

Abstract
O6-Alkylguanine-DNA alkyltransferase (AGT) mediates tumor resistance to alkylating agents that generate guanine O6-chloroethyl (Onrigin™ and carmustine) and O6-methyl (temozolomide) lesions; however, the relative efficiency of AGT protection against these lesions and the degree of resistance to these agents that a given number of AGT molecules produces are unclear. Measured from differential cytotoxicity in AGT-ablated and AGT-intact HL-60 cells containing 17,000 AGT molecules/cell, AGT produced 12- and 24-fold resistance to chloroethylating (90CE) and methylating (KS90) analogs of Onrigin™, respectively. For 50% growth inhibition, KS90 and 90CE generated 5,600 O6-methylguanines/cell and ~300 O6-chloroethylguanines/cell, respectively. AGT repaired O6-methylguanines until the AGT pool was exhausted, while its repair of O6-chloroethylguanines was incomplete due to progression of the lesions to AGT-irreparable interstrand DNA cross-links. Thus, the smaller number of O6-chloroethylguanine lesions needed for cytotoxicity accounted for the marked degree of resistance (12-fold) to 90CE produced by AGT. Transfection of human or murine AGT into AGT deficient transplantable tumor cells (i.e., EMT6, M109 and U251) generated transfectants expressing AGT ranging from 4,000 to 700,000 molecules/cell. In vitro growth inhibition assays using these transfectants treated with 90CE revealed that AGT caused a concentration dependent resistance up to a level of ~10,000 AGT molecules/cell. This finding was corroborated by in vivo studies where expression of 4,000 and 10,000 murine AGT molecules/cell rendered EMT6 tumors partially and completely resistant to Onrigin™, respectively. These studies imply that the antitumor activity of Onrigin™ stems from guanine O6-chloroethylation and define the threshold concentration of AGT that negates its antineoplastic activity.
Keywords: Onrigin™ (laromustine; cloretazine; VNP40101M; 101M), O6-Alkylguanine-DNA alkyltransferase (AGT), O6-Benzylguanine, Guanine O6-chloroethyl and O6-methyl lesions, Carmustine (BCNU), Temozolomide
1. Introduction
Onrigin™ (laromustine; cloretazine; VNP40101M; 101M; 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]hydrazine) is an active antitumor agent in humans, designed and synthesized in our laboratory as a prodrug that upon base catalyzed fragmentation releases chloroethylating (alkylating) and carbamoylating species (Fig. 1) [1]. Onrigin™ was conceived through incorporation of a methylaminocarbonyl residue into the prototype chloroethylating molecule 90CE (1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine) (Fig. 1A) [2]. The methylaminocarbonyl residue in which carbamoylating activity resides, acts as a masking group, thereby delaying the rapid fragmentation reactions observed with 90CE. The half-lives of 90CE and Onrigin™ at physiological pH and 37°C are ~0.5 and ~60 min, respectively [3]. This modification results in a dramatic increase in antitumor activity in preclinical tumor models, presumably due to improved distribution in vivo [1–4].
Fig. 1.
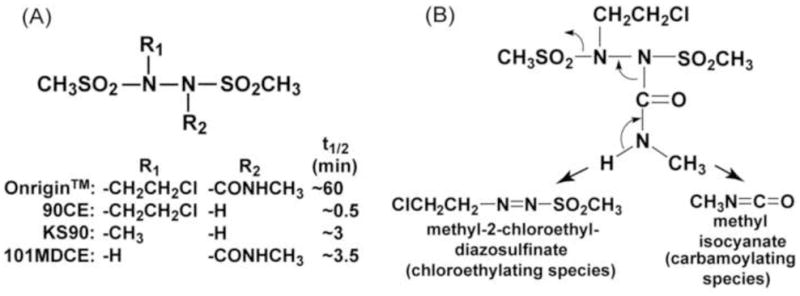
The structures of Onrigin™ and its analogs used in this study (A) and the fragmentation pattern of Onrigin™ generating chloroethylating and carbamoylating species (B). 90CE and KS90 are chloroethylating and methylating agents, respectively, without the carbamoylating moiety. 101MDCE is a carbamoylating agent without the alkylating moiety. The half-lives (t1/2) of these compounds measured in phosphate buffer, pH 7.4 at 37°C are listed.
Alkylating agents have occupied an important position in cancer chemotherapy, being among the most extensively used anticancer agents [5]. However, alkylating agents such as the nitrogen mustards whose antitumor effects are largely attributable to their reactivity with the N-7 position of guanine in DNA, are exceedingly toxic resulting in relatively low therapeutic indices [5,6]. Onrigin™ appears to be an exception in that, in phase II clinical studies, Onrigin™ as a single agent produced 28% complete response rates in elderly high-risk myelodysplasia and acute myelogenous leukemia patients with limited extramedullary toxicity [7]. Defining the underlying mechanisms of tumor selectivity manifested by Onrigin™ in this subset of patients [6,8,9] has been a major focus of our laboratory.
Using Onrigin™, 90CE and the carbamoylating-only analog 101MDCE (1,2-bis(methylsulfonyl)-1-[(methylamino)carbonyl]hydrazine) that lacks the alkylating moiety (Fig. 1A), the mode of action of the chloroethylating and carbamoylating species of Onrigin™ has been dissected [10–12]. As exemplified by the chloroethylating agent carmustine (BCNU; N,N′-bis(2-chloroethyl)-N-nitrosourea) [13], the antitumor activity of Onrigin™ derives from its ability to chloroethylate the O-6 position of guanine, ultimately yielding interstrand DNA cross-links between complementary G-C base pairs, a proposition supported by a series of observations. First, interstrand DNA cross-link formation by 90CE in vitro can be essentially prevented by the presence of the repair protein O6-alkylguanine-DNA alkyltransferase (AGT) [3,14] which transfers guanine O6-alkyl groups to the AGT active site cysteine and restores the O-6 position of guanine to the native state [15,16]. In addition, interstrand DNA cross-link formation is demonstrated in cells exposed to Onrigin™ and 90CE, but not to 101MDCE, in alkaline comet assays [12]. Second, the cytotoxicity of Onrigin™ and 90CE is markedly attenuated by expression of AGT in cultured cells [11,17].
The carbamoylating species generated from Onrigin™ is methyl isocyanate with affinity for nucleophiles such as sulfhydryls and amines in amino acid side chains of cellular proteins [18]. Consistent with this reactivity, the carbamoylating-only agent 101MDCE causes inhibition of DNA, RNA and protein syntheses in a non-selective manner and is cytotoxic by itself [12]. In AGT negative cells, the chloroethylating agent 90CE is more potent than 101MDCE in producing cytotoxicity [11].
Convincing evidence has demonstrated that AGT is a major factor in the production of resistance to the clinically active antitumor alkylating agents that generate chloroethyl (Onrigin™ and carmustine) and methyl (temozolomide) adducts at the O-6 position of guanine [19]. However, AGT is likely to repair guanine O6-chloroethyl and O6-methyl lesions with different degrees of effectiveness. Moreover, these guanine O6-targeting agents are known to exert their cytotoxicities by different mechanisms, with chloroethylating agents through generation of interstrand DNA cross-links and methylating agents via an intact mismatch repair system [19,20]. For better understanding of the role of AGT in conferring resistance to Onrigin™, we compared the effectiveness of AGT repair of guanine O6-chloroethyl and O6-methyl lesions.
AGT is a unique DNA damage repair protein in that the transfer of an alkyl group to an AGT active site cysteine results in a stoichiometric irreversible inactivation of the protein [15,16]. Thus, pretreatment of AGT positive cells with the relatively potent pseudosubstrate inhibitor of AGT O6-benzylguanine (O6-BG, IC50 = 0.05 μM) [21,22] was used to create AGT-ablated conditions in cultured cells. Two additional approaches unique to this laboratory were employed. The first was the availability of the chloroethylating agent 90CE and the methylating agent KS90 (1,2-bis(methylsulfonyl)-1-methylhydrazine) [23], analogs of Onrigin™ differing in the alkylating structure in the absence of the carbamoylating moiety (Fig. 1A), enabling exclusive analyses on these alkylating functions. The second was the availability of an AGT assay based upon the covalent transfer of the benzyl moiety from [benzene-3H]O6-benzylguanine to AGT developed in this laboratory [24]. Unlike conventional AGT assays using O6-methylguanine containing DNA as substrates, this assay employed the small chemical substrate O6-BG, enabling (a) multiple assays to be conducted using a relatively small number (2 × 106 cells/assay) of intact cells, (b) direct measurement of AGT activity as the number of AGT molecules/cell, and (c) quantification of guanine O6-alkyl adducts generated in cells by guanine O6-targeting agents without DNA extraction and chromatographic analyses. These analyses have uncovered the distinct ways in which AGT produces resistance to chloroethylating and methylating agents.
The present report measures the relationship between tumor AGT content and AGT mediated tumor resistance to Onrigin™ both in cultured cells and in a murine tumor model, using tumor cell lines expressing AGT at various levels, AGT negative transplantable tumor cell lines and those transfected with human or murine AGT. These studies demonstrated that maximum resistance to Onrigin™ is reached by AGT at a level of ~10,000 molecules/cell both in vitro and in vivo. These findings underscore the importance of determining the tumor AGT content prior to treatment with Onrigin™ and analyzing the relationship between the tumor AGT level and effectiveness of Onrigin™ in clinical trials.
2. Materials and Methods
2.1. Chemicals
Onrigin™, 90CE, 101MDCE and KS90 were synthesized in our laboratory as previously described [1–3,23]. Carmustine (C0400) and O6-BG (B2292) were obtained from Sigma (St. Louis, MO). Temozolomide was purchased from LKT Laboratories, Inc. (St. Paul, MN). All agents were dissolved in anhydrous DMSO; Onrigin™, 90CE, 101MDCE, carmustine and temozolomide at 200 mM, KS90 at 2 M and O6-BG at 20 mM. These stock solutions were serially diluted with DMSO and the same volume of DMSO or drug solution was added to control or treated samples to produce a constant final DMSO concentration in all assays. The half-lives of Onrigin™ (~60 min), 90CE (~0.5 min), 101MDCE (~3.5 min) and KS90 (~3 min) in phosphate buffer, pH 7.4, at 37°C have been described elsewhere [3,25]. The half-lives of carmustine (50 min) and temozolomide (74 min) at physiological pH have also been reported [26,27].
2.2. Tumor cell lines
HL-60 human promyelocytic leukemia cells and EMT6 murine mammary carcinoma cells were obtained from Drs. Robert C. Gallo and Sara C. Rockwell, respectively. Madison 109 (M109) murine lung carcinoma cells and U251 human glioblastoma cells were from the DCTD Tumor Repository, National Cancer Institute at Frederick (Frederick, MD). Human carcinoma cell lines, HCT 116 (colon), A549 (lung), HeLa S3 (cervix) and DU145 (prostate) were obtained from the American Type Culture Collection (Manassas, VA). Suspension and attached cell lines were maintained in RPMI 1640 and McCoy’s 5A media, respectively, supplemented with 10% fetal bovine serum (FBS) in a humidified 5% CO2 incubator.
2.3. Growth inhibition assays
Growth inhibition assays were conducted based upon cell number using 24-well plates in a volume of 1 ml/well. HL-60 cells at an initial density of 7 × 104 cells/ml, with or without pretreatment with 10 μM O6-BG for 1 hour, were incubated with various guanine O6-targeting agents for 3 days and cell numbers were determined using a Beckman Coulter Counter (Hialeah, FL). For attached cell lines (U251, HCT 116, A549, HeLa S3, DU145, EMT6 and M109), 2 × 104 cells (except for 3 × 104 cells for U251) were seeded per well and cultured overnight prior to drug exposure. O6-BG (20 mM) was added to cultures at 0.5 μl/well. Guanine O6-targeting agents were added to cultures at 1 μl/well. The final concentration of DMSO in control and treated cultures was 0.15%. DMSO at this level had no effect on cell growth. The percent growth inhibition was calculated using the formula: [log(final density of the control culture) − log(final density of the treated culture)]/[(log(final density of the control culture) − log(initial density of the control culture)] × 100.
2.4. AGT assays
AGT levels were measured using [benzene-3H]O6-benzylguanine (3H-BG) as a substrate as described previously [24]. Briefly, 2 × 106 intact cells were incubated with 1 μCi of 3H-BG in a volume of 100 μl at 37°C for 2 hours, and radioactivity in 70% methanol precipitates was measured after extensive washing. The cellular AGT content was expressed as the number of molecules/cell [24]. In AGT inactivation assays, 2 × 106 HL-60 cells containing 17,000 AGT molecules/cell were treated with AGT inactivators (0.5 μl) in a total volume of 100 μl at 37°C. The final DMSO concentration in control and treated samples was 0.5%. Cells were then incubated with 1 μCi of 3H-BG for 1 hour to measure the remaining levels of AGT.
2.5. Transfection of human and murine AGTs into AGT negative tumor cells
Human and murine AGT cDNAs (BC000824 and BC031888, respectively) in pCMV-SPORT6 were obtained from Open Biosystems (Huntsvill, AL). Each AGT coding region was amplified by PCR and inserted into pCRII-TOPO by TA cloning (Invitrogen, Carlsbad, CA). The fragments generated by PCR were verified by DNA sequencing. Human and murine AGT coding sequences were subcloned into mammalian expression vector p75/15 [28] containing the human metallothionein IIA promoter using BamHI (5′) and XbaI (3′) sites.
The vector p75/15 contained the neor selection marker [28]. Nontransfected cell lines, EMT6, M109 and U251, were 100% non-viable at 0.8 to 1.0 mg/ml of G418. For transfection, 5–7.5 × 105 cells were plated in 25 cm2 flasks and cultured overnight. Cells were then washed with Opti-MEM (Invitrogen) and exposed to a mixture of 4.8 μg of plasmid DNA and 30 μl of lipofectamine (Invitrogen) in 3.6 ml of Opti-MEM for 3 hours. After incubation in serum-containing medium for 1 day for expression, cells were plated in 6-well plates for selection at densities of 1 × 104/6 wells, 4 × 104/6 wells and 1.6 × 105/6 wells in the presence of 0.8 to 1 mg/ml of G418. After selection for 7 (EMT6 and M109) and 12 (U251) days, well-separated colonies were isolated, expanded and subjected to AGT assays.
2.6. Treatment of EMT6 tumors by Onrigin™ in vivo
Animal experiments were reviewed and approved by Yale University’s Institutional Animal Care and Use Committee. EMT6/wild-type, EMT6/mAGT4 and EMT6/mAGT10 cells were implanted by s.c. inoculation of 0.1 mL of Dulbecco’s phosphate buffered saline containing 105 tumor cells into the flank of 8–10 week-old female BALB/c mice (Charles River Laboratories, Wilmington, MA). When tumor volumes reached 100 to 120 mm3 after 7 days of tumor inoculation, mice were randomized into treatment groups, each consisting of 5 mice, and treatment with Onrigin™ (10 mg/kg, i.p., q2d × 10) was initiated. Onrigin™ was dissolved in anhydrous DMSO at 50 mg/ml, freshly diluted 10-fold with water and administered at 20 μl/10 g of body weight using insulin syringes. A vehicle control (2 μl of DMSO in 18 μl of water/10 g of body weight, i.p., q2d × 10) did not produce weight loss in host animals; the LD50 for a single i.p. injection of DMSO is 126 μl/10g in mice [29]. Tumor volume was estimated from caliper measurements of the length and width using the formula: V = (l × w2)/2.
2.7. Statistical and Mathematical Analyses
AGT assays and growth inhibition assays were repeated at least three times and values of IC50 (mean) ± standard deviations are shown. IC50 values were derived from logistic 3-parameter regression analyses using KaleidaGraph software (Synergy Software, PA).
3. Results
3.1. Cytotoxicity of chloroethylating and carbamoylating moieties of Onrigin™ in AGT-intact and AGT-ablated HL-60 cells and rationale for using 90CE and KS90 to analyze AGT mediated resistance
AGT transfers a guanine O6-alkyl group onto an AGT active site resulting in a stoichiometric (1:1) inactivation of the protein [15,16]. Since regeneration of the cellular AGT pool is a relatively slow process dependent upon new de novo protein synthesis [15], pretreatment of human AGT positive cells with the pseudosubstrate O6-BG, whose IC50 value for AGT inhibition is 0.073 μM in our assay (Fig. 2B), at a level of 10 μM for 1 hour and subsequent growth inhibition assays in the continuous presence of O6-BG are capable of generating a complete AGT-ablated environment.
Fig. 2.
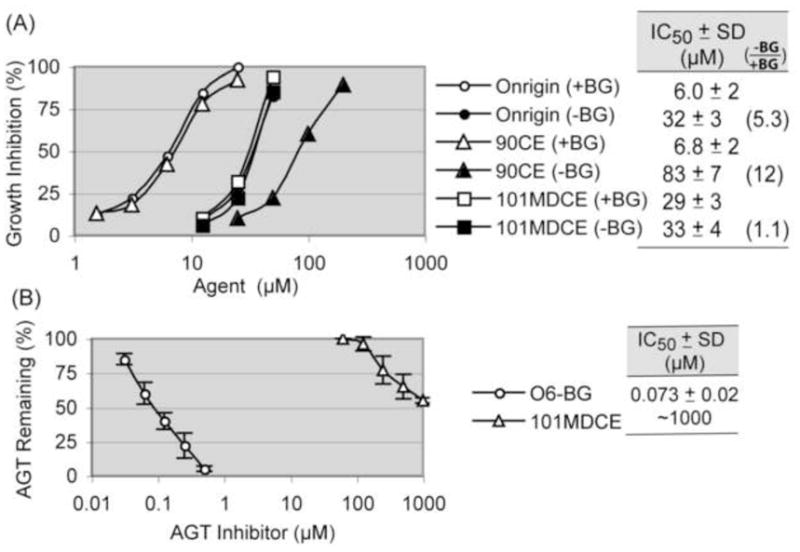
Dissection of the cytotoxicity stemming from chloroethylating and carbamoylating species of Onrigin™ using its mono-functional analogs (A) and AGT inhibition caused by O6-BG and 101MDCE (B). Panel A, HL-60 cells containing 17,000 AGT molecules/cell were pretreated with either vehicle (−BG) or 10 μM O6-BG (+BG) for 1 hour. Cells were then exposed to Onrigin™, 90CE, or 101MDCE for 3 days and cell numbers were determined. Growth inhibition (%) was calculated using the log of the cell number. Standard deviation (SD) bars at each drug concentration are omitted in the graph for clarity, as IC50 value ± SD for each agent is provided. Panel B, HL-60 cells were exposed to O6-BG or 101MDCE for 1 hour. Thereafter, cells were incubated with 3H-BG for 1 hour to measure AGT levels.
The cytotoxicities of the two electrophiles (i.e., chloroethylating and carbamoylating species) generated from Onrigin™ were evaluated using HL-60 human promyelocytic leukemia cells expressing 17,000 AGT molecules/cell [24] exposed to Onrigin™, the chloroethylating-only agent 90CE, or the carbamoylaing-only agent 101MDCE for 3 days with or without pretreatment with O6-BG. The AGT ablating treatment sensitized HL-60 cells to Onrigin™ (IC50 values from 32 to 6.0 μM) and to 90CE (IC50 values from 83 to 6.8 μM), but not appreciably to 101MDCE (IC50 values from 33 to 29 μM) (Fig. 2).
Based upon the finding that Onrigin™ (IC50: 32 μM) produced more cytotoxicity than 90CE (IC50: 83 μM) in AGT-intact cells, we suggested earlier the possibility that the carbamoylating species derived from Onrigin™ inactivated AGT via carbamoylation of the AGT active site cysteine thiol and sensitized AGT positive cells to the chloroethylating species of Onrigin™ [10,14]. We addressed this possibility by directly measuring AGT levels in HL-60 cells exposed to 101MDCE for 1 hour (Fig. 2B). 101MDCE inactivated AGT at well above 100 μM (IC50: ~1000 μM), a level much greater than that required for the growth inhibitory effect (i.e., IC50: 33 μM). Since the IC50 values for Onrigin™ and 101MDCE in AGT-intact conditions (−BG) were equivalent (32 and 33 μM, respectively), while the IC50 value for 90CE in AGT-intact conditions was much higher (83 μM), these results collectively implied that the carbamoylating species of Onrigin™ became the predominant cytotoxic entity in AGT-intact HL-60 cells, and that sensitization to the chloroethylating species of Onrigin™ through AGT inhibition by the carbamoylating species did not occur.
Because measurement of AGT mediated resistance to the alkylating function of Onrigin™ was not possible in the co-presence of the carbamoylating species that caused cytotoxicity by itself, the chloroethylating agent 90CE and the methylating agent KS90, analogs of Onrigin™ that differed in alkylating activity in the absence of the carbamoylating moiety (Fig. 1A) were employed. Since the half-lives of 90CE and KS90 in aqueous solution were less than 5 min (Fig. 1A), these agents provided a practical advantage for in vitro experiments, in that their alkylation reactions were completed within a short incubation time.
3.2. Quantification of guanine O6-alkyl lesions generated by 90CE and KS90 using AGT inactivation assays
Because (a) the reaction of AGT with guanine O6-alkyl adducts generated by guanine O6-targeting agents leads to stoichiometric (1:1) inactivation of AGT [15,16], (b) unlike bacterial AGT, mammalian AGT does not repair O4-methylated thymine effectively [30,31], and (c) thymine O4-methylation (0.1%) is much less frequent than guanine O6-methylation (7.5%) by N-methyl-N-nitrosouurea [32], the number of AGT molecules inactivated roughly reflects the number of DNA guanine O6-alkylations generated by guanine O6-targeting agents. HL-60 cells expressing 17,000 AGT molecules/cell were incubated with the chloroethylating agent 90CE or the methylating agent KS90 for 1 hour to allow guanine O6-alkylation and AGT repair, and then subjected to AGT assays for 1 hour to measure remaining AGT levels. As shown in Fig. 3A, the methylator KS90 (IC50: 53 μM) generated guanine O6-alkylations 5.5 times more effectively than the chloroethylator 90CE (IC50: 290 μM).
Fig. 3.
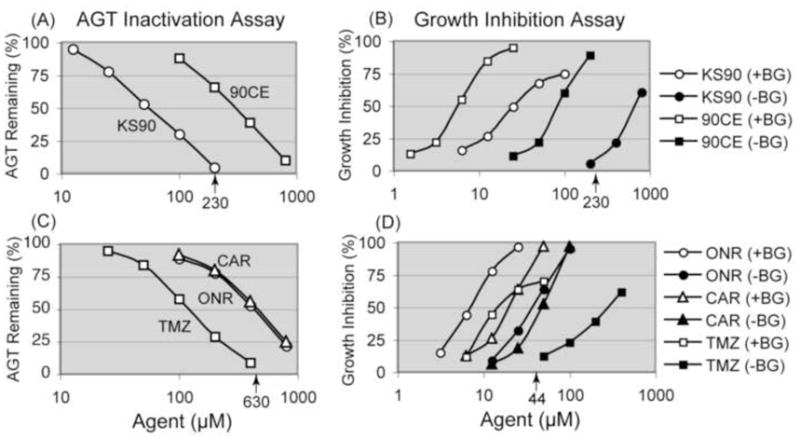
AGT inactivation assays to measure guanine O6-alkyl lesions generated by guanine O6-targeting agents and growth inhibition assays to determine IC50 values for these agents in AGT-intact and AGT-ablated HL-60 cells. Panels A and B show the results with 90CE and KS90. Panels C and D show the results with Onrigin™ (ONR), carmustine (CAR) and temozolomide (TMZ). For AGT inactivation assays, HL-60 cells were exposed to 90CE or KS90 for 1 hour, to ONR or CAR for 2 hours, and to TMZ for 4 hours. Cells were then subjected to AGT assays to measure remaining AGT levels. For growth inhibition assays, HL-60 cells, with or without pretreatment with 10 μM O6-BG for 1 hour, were treated with various agents for 3 days. The bold numbers with arrows on the x-axis indicate 100% AGT depleting concentrations in AGT inactivation assays and growth inhibition initiating concentrations in AGT-intact condition (−BG) in growth inhibition assays for KS90 and TMZ. SD bars at each drug concentration are omitted in the graph for clarity, as IC50 value ± SD for each agent is provided in Table 1.
Using this methodology, guanine O6-alkylations generated by the clinically active chloroethylating (Onrigin™ and carmustine) and methylating (temozolomide) agents were also estimated. The half-lives of Onrigin™, carmustine and temozolomide in aqueous solution are 60, 50 and 74 min, respectively [3,26,27]; therefore, longer incubation periods (2 hours for Onrigin™ and carmustine and 4 hours for temozolomide) were employed. As shown in Fig. 3C, temozolomide (IC50: 120 μM) generated guanine O6-alkylations ~3.5 times more effectively than Onrigin™ (IC50: 410 μM) and carmustine (IC50: 440 μM). The IC50 values for all of the guanine O6-targeting agents used in AGT inactivation assays are summarized in Table 1.
Table 1.
Guanine O6-alkylation events at the IC50 values and repair efficiency of AGT for guanine O6-methyl and guanine O6-chloroethyl lesions estimated in HL-60 cells
| Agent | AGT inactivationa IC50 (μM) | Growth inhibitiona IC50 (μM) | Alkyl events at IC50 (sites/cell) | AGT protective repair efficiency (alkyl site:AGT) | ||
|---|---|---|---|---|---|---|
| +BG | −BG | (−BG/+BG) | ||||
| 90CE | 290 ± 24 | 6.8 ± 2 | 83 ± 7 | (12) | ~300 | 1: 19b |
| KS90 | 53 ± 7 | 28 ± 3 | 670 ± 45 | (24) | 5,600 | 1:1 |
| ONR | 410 ± 42 | 6.0 ± 2 | 32 ± 3 | (5.3) | ||
| CAR | 440 ± 61 | 19 ± 4 | 45 ± 4 | (2.4) | ||
| TMZ | 120 ± 10 | 19 ± 5 | 280 ± 31 | (15) | ||
The IC50 values are derived from the data shown in Fig. 2.
The AGT protective repair efficiency (1: 19) for 90CE was estimated from a ratio of alkyl events at IC50 for KS90 and 90CE (5,600/300 = 19).
Abbreviations: ONR, onrigin™; CAR, carmustine; TMZ, temozolomide.
3.3. Determination of IC50 values for guanine O6-alkylators in AGT-intact and AGT-ablated HL-60 cells
HL-60 cells were treated with the chloroethylator 90CE or the methylator KS90 for 3 days with or without O6-BG pretreatment and degrees of growth inhibition caused by these agents under AGT-ablated (+BG) and AGT-intact (−BG) conditions were measured (Fig. 3B). The AGT ablative treatment increased the cytotoxicity of 90CE by 12-fold (IC50 values from 83 to 6.8 μM) and the cytotoxicity of KS90 by 24-fold (IC50 values from 670 to 28 μM) (Table 1). The AGT ablative treatment also increased the sensitivity to the clinically active agents Onrigin™ by 5.3-fold (IC50 values from 32 to 6.0 μM), carmustine by 2.4-fold (IC50 values from 45 to 19 μM) and temozolomide by 15-fold (IC50 values from 280 to 19 μM) (Fig. 3D and Table 1).
3.4. Estimation of the numbers of guanine O6-chloroethylations and guanine O6–methylations necessary to produce 50% growth inhibition
Calculations were made from the % inactivation values obtained from AGT inactivation assays (Fig. 3A) and the IC50 values derived from growth inhibition assays (Fig. 3B) for 90CE and KS90. The methylating agent KS90 caused 50% growth inhibition at 28 μM under AGT-ablated conditions (+BG) in HL-60 cells. Since AGT assays demonstrated that KS90 caused 33% inactivation of AGT at this concentration in HL-60 cells containing 17,000 AGT molecules/cell, the combined results implied that 5,600 guanine O6-methylations/cell were needed by KS90 to produce 50% growth inhibition. In contrast, the chloroethylating agent 90CE caused 50% growth inhibition at 8.2 μM under AGT-ablated conditions (+BG). Because 90CE (IC50: 290 μM) was 5.5 times less effective in producing AGT-depleting guanine O6-alkylations than KS90 (IC50: 53 μM) in AGT inactivation assays, but 90CE (IC50: 8.2 μM) was 3.4 times more growth inhibitory than KS90 (IC50: 28 μM) under AGT-ablated conditions (+BG) in growth inhibition assays, the combined results suggested that only ~300 AGT depleting guanine O6-alkylations (5,600/5.5/3.4) were required to produce 50% growth inhibition by 90CE.
3.5. Comparative efficiencies of AGT repair of guanine O6-methyl lesions and guanine O6-chloroethyl lesions
KS90 in AGT-intact conditions (−BG) became growth inhibitory at 230 μM (Fig. 3B), the concentration corresponding to that producing 100% inactivation in AGT inactivation assays (Fig. 3A), indicating that AGT repaired guanine O6-methyl lesions until the AGT pool (17,000 AGT molecules/cell in HL-60 cells) was exhausted before KS90 became growth inhibitory. Compared to the enormous efficiency of AGT repair of guanine O6-methyl lesions, AGT mediated removal of guanine O6-alkyl lesions generated by 90CE was inefficient, because the initial chloroethyl lesion progressed through the formation of a cyclic intermediate to an AGT irreparable form, i.e., an interstrand DNA cross-link that does not involve the O-6 position of guanine (Fig. 4). Thus, in contrast to AGT repairing the O6-methylguanine damage at a ratio of 1:1, the protective repair efficiency of AGT for guanine O6-chloroethyl lesions, i.e., the number of AGT molecules needed to guard against the damage from one guanine O6-chloroethylation was estimated to be 19 (5600/300), taking from a ratio of alkyl events at the IC50 values for KS90 and 90CE (Table 1).
Fig. 4.
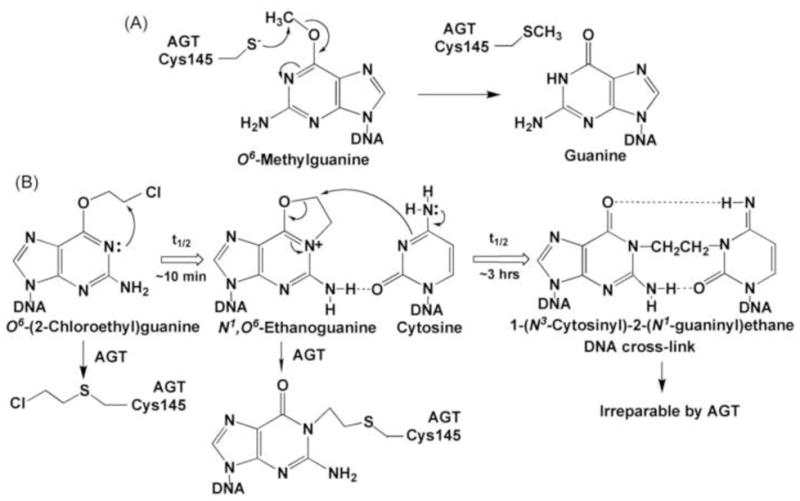
AGT repair of guanine O6-methyl and guanine O6-chloroethyl lesions. Panel A, direct reversal of a guanine O6-methyl lesion by AGT. Panel B, progression of a guanine O6-chloroethyl lesion via the formation of a cyclic intermediate, N1,O6-ethanoguanine, then to a interstrand DNA G-C cross-link and AGT reparability of these lesions. Note that repair of the cyclic intermediate by AGT yields AGT tethered to guanine N-1 and that AGT does not produce repair of the DNA cross-link, because the ethylene linkage between the two DNA strands does not involve guanine O-6.
Unlike KS90, whose 100% AGT inhibitory concentration in AGT inactivation assays coincided with its growth inhibition initiating concentration under AGT-intact conditions in growth inhibition assays, the methylating agent temozolomide exhibited its growth inhibitory activity in AGT-intact conditions at 44 μM (Fig. 3D), a concentration much below that causing 100% AGT inhibition (630 μM) in AGT inactivation assays (Fig. 3C). This result suggests that the cytotoxicity of temozolomide is derived from a mixture of events dependent upon and independent of guanine O6-methylation.
3.6. Generation of tumor cell lines transfected with human or murine AGT and the relationship between AGT content and the sensitivity to 90CE in vitro
The growth inhibition studies with five human tumor cell lines with naturally occurring AGT levels ranging from none (U251) to 42,000 (DU145) molecules/cell clearly demonstrated an inverse relationship between the content of AGT and sensitivity to 90CE (Fig. 5A). Since this relationship was more accurately examined in an isogenic background, AGT was transfected into three transplantable AGT negative tumor cell lines, i.e., EMT6 murine mammary carcinoma, M109 murine lung carcinoma and U251 human glioblastoma. Both human and murine AGTs were transfected into EMT6 and M109 cells to examine species differences in the ability to repair guanine O6-chloroethyl lesions. Transfectants were denoted as cell type/mAGT for murine AGT or cell type/hAGT for human AGT and the AGT content expressed as the number of molecules × 10−3/cell. Transfection of AGT generated clones expressing AGT at levels ranging from 4,000 (EMT6/mAGT4) to 700,000 (U251/hAGT700) molecules/cell. The rate of cell growth was not significantly altered by the expression of human or murine AGT at any level in any cell type with the doubling times of EMT6, M109, and U251 cells being ~10, ~10 and ~17 hours, respectively. AGT expression in the transfectants was extremely stable upon repeated subcultures in the absence of G418 for more than 6 months.
Fig. 5.
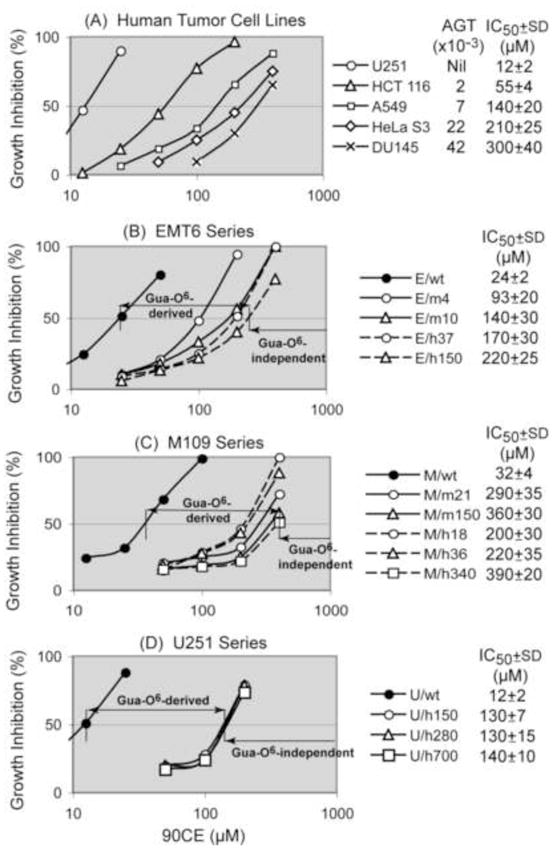
Relationship between the cellular AGT content and the sensitivity to the chloroethylating agent 90CE in vitro. Cells were treated with 90CE for 3 days and the % growth inhibition was calculated using the log of the cell number. Transfectants were denoted as E for EMT6, M for M109 or U for U251, and m or h to represent murine or human AGT followed by the AGT level expressed as the number of molecules × 10−3/cell. In all three types of cells, growth inhibition curves of non-transfected (wild-type) and empty vector-transfected cells were indistinguishable. Hence, results using wild-type cells are shown.
Growth inhibition assays for 90CE using AGT transfectants from EMT6, M109 and U251 cells are shown in Fig. 5B to 5D. The degree of resistance to 90CE caused by the expression of AGT, based upon the lowest and highest IC50 values, were approximately an order of magnitude in an isogenic background, 9.2 (220/24) for EMT6, 12 (390/32) for M109 and 12 (140/12) for U251. As shown in Fig. 5B, resistance to 90CE was proportional to the AGT level up to approximately 10,000 molecules/cell and AGT levels above this concentration resulted in only a relatively minor further increase in the degree of resistance in EMT6 cells. AGT levels of M109 and U251 transfectants were all above 18,000 molecules/cell (M109/hAGT18); these transfectants demonstrated semi-saturability in AGT mediated resistance to 90CE at extremely high AGT levels (Fig. 5C and 5D). These findings implied that the cytotoxicity of 90CE at high concentrations was derived from alkylations at positions other than the O-6 position of guanine in DNA. Thus, these studies defined the cytotoxic ranges of 90CE dependent upon and independent of DNA guanine O6-alkylations in EMT6, M109 and U251 cells as depicted in Fig. 5B to 5D.
Resistance to 90CE caused by 21,000 murine AGT molecules/cell (M/m21) and by 18,000 human AGT molecules/cell (M/h18) was both in the semi-saturable range in M109 transfectants (Fig. 5C), suggesting human and murine AGTs repaired guanine O6-alkyl adducts generated by 90CE with comparable efficiencies.
3.7. Relationship between the tumor AGT content and resistance to Onrigin™ in vivo
To examine the relationship between the content of AGT and tumor resistance to Onrigin™ treatment in vivo, BALB/c mice were transplanted with wild-type EMT6, EMT6/mAGT4 and EMT6/mAGT10, expressing murine AGT at levels of 0, 4,000 and 10,000 molecules/cell, respectively. The treatment schedule of Onrigin™ employed was 10 mg/kg, i.p., q2d × 10. AGT caused tumor resistance to Onrigin™ in an AGT concentration dependent manner in vivo, with tumor growth delays produced by Onrigin™ being 11, 3 and 0 days in wild-type EMT6, EMT6/mAGT4, and EMT6/mAGT10 tumors, respectively (Fig. 6).
Fig. 6.
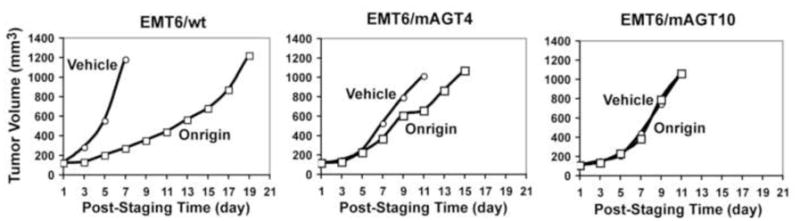
Relationship between tumor AGT content and resistance to Onrigin™ in vivo. EMT6/wt, EMT6/mAGT4 and EMT6/mAGT10 cells were implanted into the flank of BALB/c mice. Onrigin™ treatment was initiated when average tumor volumes reached 100 to 120 mm3 (Day 1). Non-transfected (wild-type) and empty vector transfected EMT6 tumors exhibited equal sensitivity to Onrigin™ and thus only results with wild-type EMT6 tumors are shown.
Discussion
In this report, we analyzed the quantitative relationships between tumor AGT content and the degree of tumor resistance to the prodrug Onrigin™ in vitro and in vivo. Onrigin™ is composed of both chloroethylating and carbamoylating functions. Using the mono-functional analogs of Onrigin™, i.e., the chloroethylating-only agent 90CE and the carbamoylating-only agent 101MDCE, we demonstrated that the carbamoylating species became the dominant cytotoxic entity in AGT-intact HL-60 cells containing 17,000 AGT molecules/cell. Because quantification of AGT mediated resistance to the chloroethylating function of Onrigin™ was not possible in the co-presence of the cytotoxic carbamoylating species, 90CE instead of Onrigin™ was used for in vitro growth inhibition studies.
Using human tumor cell lines with natural AGT levels ranging from 0 to 42,000 AGT molecules/cell and AGT negative transplantable tumor cells transfected with human or murine AGT at levels ranging from 4,000 to 700,000 AGT molecules/cell, we demonstrated that AGT caused resistance to 90CE in a concentration dependent manner up to approximately 10,000 AGT molecules/cell and that further increases in the level of AGT resulted in only minor increases in the degree of resistance. These findings were consistent with those from in vivo studies where the expression of 4,000 and 10,000 murine AGT molecules/cell in EMT6 tumors conferred partial and complete tumor resistance to Onrigin™, respectively. These studies provide evidence that the antitumor activity of Onrigin™ stems from its ability to chloroethylate the O-6 position of guanine in DNA. They also demonstrate that the antineoplastic effectiveness of Onrigin™ is compromised by the presence of even low levels of AGT, emphasizing the advantage of determining tumor AGT levels in humans prior to treatment with Onrigin™ and the need to select patients with AGT negative tumors for optimum results.
In phase II clinical studies, Onrigin™ (cloretazine) as a single agent was reported to produce a 28% complete response rate in elderly high-risk myelodysplasia and acute myelogenous leukemia patients with modest extramedullary toxicity [7]. AGT assays conducted for randomly chosen human leukemia cell lines revealed that 3 out of 9 lines (33%) completely lacked AGT activity [24]. Therefore, a retrospective study is necessary to determine (a) the range of tumor AGT levels and frequency of AGT negative tumors in these disease categories, (b) the relationship between the tumor AGT content and the clinical effectiveness of Onrigin™, (c) whether the upper threshold concentration of AGT that causes maximum resistance to Onrigin™ is 10,000 molecules/cell, corresponding to 100 fmoles/mg of protein based upon the protein content of HL-60 cells being 160 pg/cell, and (d) whether tumor selectivity manifested by Onrigin™ in responsive patients [6,8,9] is due to a differential AGT content in tumor and host tissues.
The finding that AGT levels above 10,000 AGT molecules/cell did not result in a further increase in resistance to 90CE in cultured cells implies that the cytotoxicity of 90CE at high drug concentrations is caused by lesions other than guanine O6-alkyl adducts. Kaina et al. [33] also observed the saturability of AGT mediated resistance to both the methylating agent N-methyl-N′-nitro-N-nitrosoguanidine and the chloroethylating agent N-hydroxyethyl-N-chloroethylnitrosourea using Chinese hamster ovary cells transfected with human AGT at various levels. These results collectively suggest that both guanine O6-targeting chloroethylating and methylating agents are capable of causing cytotoxicity through the formation of DNA guanine O6-alkyl lesions at relatively low drug concentrations in the absence of AGT and that they cause cytotoxicity through AGT irreparable lesions such as N-alkylpurines and alkylphosphotriesters at high drug concentrations irrespective of the presence or absence of AGT.
Using the chloroethylating agent 90CE and the methylating agent KS90 in the absence of the carbamoylating moiety, we conducted analyses on the effectiveness of the repair of the guanine O6-chloroethyl lesion by AGT employing the repair of the guanine O6-methyl lesion by AGT as a reference. AGT inactivation assays combined with growth inhibition assays revealed that as many as 5,600 guanine O6-methylations/cell and only ~300 initial guanine O6-chloroethylations/cell were needed to produce a 50% growth inhibition. Using human AGT negative cells exposed to [methyl-3H]N-methyl-N′-nitro-N-nitrosoguanidine followed by DNA extraction and modified base analyses, Rasouli-Nia et al. [34] reported a value of 6,650 guanine O6-methylations/cell for 1 lethal event (the effect producing 63% lethality). This value is roughly comparable to that derived from the present study using AGT inactivation assays (5,600 guanine O6-methylations/cell for 50% growth inhibition of AGT-ablated HL-60 cells). For chloroethylating agents, to our knowledge, this is the first report to show the number of AGT depleting guanine O6-alkylations necessary for cytotoxicity (~300 initial guanine O6-chloroethylations/cell for 50% growth inhibition of AGT-ablated HL-60 cells).
Structural studies demonstrate that AGT catalyzes a unique, single-step, direct DNA damage reversal repair by flipping an O6-alkylguanine moiety out of the DNA base stack into the active site of AGT and transferring the alkyl group to an internal cysteine residue in an SN2 reaction [16]. Although AGT is capable of producing substantial resistance to both the chloroethylating agent 90CE (12-fold) and the methylating agent KS90 (24-fold) in HL-60 cells, the underlying mechanisms involved are quite different. As shown in Fig. 4, the repair of guanine O6-methyl lesions by AGT is relatively simple, since this lesion is a “dead end” target for AGT. Thus, in HL-60 cells expressing 17,000 AGT molecules/cell, AGT inactivation assays indicated that KS90 generated 17,000 guanine O6-methyl lesions at 230 μM, while the growth inhibition assay indicated that the growth inhibitory activity of KS90 started at 230 μM in AGT-intact conditions. These results implied that AGT repaired guanine O6-methyl lesions until the 17,000 AGT molecules existing in HL-60 cells were entirely consumed. In contrast, the repair of guanine O6-chloroethyl lesions by AGT is complex, since the initial guanine O6-chloroethyl lesion consecutively progresses to N1,O6-ethanoguanine and then to an AGT irreparable interstrand G-C cross-link (Fig. 4). Therefore, AGT is relatively ineffective in protecting cells from guanine O6-chloroethylation induced damage, because the initial guanine O6-chloroethyl lesion is a “moving” target for AGT and DNA cross-link formation competes with the repair of N1,O6-ethanoguanine by AGT. However, since only a relatively small number of initial guanine O6-chloroethylations is necessary to produce cytotoxicity, AGT is capable of producing significant resistance to guanine O6-chloroethylating agents. AGT repair of N1,O6-ethanoguanine results in AGT tethered at the N-1 position of guanine [35]; although this repair does not regenerate the native form of guanine, it prevents N1,O6-ethanoguanine from progressing to a deadly interstrand DNA cross-link.
We estimated the protective repair efficiency of AGT for guanine O6-chloroethyl lesions to be 19 by using the ratio of alkyl events at the IC50 values for KS90 and 90CE (5,600/300, Table 1). Although the number of AGT molecules needed to guard against the damage induced by one O6-chloroethylguanine is high, the ratio of AGT molecules to guanine O6-alkyl lesions engaged in the actual repair process resulting in inactivation of AGT protein remains 1:1. Since the number of O6-methylguanines required to produce 50% growth inhibition (5,600/cell) is 19 times greater than that of O6-chloroethylguanines (~300/cell), these findings explain differences in the clinical outcome of cellular AGT levels before and after treatment with methylating and chloroethylating agents in that, at therapeutic doses, the methylating agent temozolomide causes a sizable depletion of the AGT pool [36]; whereas, the chloroethylating agent Onrigin™ has been reported to not produce measurable AGT depletion in humans [37].
Onrigin™ bears a functional resemblance to carmustine in that both agents generate chloroethylating and carbamoylating species, and produce interstrand DNA cross-links as their primary mechanism of action. However, carmustine undergoes complex decomposition patterns generating chloroethylating, hydroxylating and vinylating species with each pathway accompanied by production of the carbamoylating species, chloroethyl isocyanate [38]. Moreover, greater than 90% of the DNA adducts generated by carmustine occur at the N-7 position of guanine (55% N7-hydroxyethylguanine and 38% N7-chloroethylguanine) [39], base modifications known to result in spontaneous and enzymatic depurination [40]. In contrast, Onrigin™ was shown to have considerably greater specificity for the O-6 position of guanine than carmustine [3]. Furthermore, Onrigin™ consistently displayed much greater differential cytotoxicity in AGT-intact and AGT-ablated HL-60 cells than carmustine (5.3-fold for Onrigin™ versus 2.4-fold for carmustine), although the contribution of cytotoxicity from the carbamoylating species (methyl isocyanate for Onrigin™ and chloroethyl isocyanate for carmustine) to the overall cytotoxicity of each agent was not taken into consideration. In preclinical tumor models, Onrigin™ was found to be more potent, more efficacious and less toxic than carmustine [4].
Unlike KS90, whose 100% AGT inhibitory concentration coincided with its growth inhibition initiating concentration in AGT-intact HL-60 cells, temozolomide initiated growth inhibition at 44 μM in AGT-intact HL-60 cells (Fig. 3D), much below its 100% AGT inhibitory concentration (630 μM) (Fig. 3C). This result suggests that the cytotoxicity of temozolomide stems from a combination of guanine O6-methyl lesions and lesions unrelated to O6-methylguanine. Temozolomide generates major adducts at guanine N-7 (70%) and at adenine N-3 (9%), base modifications subject to base excision repair; treatment with temozolomide in the presence of an inhibitor of various components of the base excision repair system has been shown to bypass resistance caused by AGT and a defect in the mismatch repair system [41].
In this report, we also ruled out the intriguing possibility that Onrigin™ can bypass AGT mediated resistance through inhibition of the AGT protein by its carbamoylating species. The findings that (a) 101MDCE inactivated AGT in HL-60 cells at concentrations greater than 100 μM, with an IC50 value of ~1,000 μM and (b) Onrigin™ and 101MDCE caused growth inhibition of HL-60 cells with comparable IC50 values (32 and 33 μM, respectively), indicated that sensitization to the chloroethylating species of Onrigin™ through inhibition of AGT by the carbamoylating species did not occur.
It is noteworthy that even among AGT negative cell lines, considerable variability existed in the basal level of sensitivity to the chloroethylating agent 90CE. For example, the IC50 values for 90CE are 6.5 μM in L1210 cells [11], 6.8 μM in HL-60 cells measured under AGT-ablated conditions, 12 μM in U251 cells, 24 μM in EMT6 cells and 32 μM in M109 cells. On the same treatment schedule in vivo, Onrigin™ is capable of producing complete tumor regression of the “relatively sensitive” U251 tumor xenograft in nude mice [4], while the effectiveness of Onrigin™ is recognized as tumor growth delay in “relatively resistant” EMT6 and M109 [4] syngeneic mouse tumor models. Elucidation of the mechanism(s) underlying variable sensitivity/resistance to the chloroethylating agent intrinsic to each cell type in the absence of AGT is currently underway.
Acknowledgments
This work was supported in part by USPHS grants CA-090671, CA-122112 and CA-129186 from the National Cancer Institute and a grant from the National Foundation for Cancer Research. We express our gratitude to Dr. Rick A. Finch for his helpful advice on animal experiments and to Dr. Kevin P. Rice for his help in using KaleidaGraph software to obtain IC50 values.
Abbreviations
- AGT
O6-alkylguanine-DNA alkyltransferase
- 3H-BG
[benzene-3H]O6-benzylguanine
- O6-BG
O6-benzylguanine
- IC50
concentration giving 50% inhibition
- 90CE
1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazine
- 101MDCE
1,2-bis(methylsulfonyl)-1-[(methylamino)carbonyl]hydrazine
- KS90
1,2-bis(methylsulfonyl)-1-methylhydrazine
Footnotes
Conflict of Interest statement
The potential anticancer agent Onrigin™, designed and synthesized in Dr. Sartorelli’s laboratory, had been licensed to Vion Pharmaceuticals, Inc., no longer a viable company involved in the development of Onrigin™, by Yale University. Dr. Sartorelli in the past served as a Director and Chairman of the Scientific Advisory Board of this company, had common stock in Vion and, several years ago, his laboratory received gift monies in support of new research. In addition to Dr. Sartorelli, two of the other authors (K. Shyam and P.G. Penketh) also owned stock in Vion in the past.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shyam K, Penketh PG, Loomis RH, Rose WC, Sartorelli AC. Antitumor 2- (aminocarbonyl)-1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazines. J Med Chem. 1996;39:796–801. doi: 10.1021/jm9505021. [DOI] [PubMed] [Google Scholar]
- 2.Shyam K, Penketh PG, Divo AA, Loomis RH, Patton CL, Sartorelli AC. Synthesis and evaluation of 1,2,2-tris(sulfonyl)hydrazines as antineoplastic and trypanocidal agents. J Med Chem. 1990;33:2259–64. doi: 10.1021/jm00170a033. [DOI] [PubMed] [Google Scholar]
- 3.Penketh PG, Shyam K, Sartorelli AC. Comparison of DNA lesions produced by tumor-inhibitory 1,2-bis(sulfonyl)hydrazines and chloroethylnitrosoureas. Biochem Pharmacol. 2000;59:283–91. doi: 10.1016/s0006-2952(99)00328-7. [DOI] [PubMed] [Google Scholar]
- 4.Finch RA, Shyam K, Penketh PG, Sartorelli AC. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-(methylamino)carbonylhydrazine (101M): a novel sulfonylhydrazine prodrug with broad-spectrum antineoplastic activity. Cancer Res. 2001;61:3033–8. [PubMed] [Google Scholar]
- 5.Colvin M, Chabner BA. Alkylating agents. In: Chabner BA, Collins JM, editors. Cancer chemotherapy: principles and practice. Philadelphia: J.B. Lippincott Company; 1990. pp. 276–313. [Google Scholar]
- 6.Giles FJ. Bendamustine and cloretazine: alkylators with sharply contrasting activity in AML. Leuk Lymphoma. 2007;48:1064–6. doi: 10.1080/10428190701332464. [DOI] [PubMed] [Google Scholar]
- 7.Giles F, Rizzieri D, Karp J, Vey N, Ravandi F, Faderl S, et al. Cloretazine (VNP40101M), a novel sulfonylhydrazine alkylating agent, in patients age 60 years or older with previously untreated acute myeloid leukemia. J Clin Oncol. 2007;25:25–31. doi: 10.1200/JCO.2006.07.0961. [DOI] [PubMed] [Google Scholar]
- 8.Giles FJ. Laromustine: the return of alkylators to non-myeloablative therapy of AML. Leuk Res. 2009;33:1022–3. doi: 10.1016/j.leukres.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Vey N, Giles F. Laromustine (cloretazine) Expert Opin Pharmacother. 2010;11:657–67. doi: 10.1517/14656561003621232. [DOI] [PubMed] [Google Scholar]
- 10.Baumann RP, Seow HA, Shyam K, Penketh PG, Sartorelli AC. The antineoplastic efficacy of the prodrug Cloretazine is produced by the synergistic interaction of carbamoylating and alkylating products of its activation. Oncol Res. 2005;15:313–25. doi: 10.3727/096504005776404553. [DOI] [PubMed] [Google Scholar]
- 11.Ishiguro K, Shyam K, Penketh PG, Sartorelli AC. Role of O6-alkylguanine-DNA alkyltransferase in the cytotoxic activity of cloretazine. Mol Cancer Ther. 2005;4:1755–63. doi: 10.1158/1535-7163.MCT-05-0169. [DOI] [PubMed] [Google Scholar]
- 12.Ishiguro K, Seow HA, Penketh PG, Shyam K, Sartorelli AC. Mode of action of the chloroethylating and carbamoylating moieties of the prodrug cloretazine. Mol Cancer Ther. 2006;5:969–76. doi: 10.1158/1535-7163.MCT-05-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludlum DB. The chloroethylnitrosoureas: sensitivity and resistance to cancer chemotherapy at the molecular level. Cancer Invest. 1997;15:588–98. doi: 10.3109/07357909709047601. [DOI] [PubMed] [Google Scholar]
- 14.Penketh PG, Shyam K, Baumann RP, Remack JS, Brent TP, Sartorelli AC. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]hydrazine (VNP40101M): I. Direct inhibition of O6-alkylguanine-DNA alkyltransferase (AGT) by electrophilic species generated by decomposition. Cancer Chemother Pharmacol. 2004;53:279–87. doi: 10.1007/s00280-003-0740-7. [DOI] [PubMed] [Google Scholar]
- 15.Pegg AE. Repair of O6-alkylguanine by alkyltransferases. Mutat Res. 2000;462:83–100. doi: 10.1016/s1383-5742(00)00017-x. [DOI] [PubMed] [Google Scholar]
- 16.Tubbs JL, Pegg AE, Tainer JA. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair (Amst) 2007;6:1100–15. doi: 10.1016/j.dnarep.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumann RP, Shyam K, Penketh PG, Remack JS, Brent TP, Sartorelli AC. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]hydrazine (VNP40101M): II. Role of O6-alkylguanine-DNA alkyltransferase in cytotoxicity. Cancer Chemother Pharmacol. 2004;53:288–95. doi: 10.1007/s00280-003-0739-0. [DOI] [PubMed] [Google Scholar]
- 18.Rice KP, Penketh PG, Shyam K, Sartorelli AC. Differential inhibition of cellular glutathione reductase activity by isocyanates generated from the antitumor prodrugs Cloretazine and BCNU. Biochem Pharmacol. 2005;69:1463–72. doi: 10.1016/j.bcp.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 19.Gerson SL. Clinical relevance of MGMT in the treatment of cancer. J Clin Oncol. 2002;20:2388–99. doi: 10.1200/JCO.2002.06.110. [DOI] [PubMed] [Google Scholar]
- 20.Margison GP, Santibanez-Koref MF. O6-alkylguanine-DNA alkyltransferase: role in carcinogenesis and chemotherapy. Bioessays. 2002;24:255–66. doi: 10.1002/bies.10063. [DOI] [PubMed] [Google Scholar]
- 21.Dolan ME, Moschel RC, Pegg AE. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc Natl Acad Sci U S A. 1990;87:5368–72. doi: 10.1073/pnas.87.14.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moschel RC, McDougall MG, Dolan ME, Stine L, Pegg AE. Structural features of substituted purine derivatives compatible with depletion of human O6-alkylguanine-DNA alkyltransferase. J Med Chem. 1992;35:4486–91. doi: 10.1021/jm00101a028. [DOI] [PubMed] [Google Scholar]
- 23.Shyam K, Hrubiec RT, Furubayashi R, Cosby LA, Sartorelli AC. 1,2-Bis(sulfonyl)hydrazines. 3. Effects of structural modification on antineoplastic activity. J Med Chem. 1987;30:2157–61. doi: 10.1021/jm00394a040. [DOI] [PubMed] [Google Scholar]
- 24.Ishiguro K, Shyam K, Penketh PG, Sartorelli AC. Development of an O6-alkylguanine-DNA alkyltransferase assay based on covalent transfer of the benzyl moiety from [benzene-3H]O6-benzylguanine to the protein. Anal Biochem. 2008;383:44–51. doi: 10.1016/j.ab.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Penketh PG, Shyam K, Sartorelli AC. Studies on the mechanism of decomposition and structural factors affecting the aqueous stability of 1,2-bis(sulfonyl)-1-alkylhydrazines. J Med Chem. 1994;37:2912–7. doi: 10.1021/jm00044a012. [DOI] [PubMed] [Google Scholar]
- 26.Wiencke JK, Wiemels J. Genotoxicity of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) Mutat Res. 1995;339:91–119. doi: 10.1016/0165-1110(95)90005-5. [DOI] [PubMed] [Google Scholar]
- 27.Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987;47:5846–52. [PubMed] [Google Scholar]
- 28.Koay DC, Sartorelli AC. Functional differentiation signals mediated by distinct regions of the cytoplasmic domain of the granulocyte colony-stimulating factor receptor. Blood. 1999;93:3774–84. [PubMed] [Google Scholar]
- 29.Bartsch W, Sponer G, Dietmann K, Fuchs G. Acute toxicity of various solvents in the mouse and rat. LD50 of ethanol, diethylacetamide, dimethylformamide, dimethylsulfoxide, glycerine, N-methylpyrrolidone, polyethylene glycol 400, 1,2-propanediol and Tween 20. Arzneimittelforschung. 1976;26:1581–3. [PubMed] [Google Scholar]
- 30.Brent TP, Dolan ME, Fraenkel-Conrat H, Hall J, Karran P, Laval L, et al. Repair of O-alkylpyrimidines in mammalian cells: a present consensus. Proc Natl Acad Sci U S A. 1988;85:1759–62. doi: 10.1073/pnas.85.6.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sassanfar M, Dosanjh MK, Essigmann JM, Samson L. Relative efficiencies of the bacterial, yeast, and human DNA methyltransferases for the repair of O6-methylguanine and O4-methylthymine. Suggestive evidence for O4-methylthymine repair by eukaryotic methyltransferases. J Biol Chem. 1991;266:2767–71. [PubMed] [Google Scholar]
- 32.Pegg AE. Methylation of the O6 position of guanine in DNA is the most likely initiating event in carcinogenesis by methylating agents. Cancer Invest. 1984;2:223–31. doi: 10.3109/07357908409104376. [DOI] [PubMed] [Google Scholar]
- 33.Kaina B, Fritz G, Mitra S, Coquerelle T. Transfection and expression of human O6-methylguanine-DNA methyltransferase (MGMT) cDNA in Chinese hamster cells: the role of MGMT in protection against the genotoxic effects of alkylating agents. Carcinogenesis. 1991;12:1857–67. doi: 10.1093/carcin/12.10.1857. [DOI] [PubMed] [Google Scholar]
- 34.Rasouli-Nia A, Sibghat U, Mirzayans R, Paterson MC, Day RS., 3rd On the quantitative relationship between O6-methylguanine residues in genomic DNA and production of sister-chromatid exchanges, mutations and lethal events in a Mer- human tumor cell line. Mutat Res. 1994;314:99–113. doi: 10.1016/0921-8777(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 35.Brent TP, Remack JS. Formation of covalent complexes between human O6-alkylguanine-DNA alkyltransferase and BCNU-treated defined length synthetic oligodeoxynucleotides. Nucleic Acids Res. 1988;16:6779–88. doi: 10.1093/nar/16.14.6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spiro TP, Liu L, Majka S, Haaga J, Willson JK, Gerson SL. Temozolomide: the effect of once- and twice-a-day dosing on tumor tissue levels of the DNA repair protein O6-alkylguanine-DNA-alkyltransferase. Clin Cancer Res. 2001;7:2309–17. [PubMed] [Google Scholar]
- 37.Gururangan S, Turner CD, Stewart CF, O’Shaughnessy M, Kocak M, Poussaint TY, et al. Phase I trial of VNP40101M (Cloretazine) in children with recurrent brain tumors: a pediatric brain tumor consortium study. Clin Cancer Res. 2008;14:1124–30. doi: 10.1158/1078-0432.CCR-07-4242. [DOI] [PubMed] [Google Scholar]
- 38.Lown JW, Chauhan SM. Mechanism of action of (2-haloethyl)nitrosoureas on DNA. Isolation and reactions of postulated 2-(alkylimino)-3-nitrosooxazolidine intermediates in the decomposition of 1,3-bis(2-chloroethyl)-, 1-(2-chloroethyl)-3-cyclohexyl-, and 1-(2-chloroethyl)-3-(4′-trans-methylcyclohexyl)-1-nitrosourea. J Med Chem. 1981;24:270–9. doi: 10.1021/jm00135a007. [DOI] [PubMed] [Google Scholar]
- 39.Tong WP, Kohn KW, Ludlum DB. Modifications of DNA by different haloethylnitrosoureas. Cancer Res. 1982;42:4460–4. [PubMed] [Google Scholar]
- 40.Liu L, Yan L, Donze JR, Gerson SL. Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol Cancer Ther. 2003;2:1061–6. [PubMed] [Google Scholar]
- 41.Tentori L, Graziani G. Recent approaches to improve the antitumor efficacy of temozolomide. Curr Med Chem. 2009;16:245–57. doi: 10.2174/092986709787002718. [DOI] [PubMed] [Google Scholar]