

Abstract
Bone marrow stromal antigen 2 (BST-2/tetherin) is a cellular membrane protein that inhibits the release of HIV-1. We show for the first time, using infectious viruses, that BST-2 also inhibits egress of arenaviruses but has no effect on filovirus replication and spread. Specifically, infectious Lassa virus (LASV) release significantly decreased or increased in human cells in which BST-2 was either stably expressed or knocked down, respectively. In contrast, replication and spread of infectious Zaire ebolavirus (ZEBOV) and Lake Victoria marburgvirus (MARV) were not affected by these conditions. Replication of infectious Rift Valley fever virus (RVFV) and cowpox virus (CPXV) was also not affected by BST-2 expression. Elevated cellular levels of human or murine BST-2 inhibited the release of virus-like particles (VLPs) consisting of the matrix proteins of multiple highly virulent NIAID Priority Pathogens, including arenaviruses (LASV and Machupo virus [MACV]), filoviruses (ZEBOV and MARV), and paramyxoviruses (Nipah virus). Although the glycoproteins of filoviruses counteracted the antiviral activity of BST-2 in the context of VLPs, they could not rescue arenaviral (LASV and MACV) VLP release upon BST-2 overexpression. Furthermore, we did not observe colocalization of filoviral glycoproteins with BST-2 during infection with authentic viruses. None of the arenavirus-encoded proteins rescued budding of VLPs in the presence of BST-2. Our results demonstrate that BST-2 might be a broad antiviral factor with the ability to restrict release of a wide variety of human pathogens. However, at least filoviruses, RVFV, and CPXV are immune to its inhibitory effect.
The host innate immune response acts as a first line of defense against viral infections, preventing virus invasion or replication before more specific protection is generated by the adaptive immune system (23). Viral infection or recognition of viral nucleic acids initiates signaling pathways that lead to the synthesis of multiple cytokines, including type I interferons (IFNs), such as IFN-α and IFN-β, which evoke coordinated antiviral responses in the host. Viruses have evolved multiple strategies to counter the IFN system by suppressing IFN production, signaling, or IFN antiviral effector proteins, thereby facilitating infection (23).
Bone marrow stromal antigen 2 (BST-2; also called CD317, HM1.24, or tetherin) is a glycosylphosphatidylinositol-anchored type II transmembrane protein that is upregulated on most cell types upon stimulation with type I IFNs or IFN-γ (8, 24, 33). BST-2 shuttles between the plasma membrane, where it is present predominantly in lipid rafts, and the trans-Golgi network, via a clathrin-mediated pathway (33, 55).
Several groups have described the antiviral role of BST-2 as an inhibitor of retrovirus release (28, 49, 63), and demonstrated that human immunodeficiency virus type 1 (HIV-1) Vpu can antagonize this function. Specifically, cell lines that endogenously expressed BST-2 or were stimulated with IFN-α exhibited markedly decreased budding of retroviral virus-like particles (VLPs) or Vpu-defective HIV-1 compared to BST-2-negative cells. Virions produced in BST-2-expressing cells were retained on the cell surface and subsequently internalized into BST-2-positive endosomal compartments. This restriction could be overcome by HIV-1 Vpu, which downregulates BST-2 from the cell surface via β-TrCP and endolysosomal trafficking (16, 45).
Lake Victoria marburgvirus (MARV), Zaire ebolavirus (ZEBOV), Lassa virus (LASV), Machupo virus (MACV), and Nipah virus (NiV) are highly virulent NIAID Priority Pathogens. Marburgviruses and ebolaviruses, members of the family Filoviridae, as well as Lassa and Machupo viruses, members of the family Arenaviridae, cause severe hemorrhagic fevers in humans and nonhuman primates with extraordinarily high case-fatality rates (22, 31). NiV, an emerging zoonotic paramyxovirus (15), was implicated as the cause of a rapidly fatal, febrile human encephalitis. Currently, U.S. Food and Drug Administration-approved vaccines or antivirals are not available for preventing or treating the diseases caused by any of these agents (9, 43).
ZEBOV, MARV, LASV, MACV, and NiV are enveloped RNA viruses that egress from their host cells by membrane protrusion (budding). Expression of the matrix proteins of these pathogens—VP40 (ZEBOV and MARV) (6, 25, 60), Z (LASV and MACV) (59), or M (NiV) (52, 53)—mimics this process and results in formation of VLPs that are released from the cell surface in a fashion similar to that of the HIV-1 matrix protein Gag (11, 47). These VLPs structurally resemble infectious virions (6, 19, 52) and therefore represent a suitable tool for both molecular-biological studies of viral budding and the development of vaccines (20, 38).
Recent reports have suggested a broad antiviral activity of BST-2. In addition to retroviruses, BST-2 can also inhibit release of VLPs generated by expression of the matrix proteins of ZEBOV, MARV, or LASV (28, 29, 56). Furthermore, the ZEBOV glycoprotein, GP1,2, was found to antagonize the antiviral activity of BST-2 in the context of VLPs, similarly to HIV-1 Vpu (29).
We examined here the hypothesis that BST-2 might be a general inhibitor of enveloped virus release. Only egress of infectious arenaviruses was affected by elevated BST-2 levels. The filovirus-encoded glycoprotein GP1,2 could counteract the antiviral activity of BST-2 and rescued replication of infectious filoviruses, whereas none of the arenavirus-encoded proteins antagonized BST-2. We also demonstrate that BST-2 can significantly decrease the release of VLPs of pathogenic viruses from three different families. Specifically, BST-2 overexpression inhibited the release of filoviral (ZEBOV and MARV), arenaviral (LASV and MACV), and paramyxoviral (NiV) VLPs.
MATERIALS AND METHODS
Plasmids, antibodies, and reagents.
Genes encoding human or murine FLAG-BST-2, HIV-1 Vpu-V5, ZEBOV (isolate Mayinga) NP-V5, VP35-V5, VP30-V5, V5-VP24, HA-VP40, GP1,2, GP1,2-V5 lacking the mucin-like domain (GP1,2ΔMLD-V5), sGP-V5, ssGP-V5, and Δ-peptide-V5; MARV (isolate Musoke) HA-VP40, MACV (strain Carvallo) Z-HA, NP-V5, and L; LASV (strain Josiah) SSP-V5, Z-HA, and NP-V5; and NiV M-HA were commercially synthesized (Geneart AG or Blue Heron Biotechnology). Plasmid encoding EGFP-VPS4A E228Q was previously described (58). Filoviral HA-VP40, arenaviral Z-HA, and NiV M-HA were cloned into the pCAGGS expression plasmid (54). The remaining genes were cloned into pcDNA3.1/Hygro(+) or pcDNA3.1(+) (Invitrogen). The MACV and LASV GPC expression vectors, as well as MARV GP1,2, were previously described (32, 54). Chloramphenicol acetyltransferase (CAT) antiserum was purchased from Invitrogen, and rabbit polyclonal antibody against BST-2 was obtained from Thermo Scientific Open Biosystems.
Cells and transfections.
293T cells (ATCC, CRL-11268) and HeLa cells (ATCC, CCL-2) were maintained in Dulbecco modified Eagle medium and Eagle minimum essential medium, respectively, supplemented with 10% fetal calf serum. 293 cells stably expressing human BST-2, CAT or an empty plasmid (pCDNA5/FRT) were generated by using the Flp-In system according to the manufacturer's instructions (Invitrogen). Stable clones were selected in hygromycin B (100 μg/ml) and analyzed by quantitative reverse transcription-PCR (qRT-PCR) and Western blot assays. 293T and 293/Flp-In cells were transfected by using Lipofectamine 2000 (Invitrogen).
siRNA knockdowns.
HeLa cells were transfected in 96-well plates (8,000 cells per well) with 80 nM small interfering RNA (siRNA) targeting human BST-2 (ON-TARGETplus, catalog no. L-011817-00), control siRNA (ON-TARGETplus nontargeting siRNA, catalog no. D-001810-04-05) (Thermo Scientific Dharmacon) or in the absence of siRNA using Lipofectamine 2000 (Invitrogen). Cells were infected with ZEBOV-GFP (61), MARV isolate Ci67, or LASV strain Josiah 24 h later.
VLP release assays.
293T cells in 12-well plates were transfected with 1 μg of plasmid encoding filoviral HA-VP40, arenaviral Z-HA, or NiV M-HA, together with 1 μg of empty vector or vector expressing enhanced green fluorescent protein (EGFP)-VPS4A E228Q or human or murine FLAG-BST-2. Alternatively, 293 cells stably expressing BST-2, CAT, or an empty plasmid were transfected with 2 μg of plasmid encoding filoviral HA-VP40, arenaviral Z-HA, or NiV M-HA. In rescue experiments, 293 cells stably expressing human BST-2 were transfected with plasmids encoding (i) ZEBOV HA-VP40 together with ZEBOV NP-V5, VP35-V5, VP30-V5, V5-VP24, GP1,2, GP1,2ΔMLD-V5, sGP-V5, ssGP-V5, or Δ-peptide-V5 or HIV-1 Vpu-V5; (ii) MARV HA-VP40 together with MARV GP1,2 or HIV-1 Vpu-V5; (iii) MACV Z-HA together with MACV NP-V5, GPC, or L-FLAG or HIV-1 Vpu-V5 or filoviral GP1,2; or (iv) LASV Z-HA together with LASV NP-V5, GPC, or SSP-V5 or HIV-1 Vpu-V5 or filoviral GP1,2. Cells were washed and supplemented with growth medium 2 h posttransfection. Cells and culture supernatants were collected for analysis 48 h later. Culture supernatants from transfected cells were clarified by low-speed centrifugation and passed through a 0.22-μm-pore-size filter (Millipore), and virus particles were pelleted through a 20% sucrose cushion at 22,000 × g at 4°C for 2 h (44, 48, 49, 66). Cells were detached with cell dissociation buffer (Invitrogen), washed with phosphate-buffered saline (PBS), and lysed with radioimmunoprecipitation assay lysis and extraction buffer (Thermo Scientific Pierce) supplemented with Complete protease inhibitor cocktail (Roche). Lysates were cleared by centrifugation at 22,000 × g at 4°C for 20 min, and FLAG-, HA-, and V5-tagged proteins were immunoprecipitated with EZview Red Anti-FLAG M2 affinity gel, EZview Red Anti-HA affinity gel, or anti-V5 Agarose affinity gel, respectively (Sigma). Pelleted VLPs and corresponding cell lysate immunoprecipitates were analyzed by SDS-PAGE and Western blotting using WesternBreeze chromogenic kits (Invitrogen) and murine monoclonal anti-FLAG-, anti-HA-, or anti-V5-alkaline phosphatase antibodies (Sigma/Invitrogen). Alternatively, expression of actin, LASV GPC, ZEBOV GP1,2, MARV GP1,2, and VPS4A E228Q was determined using murine monoclonal antibodies against actin (BD Biosciences), LASV GP1 (161-6), ZEBOV GP1,2 (6D8), or MARV GP1,2 (5D7) or using rabbit polyclonal antibodies against VPS4A (Santa Cruz Biotechnology). Semiquantitative analysis of Western blots was carried out by quantifying band intensities (given in arbitrary units) associated with released VLPs using ImageJ software (W. S. Rasband, ImageJ, U.S. National Institutes of Health [NIH], Bethesda, MD [http://rsb.info.nih.gov/ij/], 1997 to 2009).
Infection assays.
siRNA-treated HeLa cells in 96-well plates were infected with LASV Josiah strain (multiplicity of infection [MOI] = 0.1), ZEBOV-GFP (MOI = 10) (61), or MARV isolate Ci67 (MOI = 3). 293 cells stably expressing human BST-2, CAT, or an empty vector in 96-well plates were infected with LASV Josiah strain or ZEBOV isolate Mayinga, at MOIs of 0.5, 1, or 5. The inocula were removed 1 h later, and the cells were washed three times with PBS. Culture supernatants were harvested in TRIzol (Invitrogen) at 48 or 72 h postinfection, and the virion yield was determined by qRT-PCR. Alternatively, cells were fixed in 10% buffered formalin (Val Tech Diagnostics) for 72 h and stained for high-content quantitative image-based analysis with murine monoclonal antibodies against ZEBOV or MARV GP1,2 (6D8 or 9G4 antibodies, respectively), followed by Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen). Cells were also infected for 1 h with Rift Valley fever virus (RVFV) MP12 vaccine strain or eGFP-expressing cowpox virus (CPXV-GFP) at different MOIs. At 24 or 48 h postinfection, the cells were fixed with formalin for 72 h. RVFV-infected cells were stained with murine monoclonal antibody against the RVFV glycoprotein (4D4) (5, 30), followed by Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen). All infected cells were stained with Hoechst 33342 and HCS CellMask Red (Invitrogen).
qRT-PCR.
Total RNA from culture supernatants of untreated cells (mock) or cells infected with LASV or ZEBOV was prepared by using a MagMax 96 RNA extraction kit (Ambion). qRT-PCR assays were performed on an ABI Prism 7900HT sequence detection system with the RNA UltraSense one-step kit (Invitrogen) and TaqMan probes (Applied Biosystems) in accordance with the manufacturers' instructions. The final concentrations used in the 20-μl reaction mix contained 5 μl of RNA, 0.4 μM concentrations of each primer, 0.2 μM probe, 4 μl of 5× reaction mix, 0.4 μl of Rox, and 1 μl of enzyme mix. The reaction was run as follows: RT at 50°C for 20 min; initial denaturation at 95°C for 2 min; and then amplification for 40 cycles at 95°C for 15 s and 60°C for 30 s. Serial 10-fold dilutions of the assayed (102 to 107 copies) virus were used as standards. The sequences of the primers and probes used for determination of the virus copy number are listed in Table 1 .
TABLE 1.
Sequences of the primers and probes used for determination of virus copy number in qRT-PCR assays
| Primer or probe | Target gene | Sequence |
|---|---|---|
| ZEBOV-Forward | GP | TGGGCTGAAAACTGCTACAATC |
| ZEBOV-Reverse | GP | CTTTGTGCACATACCGGCAC |
| ZEBOV-Probe | GP | CTACCAGCAGCGCCAGACGG |
| LASV-Forward | GPC | GCAGTGCTGAAAGGTCTGTACAA |
| LASV-Reverse | GPC | AGGAGGAAAGTGACCAAACCAA |
| LASV-Probe | GPC | TTTGCAACGTGTGGCCT |
To determine the relative expression levels of BST-2 in siRNA-treated HeLa cells, total RNA was isolated 24, 48, and 72 h after transfection by using the RNeasy Plus minikit (Qiagen). Purified RNA was then measured by using the Quant-IT RiboGreen RNA assay kit (Invitrogen). Portions (50 ng) of the RNA were used in qRT-PCR assays using the conditions described above with BST-2 and GAPDH TaqMan probes (Applied Biosystems). Relative expression levels were determined by using the comparative CT method.
Immunofluorescence and confocal microscopy.
For immunofluorescence studies, 293 cells stably expressing human BST-2 were plated on BD BioCoat eight-chamber poly-d-lysine culture slides (Fisher Scientific) and infected with ZEBOV Mayinga isolate or MARV Ci67 isolate at MOIs of 0.5 or 5, respectively. At 24 or 48 h after infection, the cells were fixed in 10% buffered formalin for 72 h and blocked in 3% bovine serum albumin-PBS for 1 h. BST-2 was stained using rabbit anti-BST-2 antibody (Open Biosystems), followed by Alexa Fluor 647-conjugated goat anti-rabbit IgG (Invitrogen). MARV and ZEBOV GP1,2 were stained with murine monoclonal antibodies against ZEBOV or MARV GP1,2 (6D8 or 9G4 antibodies, respectively), followed by Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen). Nuclei were stained with Hoechst 33342. The confocal images were collected on a Leica TCS-SP5 confocal/multiphoton microscope.
High-content quantitative imaging data were acquired and analyzed on an Opera confocal reader (model 3842; quadruple excitation high sensitivity; Perkin-Elmer), at two exposures using a ×10 air objective lens. The first exposure utilized the 488-nm and 640-nm lasers to excite the viral and cell body fluorophores, respectively. The emission light was split by a 580-nm short-pass dichroic mirror and collected on separate cameras through 562/540-nm and 690/670-nm band-pass filters. The second exposure used a 425-nm long-pass mirror to steer the UV (350 nm) to the sample to excite the nuclear stain. The visible emission was directed to the nonconfocal camera by a 475-nm long-pass filter with the emission defined with a 450/450-nm band-pass filter. The optimal z-position for the 488-nm signal was determined by using the delta correction script. Analysis of the images was accomplished within the Opera environment using standard Acapella scripts.
GenBank accession numbers.
Sequence data can be found in the GenBank data libraries under accession numbers NM_198095 for murine BST-2, D28137 for human BST-2, P05923 for HIV-1 Vpu, NC_002549 for ZEBOV, DQ217792 for MARV, NC_005079 and AY571904 for MACV, AY628202 and AY628203 for LASV, and NC_002728 for NiV.
RESULTS
LASV, MACV, ZEBOV, MARV, and NiV VLP release is significantly decreased in murine or human BST-2-expressing 293T cells.
Expression of the matrix proteins of various enveloped viruses, including filoviruses (ZEBOV and MARV VP40), arenaviruses (LASV and MACV Z), and paramyxoviruses (NiV M), results in the formation of VLPs that are released from the cells in a similar fashion to HIV-1 Gag (6, 25, 52, 53, 59, 60). To examine the ability of BST-2 to inhibit budding of such VLPs, 293T cells, in which expression levels of endogenous BST-2 are minimal (49), were transiently transfected with HA-tagged arenaviral Z, filoviral VP40, or NiV M, together with FLAG-tagged human or murine BST-2. Transfection of an empty expression vector was used as a negative control, and transfection of a vector expressing an enzymatically inactive VPS4A mutant (E228Q) (21) was used as positive control since this cellular AAA-type ATPase has previously been demonstrated to be involved in budding of different enveloped viruses (21, 34, 37, 58, 62). Release of VLPs and expression levels of the different matrix proteins in cell lysates were evaluated by Western blot analysis. As shown in Fig. 1 A, expression of either murine or human BST-2 significantly decreased release of ZEBOV, MARV, LASV, MACV, and NiV VLPs. This inhibitory effect was not due to major differences in expression levels of the different matrix proteins (Fig. 1B). Furthermore, as expected, VPS4A E228Q decreased budding of the different VLPs to various extents with the exception of NiV, as previously reported (53). Figure 1B shows the expression levels of human and murine BST-2, as well as VPS4A E228Q and actin (loading control). Of note, BST-2 appears as multiple bands on the gel with estimated sizes of 21 to 35 kDa, presumably because of heterogeneous glycosylation (24).
FIG. 1.

BST-2 inhibits budding of arenaviral, filoviral, and paramyxoviral VLPs. (A) Human 293T cells were cotransfected with plasmids encoding hemagglutinin (HA)-tagged matrix proteins of LASV, MACV, ZEBOV, MARV, or NiV, together with an empty plasmid, or plasmid encoding VPS4A-E228Q, human BST-2 (hBST-2), or murine BST-2 (mBST-2). Cell lysates and supernatants were harvested 48 h after transfection. VLPs in clarified medium were pelleted through a sucrose cushion, and HA-tagged matrix proteins in VLPs were analyzed by Western blotting. Numbers below each lane indicate values obtained with densitometric scanning using the ImageJ program (NIH). (B) Expression of the HA-tagged matrix proteins, actin, VPS4A-E228Q, hBST-2, or mBST-2 in cell lysates was determined by Western blotting. Shown is a representative Western blot from three independent experiments.
To further study the effect of BST-2 on viral release, we established 293 cell lines stably expressing human BST-2, CAT or an empty plasmid. Again, stable expression of human BST-2 inhibited the budding of ZEBOV, MARV, LASV, MACV, and NiV VLPs, whereas expression of CAT had no effect on VLP release (Fig. 2 A). Similar expression levels of the different matrix proteins were observed in the cell lysates and could not account for the observed differences in VLP release (Fig. 2B). These data demonstrate the broad antiviral effects of both human and murine BST-2, which can inhibit release of VLPs of various human pathogens belonging to three viral families.
FIG. 2.

Release of arenaviral, filoviral, and paramyxoviral VLPs is reduced in cells stably expressing human BST-2. (A) Human 293 cells stably expressing human BST-2 (FLP-BST-2), CAT (FLP-CAT), or an empty plasmid (pCDNA5/FRT, FLP) were transfected with plasmids encoding HA-tagged matrix proteins of LASV, MACV, ZEBOV, MARV, or NiV. Cell lysates and supernatants were treated as in Fig. 1, and HA-tagged matrix proteins in VLPs were analyzed by Western blotting. Numbers below each lane indicate values obtained with densitometric scanning using the ImageJ program (NIH). (B) Expression of the HA-tagged matrix proteins and actin in cell lysates was determined by Western blot analysis. Shown is a representative Western blot from three independent experiments.
Filoviral GP1,2 but not arenaviral GPC counteracts BST-2 antiviral activity.
Filoviruses, which are negative-strand RNA viruses, encode six genes in addition to VP40 (NP, VP35, GP, VP30, VP24, and L). Cotranscriptional RNA editing of the ebolavirus, but not MARV, GP gene produces four proteins: GP1,2, sGP, ssGP, and Δ-peptide (31, 57, 64, 65). We sought to examine whether one of the ebolavirus-encoded proteins has a Vpu-like function and could rescue VLP release from cells that overexpress BST-2. To this end, we utilized a 293 cell line stably expressing human BST-2. Cells were transfected with plasmid encoding the matrix protein of ZEBOV (VP40), together with plasmids encoding other ebolavirus proteins (NP, VP35, VP30, VP24, GP1,2, sGP, ssGP, or Δ-peptide). Transfection of a plasmid expressing HIV-1 Vpu or an empty plasmid served as controls. As expected, release of ZEBOV VLPs was promoted by HIV-1 Vpu (Fig. 3 A) (29). Of the eight Ebolavirus proteins, only GP1,2 rescued ZEBOV VLP release to levels similar to those observed in Vpu-expressing cells. However, as GP1,2 expression was previously shown to enhance the efficiency of VP40-driven VLP release in 293T cells (36, 50), we wanted to examine whether ZEBOV GP1,2 is a bona fide BST-2 antagonist or whether the observed increase in VLP production is due to coexpression of VP40 and GP1,2. To this end, we compared the increase in VLP release between 293 cells that express minimal levels of BST-2 (FLP) and 293 cells stably expressing BST-2 (FLP-BST-2). The increase in ZEBOV VLP production upon expression of GP1,2 in FLP-BST-2 was significantly higher than that observed in FLP cells, indicating that ZEBOV GP1,2 is indeed a BST-2 antagonist (Fig. 4).
FIG. 3.

Filoviral GP1,2, but none of the arenavirus-encoded proteins, counteract human BST-2. (A) Human 293 cells stably expressing human BST-2 were cotransfected with ZEBOV HA-VP40 DNA and plasmids encoding ZEBOV VP30-V5, VP35-V5, V5-VP24, GP1,2, GP1,2ΔMLD-V5, sGP-V5, ssGP-V5, Δ-peptide-V5, or NP-V5 or HIV-1 Vpu-V5. Alternatively, cells were transfected with MARV HA-VP40 DNA, together with plasmids encoding MARV GP1,2 or HIV-1 Vpu-V5. Cell lysates and supernatants were treated as in Fig. 1, and HA-tagged VP40 in VLPs were analyzed by Western blotting. Numbers below each lane indicate values obtained with densitometric scanning using the ImageJ program (NIH). (B) Expression of HA-tagged VP40, actin, and other filovirus-encoded proteins in cell lysates was determined by Western blotting. Expression of HIV-1 Vpu (∼16 kDa), ZEBOV VP30 (∼30 kDa), VP35 (∼35 kDa), VP24 (∼24 kDa), GP1,2 ΔMLD (∼75 kDa), sGP (∼50 kDa), ssGP (∼47 kDa), Δ-peptide (∼10 kDa), and NP (∼104 kDa) was detected using anti-V5 antibody. Expression of ZEBOV (∼140 kDa) and MARV (∼170 kDa) GP1,2 was detected using anti-GP antibodies (6D8 and 5D7, respectively). (C) Human 293 cells stably expressing human BST-2 were cotransfected with LASV Z-HA DNA and plasmids encoding LASV NP-V5, SSP-V5, or GPC or HIV-1 Vpu-V5. Alternatively, cells were transfected with MACV Z-HA and MACV NP-V5, GPC, L-FLAG, or HIV-1 Vpu-V5. Cell lysates and supernatants were treated as in Fig. 1, and HA-tagged Z proteins in VLPs were analyzed by Western blotting. Numbers below each lane indicate values obtained with densitometric scanning using the ImageJ program (NIH). (D) Expression of HA-tagged Z, actin, and other arenavirus-encoded proteins in cell lysates was determined by Western blotting. Expression of HIV-1 Vpu (∼16 kDa), arenaviral NP (∼63 kDa), and LASV SSP (∼10 kDa) was detected by using anti-V5 antibody. Expression of MACV L-FLAG (∼200 kDa) was detected using anti-FLAG antibody. Expression of LASV GPC (∼75 kDa) and GP1 (∼42 kDa) was detected with an antibody against GP1 (161-6). Shown is a representative Western blot from three independent experiments.
FIG. 4.

The increase in ZEBOV VLP release when GP1,2 is expressed in the presence of BST-2 is significantly higher than that observed in the absence of BST-2. (A) Human 293 cells stably expressing human BST-2 (FLP-BST-2) or an empty plasmid (FLP) were transfected with ZEBOV VP40, together with an empty plasmid or a plasmid encoding GP1,2, or HIV-1 Vpu. Cell lysates and supernatants were treated as in Fig. 1, and HA-tagged VP40 in VLPs was analyzed by Western blotting. Numbers below each lane indicate values obtained with densitometric scanning using the ImageJ program (NIH). (B) Expression of the HA-tagged VP40, actin, GP1,2, and HIV-1 Vpu-V5 in cell lysates was determined by Western blot analysis. Shown is a representative Western blot from three independent experiments.
We also tested a GP1,2 deletion variant lacking the mucin-like domain (GP1,2ΔMLD) for its ability to function as a BST-2 antagonist. This variant was almost as potent as wild-type GP1,2 in promoting ZEBOV VLP release, suggesting that the mucin-like domain is most likely dispensable for this function (Fig. 3A). NP, VP35, and VP24 were previously reported to influence viral release (27, 36). In this assay, however, their effect was less pronounced compared to GP1,2 (Fig. 3A). Expression of ZEBOV VP40, as well as the different ebolavirus proteins and HIV-1 Vpu in the cell lysates, are shown in Fig. 3B (upper and bottom panels, respectively).
We next examined whether MARV GP1,2 was also capable of rescuing VP40-mediated VLP release in this system. 293 cells stably expressing human BST-2 were transfected with a plasmid encoding MARV VP40, together with plasmids encoding MARV GP1,2 or HIV-1 Vpu, or an empty plasmid. As shown in Fig. 3A, MARV GP1,2, akin to ZEBOV GP1,2, could promote MARV VLP release to levels similar to those observed in Vpu-expressing cells. Overall, these results confirm that filoviral GP1,2 acts as a BST-2 antagonist (29).
We also tested arenavirus-encoded proteins for their ability to counteract BST-2. In addition to Z, arenaviruses encode three other genes: NP, GPC, and L (10, 13, 26). The arenaviral envelope glycoprotein (GPC) is the filoviral GP1,2 counterpart. The stable signal peptide (SSP) of GPC plays unique roles in the proteolytic maturation of GPC, intracellular trafficking of GPC from the ER to the cell surface, and arenaviral pH-dependent membrane fusion during entry (1, 17, 18). In contrast to filoviral GP1,2, arenaviral GPC did not promote arenaviral VLP release (Fig. 3C). Similar results were obtained with the other arenavirus-encoded proteins (LASV NP and SSP, and MACV NP and L), which, compared to HIV-1 Vpu, had less or no effect on VLP release. The expression of arenaviral Z in cell lysates is shown in Fig. 3D (upper panel), as well as the expression of other arenavirus-encoded proteins (lower panel). These results demonstrate that, unlike filoviruses and retroviruses, arenaviruses do not encode a BST-2 antagonist.
Finally, we wanted to examine whether filoviral GP1,2, similarly to HIV-1 Vpu, is a general BST-2 antagonist that can overcome the BST-2 antiviral effect not only in the context of filovirus release but also in the context of release of other BST-2-sensitive viruses. To this end, we tested the ability of filoviral GP1,2 to rescue arenavirus VLP production when BST-2 is expressed and compared it to HIV-1 Vpu. 293 cells stably expressing human BST-2 were transfected with plasmids encoding the matrix protein of arenaviruses (LASV or MACV Z), together with an empty plasmid or plasmid encoding HIV-1 Vpu, ZEBOV GP1,2, or MARV GP1,2. HIV-1 Vpu promoted arenavirus VLP release, as expected, whereas the filoviral glycoprotein had less or no effect on MACV or LASV VLP release, respectively (Fig. 5). These results suggest that, in the context of arenavirus VLP release, the filoviral glycoprotein cannot counteract the antiviral activity of BST-2. Of note, expression of all matrix proteins in cell lysates was reduced whenever HIV-1 Vpu was expressed. We believe that this is because Vpu enhances matrix protein release as VLPs.
FIG. 5.
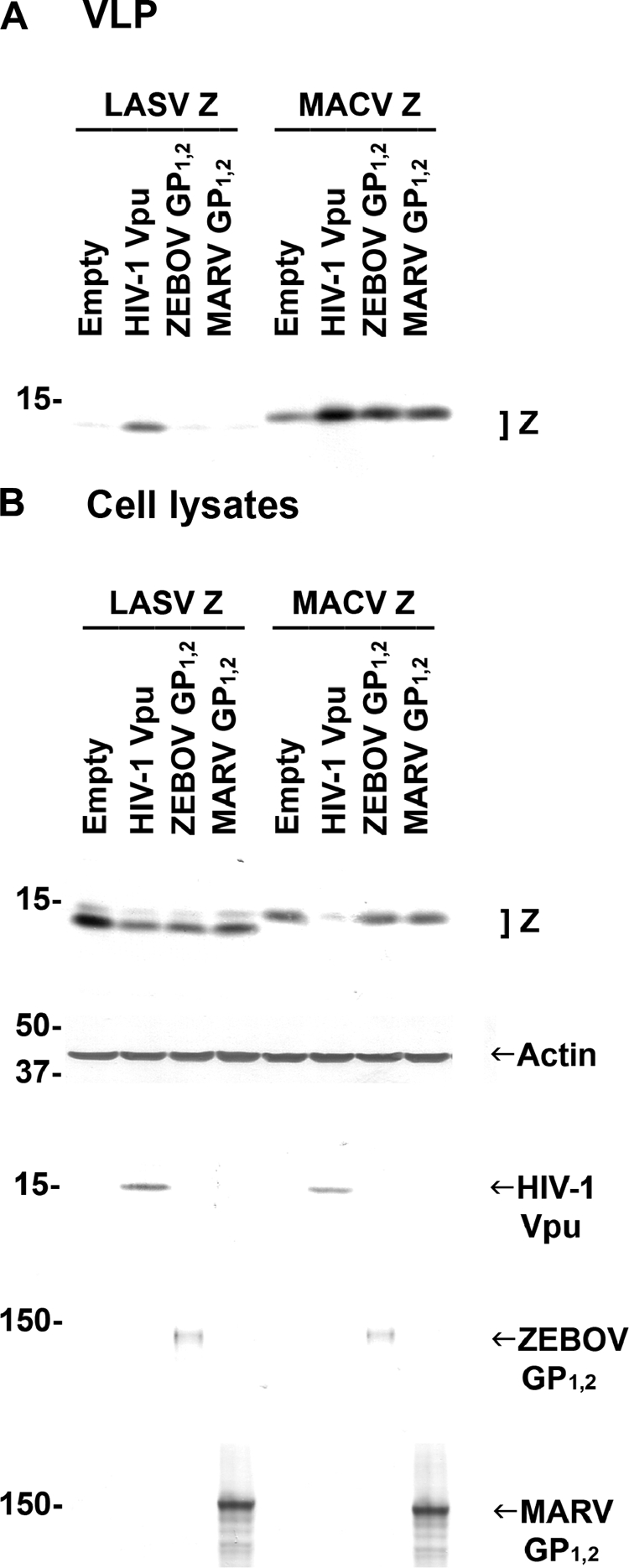
Expression of filoviral GP1,2 has little or no effect on release of arenaviral VLPs in BST-2-expressing cells. (A) Human 293 cells stably expressing human BST-2 were cotransfected with arenaviral Z-HA DNA and an empty plasmid or plasmids encoding filoviral GP1,2 or HIV-1 Vpu-V5. Cell lysates and supernatants were treated as in Fig. 1, and HA-tagged Z proteins in VLPs were analyzed by Western blotting. (B) Expression of HA-tagged Z, actin, HIV Vpu-V5, and filoviral GP1,2 in cell lysates was determined by Western blotting. Shown is a representative Western blot from three independent experiments.
Effect of human BST-2 overexpression on the release and spread of infectious LASV, ZEBOV, RVFV, and CPXV.
VLPs are a valuable system for initial studies of viral budding. However, the system is simplistic and uses only one or few viral proteins in the absence of other virus-encoded proteins or genomic elements. Therefore, any results obtained with this system should not be extrapolated until verified with infectious virus. To this end, 293 cell lines stably expressing human BST-2, CAT, or an empty plasmid were infected with one representative virus of the family Arenaviridae (LASV) or one representative virus of the family Filoviridae (ZEBOV) at different MOIs. Viral RNA was extracted from the media, and the viral copy number was determined by qRT-PCR. Alternatively, the cells were fixed in formalin and stained for high-content quantitative image-based analysis with virus-specific antibodies. Stable BST-2 expression reduced LASV virion yield by >10-fold, whereas CAT expression had no effect on LASV copy number (Fig. 6 A). Conversely, BST-2 expression had no effect on replication and the spread of infectious ZEBOV (Fig. 6B) or on virus release into the supernatant, as determined by qRT-PCR (data not shown).
FIG. 6.

Human BST-2 overexpression inhibits infectious LASV, but not ZEBOV, release and spread. (A) Human 293 cells stably expressing human BST-2 (FLP-BST-2), CAT (FLP-CAT), or an empty plasmid (FLP) were infected with LASV at the indicated MOIs. After 72 h, viral RNA was extracted from the media, and the viral copy number was determined by qRT-PCR. (B to D) Alternatively, cells were infected with ZEBOV (B), CPXV-GFP (C), or RVFV (D), fixed in formalin and stained for high-content quantitative image-based analysis with virus-specific antibodies 24 or 48 h (C and D) or 72 h (B) after infection. Error bars indicate standard deviations.
We then evaluated the antiviral activity of human BST-2 against RNA viruses that lack a structural matrix protein or DNA viruses. The effect of BST-2 overexpression on replication of RVFV, a representative RNA virus of the family Bunyaviridae, and on cowpox virus (CPXV), a representative DNA virus of the family Poxviridae, was examined. As shown in Fig. 6C and D, neither RVFV nor CPXV replication and spread was significantly affected by BST-2 overexpression.
To summarize, in the context of the viruses tested here, only infectious arenaviruses (LASV) are restricted by human BST-2. These results are not surprising since filoviral GP1,2 can counteract BST-2 antiviral activity, whereas no arenavirus-encoded protein has such a function (Fig. 3).
Effect of human BST2 knockdown on the release and spread of infectious LASV, ZEBOV, and MARV.
To further verify our overexpression studies, we chose an alternative and complementary siRNA knockdown approach to test BST-2's antiviral activity. To this end, HeLa cells, which endogenously express high levels of BST-2 (49), were transfected with siRNA targeting BST-2 or a control siRNA. As shown in Fig. 7 A, the expression of human BST-2 was effectively knocked down 24, 48, or 72 h posttransfection. siRNA targeting BST-2, but not control siRNAs, enhanced the yield of LASV from HeLa cells by almost 2-fold (Fig. 7B). In contrast, only negligible effects on filovirus replication and spread were observed under these conditions (Fig. 7C). These results further demonstrate that in the context of infectious viruses, BST-2 restricts only arenaviruses (LASV) and not filoviruses.
FIG. 7.

Knockdown of human BST-2 expression enhances infectious LASV release, but not ZEBOV or MARV spread. (A) HeLa cells were transfected with siRNA targeting BST-2 or with a control siRNA. After 24, 48, or 72 h, cellular RNA was extracted, and the relative BST-2 expression levels were determined by qRT-PCR. BST-2 protein expression levels in cell lysates were also determined by Western blotting. (B) Cells were infected with LASV (MOI = 0.1) 24 h after transfection. Viral RNA was extracted from the medium 72 h later, and the viral copy number was determined by qRT-PCR. (C) Alternatively, cells were infected with ZEBOV-GFP (MOI = 10) or MARV (MOI = 3). Cells were fixed in formalin 72 h later and stained for high-content quantitative image-based analysis with virus-specific antibodies. Error bars indicate standard deviations.
Localization of human BST2 and filoviral GP1,2 during filovirus infection.
To begin to delineate the mechanism by which filoviral GP1,2 antagonizes BST-2, 293 cells stably expressing human BST-2 were infected with MARV or ZEBOV for 24 or 48 h and stained for immunofluorescence analysis with antibodies against BST-2 and antibodies against ZEBOV or MARV GP1,2. Colocalization of BST-2 with ZEBOV or MARV GP1,2 was not observed under any of these conditions (Fig. 8). Instead, BST-2 and the filoviral glycoprotein exhibited complementary distribution at the plasma membrane. Areas with the highest BST-2 staining showed the lowest filoviral GP1,2 staining and vice versa.
FIG. 8.

BST-2 localization in relation to filoviral GP1,2. (A) Human 293 cells stably expressing human BST-2 were infected with ZEBOV. Cells were fixed in formalin 24 h later and stained with antibodies against BST-2 and ZEBOV GP1,2. (B and C) Alternatively, cells were infected with MARV for 24 h (B) or 48 h (C) and stained with antibodies against BST-2 and MARV GP1,2. Immunofluorescence of anti-filoviral GP1,2 (green) and anti-BST-2 (red) in a single confocal plain is shown as indicated. Right columns show the merged images for triple staining of BST-2 (red), filoviral GP1,2 (green), and the nucleus (blue), as well as bright-field images.
DISCUSSION
Previous studies demonstrated restriction of ZEBOV, MARV, and LASV VLP release by BST-2 using minimal viral components (28, 29, 56). However, none has examined the effect of BST-2 expression levels on the release and spread of infectious viruses. We found that knockdown or overexpression of BST-2 increased and decreased infectious LASV release, respectively (Fig. 6A and Fig. 7B). These results are in accordance with our data showing that arenaviruses do not encode a BST-2 antagonist (Fig. 3). It is interesting to speculate how arenaviruses overcome BST-2 restriction in the absence of a direct antagonism of its function. Since BST-2 is an effector protein of the IFN-induced antiviral response, one possibility is that arenaviruses target BST-2 indirectly by attenuating type I IFN production. Indeed, Martínez-Sobrido et al. showed that arenaviral nucleoprotein (NP) could inhibit the induction of type I IFN (41, 42), which could in turn prevent stimulation of BST-2 expression. Furthermore, it is plausible that arenaviruses target cells that express low levels of BST-2 in the absence of IFN induction. Human dendritic cells, macrophages, and endothelial cells are known to be such arenaviral targets (2, 39). BST-2 expression is low to undetectable in these cells but can be induced by IFN-α (12, 46). Therefore, we hypothesize that arenaviruses target cells that express minimal levels of BST-2 and prevent its further induction by NP-mediated inhibition of type I IFN production. Thus, even in the absence of a direct BST-2 antagonist, arenaviruses could still multiply in their host(s) with minimal deleterious effects of this antiviral protein. Several observations support this hypothesis and show that type I IFNs are crucial in the control of arenaviruses. First, whereas especially high arenavirus titers could be obtained from infected dendritic cells, lower titers were obtained from infected macrophages. In accordance with this, no type I IFN production was observed in dendritic cells, whereas a moderate response was observed in macrophages. In addition, exogenous activation of arenavirus-infected macrophages or dendritic cells, as well as recombinant type I IFN, strongly inhibited viral replication (3).
We also show that BST-2 inhibits the release of a variety of VLPs, generated by the expression of arenaviral (LASV and MACV), filoviral (ZEBOV and MARV), or paramyxoviral (NiV) matrix proteins (Fig. 1 and 2). BST-2's antiviral activity against retroviral VLPs and Vpu-deficient HIV-1 has been demonstrated previously (28, 49, 63). The matrix proteins of these divergent viruses show no sequence homology and are targeted to the cell membrane by different mechanisms. The only commonality shown for filoviruses, NiV, and retroviruses is that all assemble and bud from plasma membrane microdomains termed lipid rafts or detergent-resistant membranes (6, 7, 14, 35, 51). BST-2 also localizes to lipid raft microdomains (33). Overall, these data suggest that BST-2 is a broad nonspecific inhibitor of VLP release, whose effect might depend on colocalization with nascent virions in plasma membrane lipid rafts.
We found that filoviral GP1,2, similar to HIV-1 Vpu, rescued VP40-mediated VLP release from cells overexpressing human BST-2 (Fig. 3A). These results are in agreement with our studies with infectious filoviruses, showing that various expression levels of BST-2 had no or minimal effect on filoviral spread (Fig. 6B and Fig. 7C) and release (data not shown). Note that none of our assays demonstrated that filoviral GP1,2 counteracted BST-2 directly. Although our confocal microscopy studies show complementary staining of BST-2 and filoviral GP1,2 rather than colocalization, they do not necessarily rule out the possibility of such interaction (Fig. 8). A recent study by Kaletsky et al. (29) demonstrated that, reminiscent of HIV-1 Vpu (16, 49), ebolaviral GP1,2 indeed specifically interacts with BST-2. Nevertheless, the mechanism by which this filoviral protein antagonizes BST-2 remains under investigation. In the case of HIV-1 Vpu, several studies demonstrated that Vpu directs the degradation of human BST-2 via a β-TrCP-dependent mechanism. Vpu acts as an adapter molecule linking BST-2 to the β-TrCP/SCF E3 ubiquitin ligase complex to induce endolysosomal trafficking and subsequent lysosomal BST-2 degradation. Through this action, HIV-1 Vpu removes BST-2 from its site of action as a virion-tethering factor (16, 45). Studies are under way to determine whether filoviral GP1,2 also functions via a β-TrCP-dependent mechanism. If filoviral GP1,2, akin to HIV-1 Vpu, overcomes BST-2 restriction by a mechanism involving BST-2 degradation, a rather transient interaction between these two proteins could be expected that would be rather hard to detect. Such a mechanism would explain why we did not observe BST-2 where filoviral GP1,2 is localized: BST-2 in these areas might have been degraded, whereas in membrane domains where filoviral GP1,2 is absent, BST-2 could still be present.
Replication and spread of RVFV, a representative bunyavirus that lacks a matrix protein, and CPXV, a representative DNA virus, were not affected by human BST-2 overexpression (Fig. 6C and Fig. 6D). It is possible that, like HIV-1, SIV, and filoviruses (29, 49, 63, 66), these viruses encode BST-2 antagonists that rescue viral release from BST-2-mediated restriction. Recently, a DNA virus, human herpesvirus 8 (HHV-8; also known as Kaposi's sarcoma-associated virus), has been shown to encode such a BST-2 antagonist. The HHV-8-encoded K5 protein is a RING-CH ubiquitin ligase that ubiquitinylates BST-2, leading to its rapid degradation (4, 40). Furthermore, HHV-8 release is decreased in the absence of K5 in a BST-2-dependent manner, suggesting that BST-2's antiviral activity is not limited to RNA viruses. It would be of interest to examine whether other DNA viruses, such as poxviruses, encode for similar BST-2 antagonists. Alternatively, it is plausible that RVFV and CPXV bud from membranes or membrane domains that lack BST-2. Such a scenario might indicate that the intrinsic properties of viral structural proteins, in particular the specific membrane microdomains to which they are targeted and where assembly and budding takes place, might influence BST-2 sensitivity.
Acknowledgments
Work performed at the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) was supported by grant 4.10022_08_RD_B from the Joint Science and Technology Office for Chemical and Biological Defense.
The opinions, interpretations, conclusions, and recommendations included here are those of the authors and are not necessarily endorsed by the U.S. Army or the Department of Health and Human Services.
We thank Steven Bradfute (USAMRIID, Fort Detrick, Frederick, MD) for useful advice.
Footnotes
Published ahead of print on 4 August 2010.
REFERENCES
- 1.Agnihothram, S. S., J. York, and J. H. Nunberg. 2006. Role of the stable signal peptide and cytoplasmic domain of G2 in regulating intracellular transport of the Junin virus envelope glycoprotein complex. J. Virol. 80:5189-5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baize, S., J. Kaplon, C. Faure, D. Pannetier, M. C. Georges-Courbot, and V. Deubel. 2004. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 172:2861-2869. [DOI] [PubMed] [Google Scholar]
- 3.Baize, S., D. Pannetier, C. Faure, P. Marianneau, I. Marendat, M. C. Georges-Courbot, and V. Deubel. 2006. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microbes Infect. 8:1194-1202. [DOI] [PubMed] [Google Scholar]
- 4.Bartee, E., A. McCormack, and K. Fruh. 2006. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Battles, J. K., and J. M. Dalrymple. 1988. Genetic variation among geographic isolates of Rift Valley fever virus. Am. J. Trop. Med. Hyg. 39:617-631. [DOI] [PubMed] [Google Scholar]
- 6.Bavari, S., C. M. Bosio, E. Wiegand, G. Ruthel, A. B. Will, T. W. Geisbert, M. Hevey, C. Schmaljohn, A. Schmaljohn, and M. J. Aman. 2002. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 195:593-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhattacharya, J., A. Repik, and P. R. Clapham. 2006. Gag regulates association of human immunodeficiency virus type 1 envelope with detergent-resistant membranes. J. Virol. 80:5292-5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blasius, A. L., E. Giurisato, M. Cella, R. D. Schreiber, A. S. Shaw, and M. Colonna. 2006. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 177:3260-3265. [DOI] [PubMed] [Google Scholar]
- 9.Bossart, K. N., and C. C. Broder. 2006. Developments toward effective treatments for Nipah and Hendra virus infection. Expert Rev. Anti-Infect. Ther. 4:43-55. [DOI] [PubMed] [Google Scholar]
- 10.Buchmeier, M. J. 2002. Arenaviruses: protein structure and function. Curr. Top. Microbiol. Immunol. 262:159-173. [DOI] [PubMed] [Google Scholar]
- 11.Campbell, S., and V. M. Vogt. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 69:6487-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao, W., L. Bover, M. Cho, X. Wen, S. Hanabuchi, M. Bao, D. B. Rosen, Y. H. Wang, J. L. Shaw, Q. Du, C. Li, N. Arai, Z. Yao, L. L. Lanier, and Y. J. Liu. 2009. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J. Exp. Med. 206:1603-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charrel, R. N., and X. de Lamballerie. 2003. Arenaviruses other than Lassa virus. Antivir. Res. 57:89-100. [DOI] [PubMed] [Google Scholar]
- 14.Choi, D. Y., C. Kwan, M. Cerrato, and H. C. Aguilar. 2008. Determinants of viral assembly for the Nipah virus matrix, fusion, and attachment proteins. J. Invest. Med. 56:103. [Google Scholar]
- 15.Chua, K. B., W. J. Bellini, P. A. Rota, B. H. Harcourt, A. Tamin, S. K. Lam, T. G. Ksiazek, P. E. Rollin, S. R. Zaki, W. Shieh, C. S. Goldsmith, D. J. Gubler, J. T. Roehrig, B. Eaton, A. R. Gould, J. Olson, H. Field, P. Daniels, A. E. Ling, C. J. Peters, L. J. Anderson, and B. W. Mahy. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432-1435. [DOI] [PubMed] [Google Scholar]
- 16.Douglas, J. L., K. Viswanathan, M. N. McCarroll, J. K. Gustin, K. Fruh, and A. V. Moses. 2009. Vpu directs the degradation of the HIV restriction factor BST-2/tetherin via a βTrCP-dependent mechanism. J. Virol. 83:7931-7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eichler, R., O. Lenz, T. Strecker, M. Eickmann, H. D. Klenk, and W. Garten. 2003. Identification of Lassa virus glycoprotein signal peptide as a trans-acting maturation factor. EMBO Rep. 4:1084-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eichler, R., O. Lenz, T. Strecker, and W. Garten. 2003. Signal peptide of Lassa virus glycoprotein GP-C exhibits an unusual length. FEBS Lett. 538:203-206. [DOI] [PubMed] [Google Scholar]
- 19.Eichler, R., T. Strecker, L. Kolesnikova, J. ter Meulen, W. Weissenhorn, S. Becker, H. D. Klenk, W. Garten, and O. Lenz. 2004. Characterization of the Lassa virus matrix protein Z: electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP). Virus Res. 100:249-255. [DOI] [PubMed] [Google Scholar]
- 20.Garcea, R. L., and L. Gissmann. 2004. Virus-like particles as vaccines and vessels for the delivery of small molecules. Curr. Opin. Biotechnol. 15:513-517. [DOI] [PubMed] [Google Scholar]
- 21.Garrus, J. E., U. K. von Schwedler, O. W. Pornillos, S. G. Morham, K. H. Zavitz, H. E. Wang, D. A. Wettstein, K. M. Stray, M. Cote, R. L. Rich, D. G. Myszka, and W. I. Sundquist. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55-65. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez, J. P., S. Emonet, X. de Lamballerie, and R. Charrel. 2007. Arenaviruses. Curr. Top. Microbiol. Immunol. 315:253-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haller, O., G. Kochs, and F. Weber. 2006. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344:119-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishikawa, J., T. Kaisho, H. Tomizawa, B. O. Lee, Y. Kobune, J. Inazawa, K. Oritani, M. Itoh, T. Ochi, K. Ishihara, et al. 1995. Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics 26:527-534. [DOI] [PubMed] [Google Scholar]
- 25.Jasenosky, L. D., G. Neumann, I. Lukashevich, and Y. Kawaoka. 2001. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 75:5205-5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jay, M. T., C. Glaser, and C. F. Fulhorst. 2005. The arenaviruses. J. Am. Vet. Med. Assoc. 227:904-915. [DOI] [PubMed] [Google Scholar]
- 27.Johnson, R. F., P. Bell, and R. N. Harty. 2006. Effect of Ebola virus proteins GP, NP, and VP35 on VP40 VLP morphology. Virol. J. 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jouvenet, N., S. J. Neil, M. Zhadina, T. Zang, Z. Kratovac, Y. Lee, M. McNatt, T. Hatziioannou, and P. D. Bieniasz. 2009. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 83:1837-1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaletsky, R. L., J. R. Francica, C. Agrawal-Gamse, and P. Bates. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 106:2886-2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keegan, K., and M. S. Collett. 1986. Use of bacterial expression cloning to define the amino acid sequences of antigenic determinants on the G2 glycoprotein of Rift Valley fever virus. J. Virol. 58:263-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhn, J. H. 2008. Filoviruses: a compendium of 40 years of epidemiological, clinical, and laboratory studies. Archives of virology supplement, vol. 20. Springer, Vienna, Austria. [PubMed]
- 32.Kuhn, J. H., S. R. Radoshitzky, A. C. Guth, K. L. Warfield, W. Li, M. J. Vincent, J. S. Towner, S. T. Nichol, S. Bavari, H. Choe, M. J. Aman, and M. Farzan. 2006. Conserved receptor-binding domains of Lake Victoria marburgvirus and Zaire ebolavirus bind a common receptor. J. Biol. Chem. 281:15951-15958. [DOI] [PubMed] [Google Scholar]
- 33.Kupzig, S., V. Korolchuk, R. Rollason, A. Sugden, A. Wilde, and G. Banting. 2003. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4:694-709. [DOI] [PubMed] [Google Scholar]
- 34.Lambert, C., T. Doring, and R. Prange. 2007. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and gamma 2-adaptin. J. Virol. 81:9050-9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung, K., J. O. Kim, L. Ganesh, J. Kabat, O. Schwartz, and G. J. Nabel. 2008. HIV-1 assembly: viral glycoproteins segregate quantally to lipid rafts that associate individually with HIV-1 capsids and virions. Cell Host Microbe 3:285-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Licata, J. M., R. F. Johnson, Z. Han, and R. N. Harty. 2004. Contribution of Ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J. Virol. 78:7344-7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Licata, J. M., M. Simpson-Holley, N. T. Wright, Z. Han, J. Paragas, and R. N. Harty. 2003. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77:1812-1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludwig, C., and R. Wagner. 2007. Virus-like particles-universal molecular toolboxes. Curr. Opin. Biotechnol. 18:537-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lukashevich, I. S., R. Maryankova, A. S. Vladyko, N. Nashkevich, S. Koleda, M. Djavani, D. Horejsh, N. N. Voitenok, and M. S. Salvato. 1999. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: different effects on IL-8 and TNF-alpha gene expression. J. Med. Virol. 59:552-560. [PMC free article] [PubMed] [Google Scholar]
- 40.Mansouri, M., K. Viswanathan, J. L. Douglas, J. Hines, J. Gustin, A. V. Moses, and K. Fruh. 2009. Molecular mechanism of BST2/Tetherin downregulation by K5/MIR2 of Kaposi's sarcoma herpesvirus. J. Virol. 83:9672-9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Sobrido, L., P. Giannakas, B. Cubitt, A. Garcia-Sastre, and J. C. de la Torre. 2007. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 81:12696-12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez-Sobrido, L., E. I. Zuniga, D. Rosario, A. Garcia-Sastre, and J. C. de la Torre. 2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 80:9192-9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marty, A. M., P. B. Jahrling, and T. W. Geisbert. 2006. Viral hemorrhagic fevers. Clin. Lab. Med. 26:345-386. [DOI] [PubMed] [Google Scholar]
- 44.McNatt, M. W., T. Zang, T. Hatziioannou, M. Bartlett, I. B. Fofana, W. E. Johnson, S. J. Neil, and P. D. Bieniasz. 2009. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 5:e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell, R. S., C. Katsura, M. A. Skasko, K. Fitzpatrick, D. Lau, A. Ruiz, E. B. Stephens, F. Margottin-Goguet, R. Benarous, and J. C. Guatelli. 2009. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5:e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyagi, E., A. J. Andrew, S. Kao, and K. Strebel. 2009. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface downmodulation and intracellular depletion. Proc. Natl. Acad. Sci. U. S. A. 106:2868-2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morikawa, Y., T. Goto, and K. Sano. 1999. In vitro assembly of human immunodeficiency virus type 1 Gag protein. J. Biol. Chem. 274:27997-28002. [DOI] [PubMed] [Google Scholar]
- 48.Neil, S. J., S. W. Eastman, N. Jouvenet, and P. D. Bieniasz. 2006. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neil, S. J., T. Zang, and P. D. Bieniasz. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425-430. [DOI] [PubMed] [Google Scholar]
- 50.Noda, T., H. Sagara, E. Suzuki, A. Takada, H. Kida, and Y. Kawaoka. 2002. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J. Virol. 76:4855-4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ono, A., and E. O. Freed. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. U. S. A. 98:13925-13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patch, J. R., G. Crameri, L. F. Wang, B. T. Eaton, and C. C. Broder. 2007. Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol. J. 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patch, J. R., Z. Han, S. E. McCarthy, L. Yan, L. F. Wang, R. N. Harty, and C. C. Broder. 2008. The YPLGVG sequence of the Nipah virus matrix protein is required for budding. Virol. J. 5:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radoshitzky, S. R., J. Abraham, C. F. Spiropoulou, J. H. Kuhn, D. Nguyen, W. Li, J. Nagel, P. J. Schmidt, J. H. Nunberg, N. C. Andrews, M. Farzan, and H. Choe. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446:92-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rollason, R., V. Korolchuk, C. Hamilton, P. Schu, and G. Banting. 2007. Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J. Cell Sci. 120:3850-3858. [DOI] [PubMed] [Google Scholar]
- 56.Sakuma, T., T. Noda, S. Urata, Y. Kawaoka, and J. Yasuda. 2009. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 83:2382-2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanchez, A., S. G. Trappier, B. W. Mahy, C. J. Peters, and S. T. Nichol. 1996. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U. S. A. 93:3602-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Silvestri, L. S., G. Ruthel, G. Kallstrom, K. L. Warfield, D. L. Swenson, T. Nelle, P. L. Iversen, S. Bavari, and M. J. Aman. 2007. Involvement of vacuolar protein sorting pathway in Ebola virus release independent of TSG101 interaction. J. Infect. Dis. 196(Suppl. 2):S264-S270. [DOI] [PubMed] [Google Scholar]
- 59.Strecker, T., R. Eichler, J. Meulen, W. Weissenhorn, H. Dieter Klenk, W. Garten, and O. Lenz. 2003. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles [corrected]. J. Virol. 77:10700-10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swenson, D. L., K. L. Warfield, K. Kuehl, T. Larsen, M. C. Hevey, A. Schmaljohn, S. Bavari, and M. J. Aman. 2004. Generation of Marburg virus-like particles by coexpression of glycoprotein and matrix protein. FEMS Immunol. Med. Microbiol. 40:27-31. [DOI] [PubMed] [Google Scholar]
- 61.Towner, J. S., J. Paragas, J. E. Dover, M. Gupta, C. S. Goldsmith, J. W. Huggins, and S. T. Nichol. 2005. Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology 332:20-27. [DOI] [PubMed] [Google Scholar]
- 62.Urata, S., T. Noda, Y. Kawaoka, H. Yokosawa, and J. Yasuda. 2006. Cellular factors required for Lassa virus budding. J. Virol. 80:4191-4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Damme, N., D. Goff, C. Katsura, R. L. Jorgenson, R. Mitchell, M. C. Johnson, E. B. Stephens, and J. Guatelli. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Volchkov, V. E., S. Becker, V. A. Volchkova, V. A. Ternovoj, A. N. Kotov, S. V. Netesov, and H. D. Klenk. 1995. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 214:421-430. [DOI] [PubMed] [Google Scholar]
- 65.Volchkova, V. A., H. D. Klenk, and V. E. Volchkov. 1999. Delta-peptide is the carboxy-terminal cleavage fragment of the nonstructural small glycoprotein sGP of Ebola virus. Virology 265:164-171. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, F., S. J. Wilson, W. C. Landford, B. Virgen, D. Gregory, M. C. Johnson, J. Munch, F. Kirchhoff, P. D. Bieniasz, and T. Hatziioannou. 2009. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6:54-67. [DOI] [PMC free article] [PubMed] [Google Scholar]