since the consensus residue 186T was strongly associated with an increased replication capacity, changes away from consensus at this position were compared between the least-fit and fittest viruses. In the least-fit group, 9 sequences had an S and 1 had an A at position 186, while only 1 had an S at this position in the fittest group (Fisher's exact test; P = 0.01) (Fig. 5A). With the exception of 1 sequence with 186A and another sequence which had a mixture of T and S at this position, the nonconsensus amino acid at codon 186 was S. Overall, 186S was associated with a decrease in replication capacity compared with that for the 186T consensus (Student's t test; P = 0.006; n = 403) (Fig. 5B). This polymorphism is associated with HLA-B*81 and occurs in the HLA-B*81-restricted epitope TL9. The difference in numbers of variant TL9 epitopes between the low- and high-fitness groups could be attributed largely to variability at position 186. However, when only HLA-B*81-positive individuals were considered, the replication capacities of viruses with 186S and 186T were both below average and were not significantly different from one another (data not shown), indicating that other mutations are also responsible for the lower fitness of viruses from these individuals. The lack of difference in replication capacity between viruses with 186S and 186T from individuals with HLA-B*81 may also suggest that the fitness cost of 186S was compensated for in some cases.
FIG. 5.
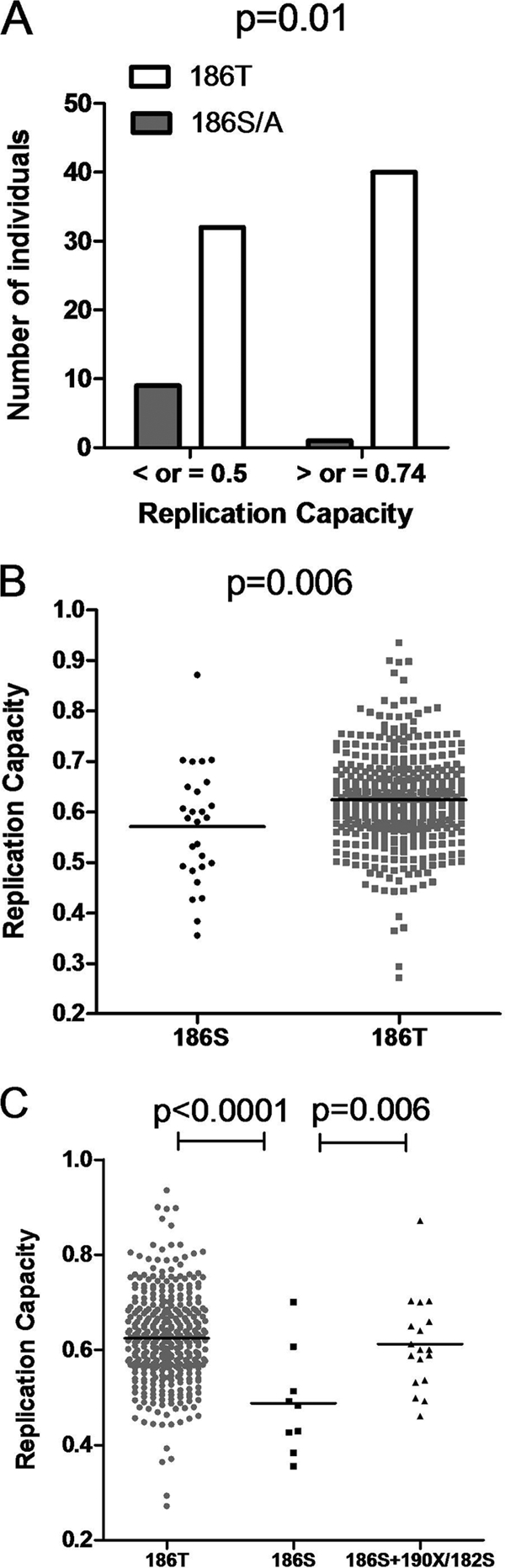
Single Gag amino acid associations with altered replication capacities of Gag-protease NL4-3 recombinant viruses. (A) Greater proportion (Fisher's exact test) of 186S/A residues in the least-fit viruses (replication capacity of ≤0.5; n = 41) than in the fittest viruses (replication capacity of ≥0.742; n = 41). (B) Lower replication capacity of viruses with 186S (n = 27) than that of viruses with the consensus 186T (n = 376; Student's t test). (C) Significantly higher replication capacities of viruses with 186T (n = 376) and with 186S with putative compensatory mutations 190X and 182S (n = 18) than those of viruses with 186S alone (n = 9; Student's t test).
(vi) Residues covarying with 186S.
Codon covariation lists were generated from the current data set as previously described (5). Amino acids positively associated with 186S and/or negatively associated with 186T included 177D, 182S, 190A, 190I, 256I, and 343I (P < 0.05; Q < 0.2). Amino acids negatively associated with 186S and/or positively associated with 186T included 65Q, 177E, 190T, 256V, and 343L (P < 0.05; Q < 0.2). Replication capacities of viruses with 186S and various numbers of associated residues (Q65X, E177X, Q182S, T190X, V256X, and L343X) were compared to assess whether these might function as compensatory mutations. The number of covarying residues present correlated positively but not significantly with replication capacity (Pearson's correlation; r = 0.26 and P = 0.19). However, on closer examination of sequences with 186S, a greater occurrence of mutations at positions 182 and 190 (but not at other covarying positions) was noted in the fitter viruses (Fig. 5C). This was statistically significant (Student's t test; P = 0.006), suggesting that 190X and 182S, which occur parallel to and on either side of residue 186 in a helix structure, might indeed be compensatory mutations.
DISCUSSION
The mechanisms underlying HIV-1 control by protective HLA alleles are not fully understood and could involve targeting of functionally important epitopes in Gag, resulting in selection of escape mutations with a fitness cost. Therefore, this study was undertaken to investigate, at the population level, the impact of HLA-mediated immune pressure in Gag on viral fitness and its impact on HIV-1 pathogenesis.
Our results showed an association between protective HLA alleles (HLA-B*57, HLA-B*5801, and HLA-B*81) and lower Gag-protease replication capacities. Since (i) protective HLA alleles were associated with lower viral loads, (ii) Gag-protease replication capacity correlated with viral loads even on removal of protective HLA alleles from the analysis and within individuals with protective alleles, and (iii) replication capacity ranked according to HLA-A, -B, and -C alleles correlated significantly with the ranks according to viral load, the possibility that HLA alleles and replication capacity are indirectly related to each other through association with viral load cannot be excluded. However, since mutations in Gag selected by the protective HLA alleles B*5703 and B*5801 were shown to significantly decrease the overall replication capacity of isolates and to confer benefits on infant and adult recipients (7, 8, 15, 33, 39), except in the presence of compensatory mutations (39), it seems very likely that a direct relationship exists between HLA alleles and Gag-protease replication capacity. Gag-protease replication capacity varied significantly between the different HLA-B but not HLA-A or HLA-C alleles, consistent with the idea that HLA alleles influence Gag-protease replication capacity through selecting mutations, as HLA-B alleles have the greatest selection pressure (20). Moreover, increasing numbers of HLA-B-associated mutations in or flanking epitopes (likely HLA-selected escape mutations) correlated with decreased HIV replication capacities. In further support of a direct relationship between protective HLA alleles and replication capacity, HLA-B*81 was by far the allele most strongly associated with lower replication capacity, even though HLA-B*5703-positive individuals had a lower average viral load than HLA-B*81-positive individuals, and 186S present in the HLA-B*81-restricted epitope TL9 (positions 180 to 188) was the mutation most strongly associated with lowered replication capacity, thereby providing a possible mechanism for the influence of HLA-B*81 on replication capacity. TL9 was previously described as one of the key Gag epitopes under strong selection pressure by a beneficial HLA allele, with variance mainly at residues 182 and 186 (both with changes predominantly to serine) (14). Interestingly, in a recent study, the number of public T-cell clonotypes specific for simian immunodeficiency virus (SIV) Gag CM9 (residues 181 to 189), which occurs in nearly the exact same position as TL9 in HIV, correlated strongly and negatively (r2 = −0.71) with the viral set point in rhesus macaques (34). Residue 186 in HIV Gag has also been classified as a site where mutations revert upon transmission to a host lacking the HLA allele that selected them, presumably due to a fitness cost (26). It should be noted, though, that differences in fitness associated with variability at position 186 did not translate into viral load differences in this chronic infection cohort (data not shown), which could suggest that the fitness cost of the 186S mutation may be compensated in some cases, and therefore not of lasting benefit, and that the balance between the fitness cost of 186S and an effective CTL response to TL9 may be important in determining the outcome. However, taking the results together, it seems likely that protective HLA alleles, in particular HLA-B*81, influence Gag-protease replication capacity through CTL selection pressure and that this may partly contribute to their protective effect. From the present data, this seems likely to be a more prominent mechanism of protection for HLA-B*81 than for HLA-B*57 and HLA-B*5801 in subtype C infection.
Given our observation that lower Gag-protease replication capacities were related to protective HLA types, lower baseline viral loads, and higher baseline CD4 counts, we wished to investigate whether viral replication capacity may also correlate with the subsequent rate of CD4 decline during chronic infection. However, such a correlation was not observed in the present study. This may be explained partly by the balance that exists between Gag CTL responses and replication capacity in influencing clinical outcomes. Accumulation of escape mutations in HIV carries a fitness cost to the virus, but the disadvantage to the virus is offset by the advantage of escaping effective CTL responses that were holding replication in check, resulting in increased viral loads and accelerated disease progression despite a replication-deficient virus (8, 19). Another consideration is that replication capacity is not static and compensatory mutations may have developed at a time point later than that measured, influencing the subsequent rate of CD4 decline. Data from the present study and previous studies suggest that mutations with a fitness cost are readily compensated. The T186S mutation was most strongly associated with decreased replication capacity, yet in the presence of covarying mutations at positions 182 and 190, the mean replication capacity was not significantly different from the mean for the entire cohort, suggesting that the possible fitness cost of this mutation was compensated in these cases. Therefore, although there may be a benefit to decreased replication capacity (as supported by cross-sectional correlations with viral loads and CD4 counts), the data do not support an enduring benefit or a lasting significant impact of Gag-protease replication capacity on the rate of disease progression, at least once the chronic infection stage has been reached. The results of Brockman et al. (submitted) are consistent with this notion. However, acute infection studies and/or longitudinal analysis of replication capacity and sequence changes, together with CTL responses, may be necessary to better assess the relative impact of each on disease progression. Site-directed mutagenesis experiments would also be necessary to confirm the suspected fitness costs and compensatory roles of some of the mutations described above.
The data support the hypothesis that mutations at conserved residues/regions, in particular in conserved Gag p24 as opposed to the less-conserved Gag p17, are more likely to result in a fitness cost: HLA-associated escape mutations at conserved sites were associated with lower replication capacities, there were significantly more variant p24 epitopes in the least-fit viruses than in the fittest viruses, and most of the mutations significantly associated with altered replication capacities in p24 decreased replication capacity, while most in p17 increased replication capacity. In agreement with these data, beneficial HLA alleles in an African cohort were associated with strong selection at key epitopes which occurred mostly in Gag p24 (14), and there is recent evidence that HLA-B*57 mediates its protective effect mainly through attenuating mutations in Gag p24 (39). Furthermore, the breadth of Gag p24, but not p17 or p15, CD8 T-cell responses in HLA-B*13-positive individuals was significantly associated with decreasing viral loads (17). Taken together, the data generally support the inclusion of conserved regions such as Gag p24 in a vaccine that is aimed at driving HIV toward a less-fit state.
Interestingly, a larger number of amino acid differences from the consensus subtype C Gag sequence were weakly but significantly associated with increasing viral fitness. The percent amino acid similarity to the consensus subtype C Gag sequence also correlated negatively with viral load and positively with CD4 count (data not shown), suggesting that more changes from consensus and increased fitness of viruses may occur with disease progression. In fact, the fitness of HIV isolates was previously shown to increase with disease progression (44). Consensus amino acids could, in some instances, be escape mutations in response to common HLA alleles, but we speculate that they represent the nonescape form in the majority of cases and that nonconsensus residues represent escape and compensatory mutations in response to CTL and non-CTL immune pressure, although they could also represent random mutations. Based on this conjecture, we suggest that more changes away from consensus likely indicate more compensation, and therefore fitter viruses. Another explanation is that the majority of mutations introduced into HIV are likely to have no or little fitness cost or to actually increase fitness. Consistent with this idea, p17 and p7 were significantly more divergent from the consensus than p24 was, i.e., significantly more mutations occurred in p17 and p7 than in p24, and the percent similarity to consensus for both p17 and p7 was negatively correlated overall with fitness, while there was no correlation for p24. The direct relationship between replication capacity and the entropy of mutated sites in the present study, as well as the recent finding that escape mutations in conserved Gag p24 carry significant fitness costs while most of the escape mutations in the highly variable env gene are fitness neutral or increase fitness (45), lends further support to this argument.
Another interesting finding was that most of the mutations in Gag associated with altered replication capacity were not HLA associated (71%). It should be noted, however, that a limitation of this study was the insertion of subtype C Gag-protease into a subtype B backbone, and therefore some Gag-protease mutations associated with altered replication capacity might represent those that interact with other components of the backbone. A significantly lower replication capacity of subtype C/B recombinants than that of subtype B recombinants was observed, which could suggest that mixing of subtypes results in suboptimal replication. Alternatively, this finding could mean that Gag-protease function is inferior in subtype C versus subtype B viruses, which may partly explain previously described fitness differences between subtypes (1). Further experiments are required to discriminate between these possibilities. Supporting the latter rather than the former possibility, convergence of subtype C Gag sequences to the consensus subtype B sequence was not associated with fitter recombinant viruses. Furthermore, the findings of the present study are in agreement with those of Brockman et al. (submitted), which show that subtype B Gag-protease NL4-3 recombinant viruses correlate with cross-sectional viral load and CD4 count data as well as with specific HLA types, strongly supporting the hypothesis that the current assay system is clinically and biologically relevant.
In summary, there is evidence that protective HLA alleles, especially HLA-B*81, influence subtype C HIV replication capacity through selection of mutations in Gag that incur a fitness cost. Moreover, mutations in conserved rather than more-variable regions of Gag are more likely to carry a fitness cost, suggesting that conserved regions such as Gag p24 should be included in a vaccine aiming to drive HIV toward a less-fit state. However, the long-term clinical impact of immune-driven fitness costs requires further investigation, given the evidence for compensation and the observation that replication capacity does not correlate with the subsequent rate of CD4 decline in chronic infection.
Supplementary Material
Acknowledgments
This research was funded by the NIH (grant ROI-AI067073, contract NOI-AI-15422), the South African AIDS Vaccine Initiative, and the Ragon Institute Fund for Innovation and New International Initiatives. J.K.W. was funded by the National Research Foundation and the Ragon Institute of Massachusetts General Hospital, Massachusetts Institute of Technology, and Harvard University. Z.L.B. was supported by a New Investigator Award from the Canadian Institutes for Health Research (CIHR). T.N. holds the South African Department of Science and Technology/National Research Foundation Research Chair in Systems Biology of HIV/AIDS.
We thank Jennifer Sela, Pamela Rosato, and Taryn Green for technical assistance; Johannes Viljoen and the Africa Centre laboratory for providing access to tissue culture and sequencing facilities; the Durban clinic staff (Sisters Kesia Ngwenya, Thandi Cele, Thandi Sikhakane, and Nokuthula Lutuli); and Isobel Honeyborne, Wendy Mphatswe, and the management of McCord Hospital for their support of the Sinikithemba cohort. Finally, we thank and acknowledge the Sinikithemba cohort study participants.
Footnotes
Published ahead of print on 11 August 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Abraha, A., I. L. Nankya, R. Gibson, K. Demers, D. M. Tebit, E. Johnston, D. Katzenstein, A. Siddiqui, C. Herrera, L. Fischetti, R. J. Shattock, and E. J. Arts. 2009. CCR5- and CXCR4-tropic subtype C human immunodeficiency virus type 1 isolates have a lower level of pathogenic fitness than other dominant group M subtypes: implications for the epidemic. J. Virol. 83:5592-5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1a.Brockman, M. A., Z. L. Brumme, C. J. Brumme, T. Miura, J. Sela, P. C. Rosato, C. M. Kadie, J. M. Carlson, T. J. Markle, H. Streeck, A. D. Kelleher, M. Markowitz, H. Jessen, E. Rosenberg, M. Altfeld, P. R. Harrigan, D. Heckerman, B. D. Walker, and T. M. Allen. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 2.Brockman, M. A., A. Schneidewind, M. Lahaie, A. Schmidt, T. Miura, I. DeSouza, F. Ryvkin, C. A. Derdeyn, S. Allen, E. Hunter, J. Mulenga, P. A. Goepfert, B. D. Walker, and T. M. Allen. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 81:12608-12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brockman, M. A., G. O. Tanzi, B. D. Walker, and T. M. Allen. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods 131:134-142. [DOI] [PubMed] [Google Scholar]
- 4.Brumme, Z., B. Wang, K. Nair, C. Brumme, C. de Pierres, S. Reddy, B. Julg, E. Moodley, C. Thobakgale, Z. Lu, M. van der Stok, K. Bishop, Z. Mncube, F. Chonco, Y. Yuki, N. Frahm, C. Brander, M. Carrington, M. Freedberg, P. Kiepiela, P. Goulder, B. Walker, T. Ndung'u, and E. Losina. 2009. Impact of select immunologic and virologic biomarkers on CD4 cell count decrease in chronic HIV-1 subtype C infection: results from Sinikithemba Cohort, Durban, South Africa. Clin. Infect. Dis. 49:956-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlson, J. M., Z. L. Brumme, C. M. Rousseau, C. J. Brumme, P. Matthews, C. Kadie, J. I. Mullins, B. D. Walker, P. R. Harrigan, P. J. R. Goulder, and D. Heckerman. 2008. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput. Biol. 4:1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrington, M., and S. J. O'Brien. 2003. The influence of HLA genotype on AIDS. Annu. Rev. Med. 54:535-551. [DOI] [PubMed] [Google Scholar]
- 7.Chopera, D. R., Z. Woodman, K. Mlisana, M. Mlotshwa, D. P. Martin, C. Seoighe, F. Treurnicht, D. Assis de Rosa, W. Hide, S. A. Karim, C. M. Gray, C. Williamson, and C. S. Team. 2008. Transmission of HIV-1 CTL escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 4:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crawford, H., W. Lumm, A. Leslie, M. Schaefer, D. Boeras, J. G. Prado, J. Tang, P. Farmer, T. Ndung'u, S. Lakhi, J. Gilmour, P. Goepfert, B. D. Walker, R. Kaslow, J. Mulenga, S. Allen, P. J. R. Goulder, and E. Hunter. 2009. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J. Exp. Med. 206:909-919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deeks, S. G., and B. D. Walker. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406-416. [DOI] [PubMed] [Google Scholar]
- 10.Derdeyn, C. A., and G. Silvestri. 2005. Viral and host factors in the pathogenesis of HIV infection. Curr. Opin. Immunol. 17:366-373. [DOI] [PubMed] [Google Scholar]
- 11.Donfack, J., F. J. Buchinsky, J. C. Post, and G. D. Ehrlich. 2006. Human susceptibility to viral infection: the search for HIV-protective alleles among Africans by means of genome-wide studies. AIDS Res. Hum. Retroviruses 22:925-930. [DOI] [PubMed] [Google Scholar]
- 12.Edwards, B. H., A. Bansal, S. Sabbaj, J. Bakari, M. J. Mulligan, and P. A. Goepfert. 2002. Magnitude of functional CD8+ T-cell responses to the Gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J. Virol. 76:2298-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frahm, N., B. Baker, and C. Brander. 2008. Identification and optimal definition of HIV-derived cytotoxic T lymphocyte (CTL) epitopes for the study of CTL escape, functional avidity and viral evolution, p. 3-24. In B. T. M. Korber, C. Brander, B. F. Haynes, R. Koup, J. P. Moore, B. D. Walker, and D. I. Watkins (ed.), HIV molecular immunology 2008. Los Alamos National Laboratory, Theoretical Biology and Biophysics, Los Alamos, NM.
- 14.Frater, A. J., H. Brown, A. Oxenius, H. F. Günthard, B. Hirschel, N. Robinson, A. J. Leslie, R. Payne, H. Crawford, A. Prendergast, C. Brander, P. Kiepiela, B. D. Walker, P. J. R. Goulder, A. McLean, R. E. Phillips, et al. 2007. Effective T-cell responses select human immunodeficiency virus mutants and slow disease progression. J. Virol. 81:6742-6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goepfert, P. A., W. Lumm, P. Farmer, P. Matthews, A. Prendergast, J. M. Carlson, C. A. Derdeyn, J. Tang, R. A. Kaslow, A. Bansal, K. Yusim, D. Heckerman, J. Mulenga, S. Allen, P. J. R. Goulder, and E. Hunter. 2008. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 205:1009-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray, C. M., M. Mlotshwa, C. Riou, T. Mathebula, D. de Assis Rosa, T. Mashishi, C. Seoighe, N. Ngandu, F. van Loggerenberg, L. Morris, K. Mlisana, C. Williamson, and S. A. Karim. 2009. Human immunodeficiency virus-specific gamma interferon enzyme-linked immunospot assay responses targeting specific regions of the proteome during primary subtype C infection are poor predictors of the course of viremia and set point. J. Virol. 83:470-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honeyborne, I., A. Prendergast, F. Pereyra, A. Leslie, H. Crawford, R. Payne, S. Reddy, K. Bishop, E. Moodley, K. Nair, M. van der Stok, N. McCarthy, C. M. Rousseau, M. Addo, J. I. Mullins, C. Brander, P. Kiepiela, B. D. Walker, and P. J. R. Goulder. 2007. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple Gag-specific CD8+ T-cell epitopes. J. Virol. 81:3667-3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, X., and J. Zhang. 1996. Methods for comparing a DNA sequence with a protein sequence. Comput. Appl. Biosci. 12:497-506. [DOI] [PubMed] [Google Scholar]
- 19.Kawada, M., H. Igarashi, A. Takeda, T. Tsukamoto, H. Yamamoto, S. Dohki, M. Takiguchi, and T. Matano. 2006. Involvement of multiple epitope-specific cytotoxic T-lymphocyte responses in vaccine-based control of simian immunodeficiency virus replication in rhesus macaques. J. Virol. 80:1949-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiepiela, P., A. J. Leslie, I. Honeyborne, D. Ramduth, C. Thobakgale, S. Chetty, P. Rathnavalu, C. Moore, K. J. Pfafferott, L. Hilton, P. Zimbwa, S. Moore, T. Allen, C. Brander, M. M. Addo, M. Altfeld, I. James, S. Mallal, M. Bunce, L. D. Barber, J. Szinger, C. Day, P. Klenerman, J. Mullins, B. Korber, H. M. Coovadia, B. D. Walker, and P. J. R. Goulder. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432:769-774. [DOI] [PubMed] [Google Scholar]
- 21.Kiepiela, P., K. Ngumbela, C. Thobakgale, D. Ramduth, I. Honeyborne, E. Moodley, S. Reddy, C. de Pierres, Z. Mncube, N. Mkhwanazi, K. Bishop, M. van der Stok, K. Nair, N. Khan, H. Crawford, R. Payne, A. Leslie, J. Prado, A. Prendergast, J. Frater, N. McCarthy, C. Brander, G. H. Learn, D. Nickle, C. Rousseau, H. Coovadia, J. I. Mullins, D. Heckerman, B. D. Walker, and P. Goulder. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46-53. [DOI] [PubMed] [Google Scholar]
- 22.Le Clerc, S., S. Limou, C. Coulonges, W. Carpentier, C. Dina, L. Taing, O. Delaneau, T. Labib, R. Sladek, A. G. Group, C. Deveau, H. Guillemain, R. Ratsimandresy, M. Montes, J. L. Spadoni, A. Therwath, F. Schächter, F. Matsuda, I. Gut, J. D. Lelièvre, Y. Lévy, P. Froguel, J. F. Delfraissy, S. Hercberg, and J. F. Zagury. 2009. Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03). J. Infect. Dis. 200:1194-1201. [DOI] [PubMed] [Google Scholar]
- 23.Leslie, A., P. C. Matthews, J. Listgarten, J. M. Carslon, C. Kadie, T. Ndung'u, C. Brander, H. Coovadia, B. D. Walker, D. Heckerman, and P. J. R. Goulder. 2010. Additive contribution of HLA class I alleles in the immune control of HIV-1 infection. J. Virol. 84:9879-9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leslie, A. J., K. J. Pfafferott, P. Chetty, R. Draenert, M. M. Addo, M. Feeney, Y. Tang, E. C. Holmes, T. Allen, J. G. Prado, M. Altfeld, C. Brander, C. Dixon, D. Ramduth, P. Jeena, S. A. Thomas, A. St. John, T. A. Roach, B. Kupfer, G. Luzzi, A. Edwards, G. Taylor, H. Lyall, G. Tudor-Williams, V. Novelli, J. Martinez-Picado, P. Kiepiela, B. D. Walker, and P. J. R. Goulder. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282-288. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Picado, J., J. G. Prado, E. E. Fry, K. Pfafferott, A. Leslie, S. Chetty, C. Thobakgale, I. Honeyborne, H. Crawford, P. Matthews, T. Pillay, C. Rousseau, J. I. Mullins, C. Brander, B. D. Walker, D. I. Stuart, P. Kiepiela, and P. Goulder. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80:3617-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matthews, P. C., A. Prendergast, A. Leslie, H. Crawford, R. Payne, C. Rousseau, M. Rolland, I. Honeyborne, J. Carlson, C. Kadie, C. Brander, K. Bishop, N. Mlotshwa, J. I. Mullins, H. Coovadia, T. Ndung'u, B. D. Walker, D. Heckerman, and P. J. R. Goulder. 2008. Central role of reverting mutations in HLA associations with human immunodeficiency virus set point. J. Virol. 82:8548-8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miura, T., M. A. Brockman, Z. L. Brumme, C. J. Brumme, F. Pereyra, A. Trocha, B. L. Block, A. Schneidewind, T. M. Allen, D. Heckerman, and B. D. Walker. 2009. HLA-associated alterations in replication capacity of chimeric NL4-3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J. Virol. 83:140-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ndongala, M. L., Y. Peretz, S. Boulet, M. Doroudchi, B. Yassine-Diab, M. R. Boulassel, D. Rouleau, C. Tremblay, R. LeBlanc, J. P. Routy, R. P. Sékaly, and N. F. Bernard. 2009. HIV Gag p24 specific responses secreting IFN-gamma and/or IL-2 in treatment-naïve individuals in acute infection early disease (AIED) are associated with low viral load. J. Clin. Immunol. 131:277-287. [DOI] [PubMed] [Google Scholar]
- 29.Ngumbela, K. C., C. L. Day, Z. Mncube, K. Nair, D. Ramduth, C. Thobakgale, E. Moodley, S. Reddy, C. De Pierres, N. Mkhwanazi, K. Bishop, M. van der Stok, N. Ismail, I. Honeyborne, H. Crawford, D. G. Kavanagh, C. Rousseau, D. Nickle, J. Mullins, D. Heckerman, B. Korber, H. Coovadia, P. Kiepiela, P. J. R. Goulder, and B. D. Walker. 2008. Targeting of a CD8 T cell Env epitope presented by HLA-B*5802 is associated with markers of HIV disease progression and lack of selection pressure. AIDS Res. Hum. Retroviruses 24:72-82. [DOI] [PubMed] [Google Scholar]
- 30.O'Brien, S. J., and G. W. Nelson. 2004. Human genes that limit AIDS. Nat. Genet. 36:565-574. [DOI] [PubMed] [Google Scholar]
- 31.Patke, D. S., S. J. Langan, L. M. Carruth, S. M. Keating, B. P. Sabundayo, J. B. Margolick, T. C. Quinn, and R. C. Bollinger. 2002. Association of Gag-specific T lymphocyte responses during the early phase of human immunodeficiency virus type 1 infection and lower virus load set point. J. Infect. Dis. 186:1177-1180. [DOI] [PubMed] [Google Scholar]
- 32.Pereyra, F., S. Palmer, T. Miura, B. L. Block, A. Wiegand, A. C. Rothchild, B. Baker, R. Rosenberg, E. Cutrell, M. S. Seaman, J. M. Coffin, and B. D. Walker. 2009. Persistent low-level viremia in HIV-1 elite controllers and relationship to immunologic parameters. J. Infect. Dis. 200:984-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prado, J. G., A. Prendergast, C. Thobakgale, C. Molina, G. Tudor-Williams, T. Ndung'u, B. D. Walker, and P. Goulder. 2010. Replicative capacity of human immunodeficiency virus type 1 transmitted from mother to child is associated with pediatric HIV-1 disease progression rate. J. Virol. 84:492-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Price, D. A., T. E. Asher, N. A. Wilson, M. C. Nason, J. M. Brenchley, I. S. Metzler, V. Venturi, E. Gostick, P. K. Chattopadhyay, M. Roederer, M. P. Davenport, D. I. Watkins, and D. C. Douek. 2009. Public clonotype usage identifies protective Gag-specific CD8+ T cell responses in SIV infection. J. Exp. Med. 206:923-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riviére, Y., M. B. McChesney, F. Porrot, F. Tanneau-Salvadori, P. Sansonetti, O. Lopez, G. Pialoux, V. Feuillie, M. Mollereau, and S. E. A. Chamaret. 1995. Gag-specific cytotoxic responses to HIV type 1 are associated with a decreased risk of progression to AIDS-related complex or AIDS. AIDS Res. Hum. Retroviruses 11:903-907. [DOI] [PubMed] [Google Scholar]
- 36.Rousseau, C. M., M. G. Daniels, J. M. Carlson, C. Kadie, H. Crawford, A. Prendergast, P. Matthews, R. Payne, M. Rolland, D. N. Raugi, B. S. Maust, G. H. Learn, D. C. Nickle, H. Coovadia, T. Ndung'u, N. Frahm, C. Brander, B. D. Walker, P. J. R. Goulder, T. Bhattacharya, D. E. Heckerman, B. T. Korber, and J. I. Mullins. 2008. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype C proteome: immune escape and viral load. J. Virol. 82:6434-6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schellens, I. M. M., J. A. M. Borghans, C. A. Jansen, I. M. De Cuyper, R. B. Geskus, D. van Baarle, and F. Miedema. 2008. Abundance of early functional HIV-specific CD8+ T cells does not predict AIDS-free survival time. PLoS One 3:e2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneidewind, A., M. A. Brockman, R. Yang, I. Adam, B. Li, S. Le Gall, C. R. Rinaldo, S. L. Craggs, R. L. Allgaier, K. A. Power, T. Kuntzen, C. S. Tung, M. X. LaBute, S. M. Mueller, T. Harrer, A. J. McMichael, P. J. Goulder, C. Aiken, C. Brander, A. D. Kelleher, and T. M. Allen. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 81:12382-12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneidewind, A., Y. Tang, M. A. Brockman, E. G. Ryland, J. Dunkley-Thompson, J. C. Steel-Duncan, M. A. St. John, J. A. Conrad, S. A. Kalams, F. Noel, T. M. Allen, C. D. Christie, and M. E. Feeney. 2009. Maternal transmission of human immunodeficiency virus escape mutations subverts HLA-B57 immunodominance but facilitates viral control in the haploidentical infant. J. Virol. 83:8616-8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Storey, J. D., and R. Tibshirani. 2003. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A. 100:9440-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takeuchi, H., and T. Matano. 2008. Host factors involved in resistance to retroviral infection. Microbiol. Immunol. 52:318-325. [DOI] [PubMed] [Google Scholar]
- 42.Tang, J., S. Tang, E. Lobashevsky, A. D. Myracle, U. Fideli, G. Aldrovandi, S. Allen, R. Musonda, R. A. Kaslow, et al. 2002. Favorable and unfavorable HLA class I alleles and haplotypes in Zambians predominantly infected with clade C human immunodeficiency virus type 1. J. Virol. 76:8276-8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thobakgale, C. F., A. Prendergast, H. Crawford, N. Mkhwanazi, D. Ramduth, S. Reddy, C. Molina, Z. Mncube, A. Leslie, J. Prado, F. Chonco, W. Mphatshwe, G. Tudor-Williams, P. Jeena, N. Blanckenberg, K. Dong, P. Kiepiela, H. Coovadia, T. Ndung'u, B. D. Walker, and P. J. R. Goulder. 2009. Impact of HLA in mother and child on disease progression of pediatric human immunodeficiency virus type 1 infection. J. Virol. 83:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Troyer, R. M., K. R. Collins, A. Abraha, E. Fraundorf, D. M. Moore, R. W. Krizan, Z. Toossi, R. L. Colebunders, M. A. Jensen, J. I. Mullins, G. Vanham, and E. J. Arts. 2005. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. J. Virol. 79:9006-9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Troyer, R. M., J. McNevin, Y. Liu, S. C. Zhang, R. W. Krizan, A. Abraha, D. M. Tebit, H. Zhao, S. Avila, M. A. Lobritz, M. J. McElrath, S. Le Gall, J. I. Mullins, and E. J. Arts. 2009. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 5:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuniga, R., A. Lucchetti, P. Galvan, S. Sanchez, C. Sanchez, A. Hernandez, H. Sanchez, N. Frahm, C. H. Linde, H. S. Hewitt, W. Hildebrand, M. Altfeld, T. M. Allen, B. D. Walker, B. T. Korber, T. Leitner, J. Sanchez, and C. Brander. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 80:3122-3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.