

Abstract
Delayed wound healing is a chronic problem in opioid drug abusers. We investigated the role chronic morphine plays on later stages of wound healing events using an angiogenesis model. Our results show that morphine treatment resulted in a significant decrease in inflammation induced angiogenesis. To delineate the mechanisms involved we investigate the role of hypoxia inducible factor 1 alpha (HIF-1 alpha), a potent inducer of angiogenic growth factor. Morphine treatment resulted in a significant decrease in the expression and nuclear translocation of HIF-1 alpha with a concurrent suppression in vascular endothelial growth factor (VEGF) synthesis. Cells of the innate immune system play a dominant role in the angiogenic process. Morphine treatment inhibited early recruitment of both neutrophils and monocytes towards an inflammatory signal with a significant decrease in the monocyte chemoattractant MCP-1. Taken together, our studies show that morphine regulates the wound repair process on multiple levels. Morphine acts both directly and indirectly in suppressing angiogenesis.
Keywords: morphine, angiogenesis, innate immune system, HIF-1 alpha, wound healing
Introduction
It is well documented in the literature that wound healing occurs in a biphasic manner. Upon tissue injury and with wound complication due to infection, pro-inflammatory cell types, including neutrophils and macrophages are sequestered to induce resolution of bacterial clearance. Once infection is resolved, the second phase/arm of wound healing entails wound closure events including fibrin matrix formation, dermal migration, collagen deposits and formation of new blood vessels. The studies carried out in this manuscript focus on the angiogenic processes of forming new blood vessels at the site of injury as a measure of the second arm to wound healing.
Several factors have been shown to play critical roles during the second phase of wound healing. Initially, following tissue injury, coagulation of platelets form a clot to ensure wound isolation and thereby induce a local hypoxic state. Isolation at this stage of healing is essential for facilitating a concentrated environment containing potent pro-inflammatory mediators (chemical gradient), as well as, deterring the dissemination of pathogens out of the wound and into the bloodstream. However, factors responding to the low oxygen environment begin to set in place processes that would promote angiogenesis. The induction of the hypoxia inducible factor 1 alpha (HIF-1α) expression and protein stability[1] acts as a potent inducer of new blood vessel formations/angiogenesis. In response to a critical drop in oxygen tension known as hypoxia, Hif-1α proteins become stable by evading ubiquitin-mediated degradation, and act to induce pro-angiogenic factors necessary for new blood vessel formations. New blood vessels are essential for carrying oxygen and nutrients required for cell survival to areas that are depleted. HIF-1α translocates to the nucleus where it interacts with constitutively expressed HIF-1β to form a heterodimer. This HIF-1αβ heterodimer complex binds to its HIF response element (HRE) on DNA to promote translation of pro-angiogenic proteins. These genes encode critical growth factors such as vascular endothelium growth factor (VEGF), insulin-like growth factor (IGF), and platelet-derived growth factor (PDGF) necessary for the promotion of new blood vessel formation[2]. Several studies have reported that these factors accelerate remodeling and neovascularization in wounds with the induction or absence of inflammation[3–10] and that the angiogenic events occur seven days post injury[11]. Although resolution of bacterial clearance must occur before angiogenesis can take place, the signals responsible for orchestrating the time by which the phases of wound healing are turned on and off has not yet been established. However, phagocytotic cell populations such as macrophages are known producers and secretors of these growth factors.
Opioids, such as morphine are the most prescribed analgesic for pain relief for both chronic pain and post surgical pain. However, chronic morphine use or abuse has been shown to result in severe immune- suppression[12] by modulating both the innate and the adaptive arm of the immune system. We have previously demonstrated that morphine's direct and indirect suppression on early innate immune responses contributes to the insufficient resolution of bacterial clearance and hence, overall detrimental effect of poor wound healing. Although wound tissues presented with a marked increase of necrosis and pus, the wounds were able to pseudo-close seven days post injury. Since both arms of wound healing are orchestrated by the same types of cells, particularly leukocytes, we set out to investigate the effects chronic morphine play in disrupting this delicate balance between pathogen resolution and repair following injury. The potent pro-angiogenic transcription factor HIF-1α, as well as the potent angiogenic growth factor VEGF were evaluated in the presence of chronic morphine and LPS. Leukocyte migration patterns, as well as, chemotactic expression during the time at which angiogenesis occurs was also evaluated. We propose that the combined suppressive effects of morphine during both phases of wound healing account for the overall detrimental effects seen in our model, as well as, clinically in opioid chronic user and abuser populations.
Materials and Methods
Animal experimental design
Following mouse anesthetization with, intraperitoneal injection, of ketamine (65 mg/kg body weight) and xylazine (5.5 mg/kg body weight), placebo or morphine (75mg) slow-release pellets were implanted s.c. in the shaved region to the left of the dorsal midline of male C57Bl/6J mice (Jackson Laboratories, Bar Harbor, ME). The morphine pellets yield a steady state serum concentration between 250–400 ng/mL. Mice were approximately 6–7 weeks of age at 20–25 g body weights.
Animals were housed four mice per cage according to treatments under controlled twelve hour light, twelve hour dark cycles and controlled temperatures (25°C). Animals received standard food and tap water freely accessible from cages. Mice subjected to placebo or morphine pellet implantation according to the protocols and models previously described, were returned to their cages and separated by experimental group classifications: (i) placebo pellet +saline:heparin matrigel implant, (ii) placebo pellet +LPS:heparin matrigel implant, (iii) morphine pellet +saline:heparin matrigel implant, and (iv) morphine pellet +LPS:heparin matrigel implant.
Discomfort, stress, and injury to vertebrate subjects were minimized. The Institutional Animal Care and Use Committee (IACUC) at the University of Minnesota have approved all protocols and surgical procedures in these studies in agreement with the guidelines set forth by the National Institute of Health Guide for Care and Use of Laboratory Animals.
Matrigel plug preparation
LPS (1μM) and heparin (20U/L) were added to a matrigel solution (400μL; BD Bioscience, San Jose, CA) for s.c. hind limb injections 72 hours following pellet implantations. Matrigel reacts with the body temperatures of mice to form a non-invasive plug. Seven days post matrigel injections; gross morphology and vascular invasion were evaluated. Mice were euthanized prior to the removal of the matrigel plug.
Matrigel plugs were snap frozen in liquid nitrogen (N2) and underwent cryostat sectioning. Five-micron thick sections of the plugs were evaluated using 100× microscopy. Sections were fixed and incubated with an anti-CD31-PE monoclonal antibody (Ab). CD31 is a known skeletonized marker of blood vessels. Quantitative analysis of vessel number, vessel branching, and vessel length were measured. Images of stained sections were captured using the Olympus BX60 Upright microscope and analyzed using the Photoshop Imaging Processing Tool kit.
ELISA analysis of VEGF protein expression
Raw 264.7 cells (ATCC, Manassas, VA), a mouse leukemic monocyte macrophage cell line, were pretreated at 37°, 5%CO2 with varying concentrations of morphine 16 hours prior to LPS stimulation. Media-containing supernatants underwent ELISA analysis (R&D Systems Quantikine VEGF kit, Minneapolis, MN) according to standard protocol.
RNA analysis of HIF-1α
Mouse macrophage RAW-264 cells (one million cells per well) were pretreated with either 1μM morphine sulfate (Ms; NIDA, Baltimore, MD) or saline 18 hours prior to 1 μM LPS (NIDA, Baltimore, MD) stimulation. Cells were placed in a modular hypoxic incubator chamber (Billups-Rothenberg, Del Mar, CA) and subjected to a 15 minute hypoxic flush using 95% N2, 5%CO2 gas mixture. Cells, in chamber, were placed at 37° for 6 hours. Cells were removed, centrifugated and underwent RNA preparation (Qiagen RNA mini prep kit, Hilden, Germany). RNA preps were DNAse treated (Promega, Madison, WI) and underwent traditional PCR analysis using the HIF-1α primers (5' GTCTCGAGA TGCAGCCAGATCTCG 3' and 5' GGTCAGATGATCAGAGTCC AAAGC 3') and TaqMan Reverse Transcriptase reagents (Invitrogen Life Technologies, Carlsbad, CA) according to the standard protocol.
HIF-1α protein preparation and Western blot analysis expression
Two 6-well plates containing 1×107 RAW 264 cells were pretreated with or without 1 μM morphine for 18 hours. Treatments were set up as follows: (i) morphine+LPS and (ii) saline+LPS. LPS was added following 18 hour morphine pretreatment. Plate 1 was placed back into 37° incubator under normal oxygen tension conditions (normoxia=N) for 6 hours. Plate 2 was placed in a hypoxic chamber and flushed for 15 minutes under hypoxic conditions (95% N2, 5%CO2; hypoxia=H). Chamber was sealed and placed at 37° for 6 hours. Plates were removed from incubator and chamber, respectively. Wells were aspirated and centrifugated (3,000 rpm, BD Bioscience, San Jose, CA). Pelleted cells underwent hypertonic fractionations using the Nu-Clear Protein extraction kit (Sigma Chemical Co., St. Louis, MO). Cytosolic and nuclear fractioned supernatants were separated on a 7.5% SDS-PAGE gel and transferred to a nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ). After incubation with Starting Block (Pierce, Rockford, IL), membranes were incubated with anti-HIF-1α Ab (Novus, Littleton, CO), followed by anti-rabbit IgG-peroxidase (Amersham Biosciences, Piscataway, NJ). Antibody complexes bound to membranes were detected by exposure to film after incubation with Supersignal Chemiluminescence Detection Substrate (Pierce, Rockford, IL) according to standard procedures.
Transient transfections and reporter assays
The luciferase reporter plasmid pGL3-VEGF-luc containing 1.5 kb of the 5′ flanking regions of the human VEGF promoter was cloned upstream of the firefly luciferase gene as described previously[13]. RAW cells were transfected with the plasmid and plated at 1×105 plated in complete growth medium treatment in the presence of 1uM morphine for 24. Identical co-transfections were performed in triplicate, using lipofectin (Gibco- BRL), with pGL3-VEGF-luc and an internal control, pRL-TK (Promega, Madison, WI, USA), renilla luciferase gene under the control of thymidine kinase promoter.
Hypoxic treatment of cells
To achieve hypoxia (~2% oxygen), cell culture dishes were placed in a modular chamber (Billups Rothenberg, Inc., Del Mar, CA) and flushed with a mix of 0% O2, 5%CO2, 95%N2 at 10L/min for 15 minutes. Chambers remained tightly sealed and placed in a 5% CO2, 37°C incubator. This method achieves pO2 levels less than 35mmHg as determined from cell culture medium analyzed using a blood gas analyzer, Rapid Lab 248 (Chiron Diagnostics Tarrytown, NY); pO2 levels of culture supernatant from cells grown in room air or normal oxygen was 150–160 mm Hg.
Harvest of Perioneal macrophages from Wt mice
Resident peritoneal macrophages were harvested by peritoneal lavage with 10 ml of ice-cold Hanks Balanced Salt Solution (HBSS) without calcium and magnesium and plated onto two 60 mm Petri dishes and adhered for 2 h at 37°C and 5% CO2, non-adherent cells were aspirated. Adherent cells were cultured in RPMI 1640 (Invitrogen Life Technologies) supplemented with 10% newborn calf serum and 1% penicillin-streptomycin (Sigma-Aldrich). The composition of the adherent populations was evaluated using FACS analysis. Flow cytometric analysis revealed that adherent cells were primarily macrophages, with almost a 5-fold enhancement of F4/80 staining compared with the total cell population. CD3+ T lymphocytes were not detected in the adherent population. The adherent cells were plated at a concentration of 1–2 ×106 cells/ml in triplicate onto 6-well culture plates.
Statistical analysis
Unpaired student T test was used to test for significant differences in the leukocyte numbers between morphine and control treatment groups at the various time points. Data is expressed as the means ± SEM was employed. Differences were considered significant when P≤0.05. Each experiment was carried out in triplicate unless otherwise stated. Statistical computer packages STATVIEW (SAS Institute, Cary, NC) and SPSS (Chicago, IL) were used.
Results
Effects of chronic morphine treatment on new blood vessel formation
Matrigel solution containing heparin and the endotoxin, LPS were subcutaneously injected into hind limbs of mice. In reaction to mouse body temperatures, a non-invasive plug formed. Seven days post injection, matrigel plugs were removed and analyzed for vascularization events. We employed the use of the endothelial cell marker PECAM/CD31 which is expressed during tubular formation of blood vessels.
The gross morphology of plugs shown in Figure 1 A is representative of those seen in the study. Mice treated with LPS alone presented with new blood vessels forming throughout the matrigel plug. Mice chronically administered morphine, however, showed a pronounced reduction of blood vessels present in or around the matrigel plug.
Figure 1A. Morphine attenuated LPS-induced new blood vessels formation in matrigel plugs.

LPS and heparin containing matrigels were injected into the hind limb of placebo or morphine pelleted mice. The non-invasive matrigel plug was removed following euthanasia. Gross morphology of matrigel plugs showed a marked presence of new blood vessels forming throughout the plugs of placebo treated mice. However, angiogenesis was attenuated following chronic morphine treatment seven days post injection.
Upon removal of the matrigel, plugs were snap frozen in liquid nitrogen, cryostat sectioned onto glass slides and stained using the nuclear marker DAPI and the angiogenic marker CD31 (Figure 1 B). In LPS alone treated mice (placebo), innervated blood vessels were seen (PE, red) throughout the matrigel plugs. CD31 binding was specific in staining blood vessels as seen in the DAPI:CD31 overlay. As predicted, a significant reduction of blood vessel innervations in plugs from chronic morphine administered mice occurred. Taking the data analysis one step further, we employed the Photoshop Imaging Processing Tool kit. This software enabled us to quantify vessel number, vessel branching, and vessel nodes of skeletonized blood vessel images. The nodes, length and branching parameters were measured in Figure 1 C. Morphine treatment not only suppressed the formation of blood vessels, but also significantly stunted vessel node, length and end formations.
Figure 1B. LPS-induced formation of new blood vessels is suppressed in the presence of morphine.
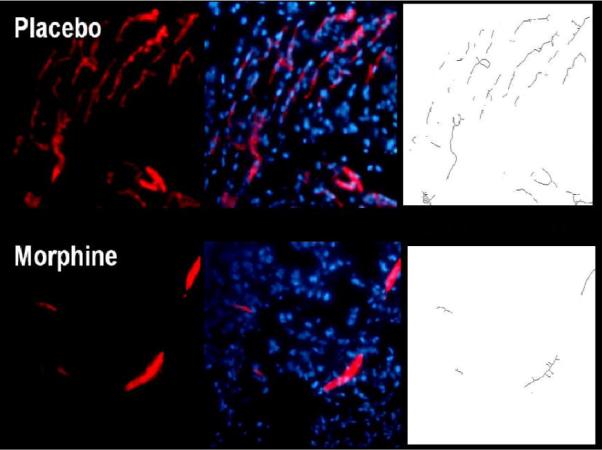
Cryostat sections from nitrogen snap frozen matrigel samples were stained using nuclear (DAPI) and PECAM (CD31) markers. In placebo treated groups, LPS-induced angiogenesis was apparent throughout matrigel plugs by Day 7. Chronic morphine treatment significantly decreased the formation of blood vessels into LPS containing plugs. Skeletonized images of newly formed blood vessels were assessed to measure vessel node, length, and end points.
Figure 1C. Morphometric analysis of angiogenic events.

A quantification of the skeletonized images shown in Figure 1B was evaluated for angiogenic events (node, length and end counts of new vessels). Plugs extracted from mice treated with morphine slow release pellets showed a significant reduction in blood vessel ends, nodes, and lengths when compared to placebo treated groups (*p<0.05, n=3).
Taken together, these studies clearly show that a LPS-induction of new blood vessel formation is suppressed when morphine is present. We next asked the question, is the suppression on vessel formation an indirect result of morphine modulating pro-angiogenic factors such as VEGF. To test this question in vitro, we used the macrophage cell line RAW 264.
Effects of chronic morphine treatment on protein expression of VEGF in macrophages
Previous studies have shown VEGF to be a potent factor necessary in the formation of new blood vessels[4]. Macrophages have been identified as critical producers and secretors of VEGF protein during angiogenesis. If morphine significantly suppresses blood vessel formation in the presence of a stimulus, we asked the question; What role would morphine have in modulating VEGF expression by macrophages? To address this question, we pre-incubated the macrophage cell line RAW 264 cells with morphine at varying concentrations. We later challenged these cells with LPS for 6 hours and measured VEGF protein expression using ELISA.
Following morphine pretreatment and LPS stimulation, LPS-induced VEGF expression was attenuated in cells pretreated with 100nM or 1μM of morphine in a dose-dependent manner (Figure 2). This suppression of VEGF expression occurred at sub-basal levels. Interestingly, morphine significantly reduced VEGF expression in the absence of LPS stimulation when compared to the levels of expression in the control group.
Figure 2. Morphine suppresses LPS-induced macrophage secretion of VEGF proteins in a concentration-dependant manner.

RAW-264 mouse macrophage cells were pretreated with varying concentrations of morphine 16h prior to LPS stimulation. Media-containing supernatants underwent ELISA analysis. VEGF expression was induced in the presence of LPS (lane 2). At 100 nM and 1uM morphine concentrations, LPS-stimulated VEGF protein expression was reduced to sub-basal levels (lanes 3 and 4). Morphine treatment, in the absence of LPS (lanes 5 and 6), significantly reduced VEGF expression as compared to its control treated group.
Taken together, we have shown morphine to act directly as a suppressor of macrophage secretion of the potent angiogenic growth factor VEGF. These studies, in line with the matrigel studies, suggest that morphine's suppressive effects on angiogenesis are attributable in part by an inhibition of macrophage secretion of the potent pro-angiogenic factor VEGF.
As mentioned previously, several factors contribute in the regulation of angiogenesis. Besides growth factor production in cells involved in innate immunity, other factors up-regulated by cues from the tissue environment also mediate the angiogenic process. HIF-1 α has been shown to play a critical role in inducing expression of pro-angiogenic factors in response to a hypoxic state. Therefore, we asked whether chronic morphine treatment would have an effect on the stabilization of HIF-1α proteins.
Effects of chronic morphine treatment on the stabilization of HIF-1α protein
HIF-α proteins are constitutively expressed at low levels in several cell types including macrophages and neutrophils. Under normal oxygen tension, prolyl hydroxylases (PHD) rapidly orchestrate the ubiquitin-mediated degradation of HIF-1α proteins by promoting interactions with HIF-1α and von Hippel-Lindau tumor suppressor proteins (vHL). Under stressful conditions such as during a hypoxic state induced by tissue injury, HIF-1α proteins become stabilized and avoid degradative processes. Stable proteins, in turn, migrate from the cytosol into the nucleus where it eventually promotes the translation of pro-angiogenic factors such as VEGF. Using the in vitro hypoxia model, we investigated whether or not HIF-α protein stability would be altered in the presence of morphine.
Primary macrophages, taken from the peritoneal cavities of wild-type mice, were pretreated with 1μM morphine 16 hours prior to hypoxia stimulation (Figure 3A). Cytosolic (upper gel) and nuclear (lower gel) extracts were analyzed using an anti-HIF-1α monoclonal antibody.
Figure 3A. Morphine suppresses hypoxia-stabilized protein levels of HIF-1α in cytosolic and nuclear fractions.

Peritoneal mouse macrophages were harvested and pretreated with 1μM morphine 16h prior to hypoxia stimulation. Cytosolic (lower gel) and nuclear (upper gel) extracts were analyzed using anti-HIF-1α monoclonal Ab. In the cytoplasmic fractions, hypoxia stabilization of HIF-1α protein was attenuated in the presence of morphine. Nuclear fractions of HIF-1α protein showed elevated levels in response to hypoxia. However, less HIF-1α was detected in the nuclear compartment following morphine treatment.
Stable HIF-1α proteins were detected in the cytoplasmic fractions following hypoxia stimulation (control, hypoxia=CH). However, a decrease in stable HIF-1α was seen when cells were pretreated with morphine (morphine, hypoxia=MH). HIF-1α proteins were significantly elevated in the nuclear fraction in response to hypoxia. Yet, Hif 1 alpha levels from morphine treated cells showed a significant reduction in these fractions.
These studies suggest that in the presence of morphine, stability of HIF-1α proteins under hypoxic conditions is suppressed and thereby, reduces the protein's availability to translocate into the nucleus to promote the transcription of pro-angiogenic factors. This decrease in protein stability by morphine may be attributable to an indirect effect on factors involved in HIF-1α stabilization (i.e. PHD or vHL activity) or a direct effect on HIF-1α messenger RNA expression.
Using our in vitro macrophage model, we measured the transcription levels of HIF-1α RNA using RAW-264 cells.
Effects of chronic morphine treatment on HIF-1α RNA message expression
Mouse macrophage RAW-264 cells were pretreated with 1μM morphine sixteen hours prior to administration of 1μM LPS. Cells were subjected to a hypoxic state (95% N2, 5% CO2) for six hours. RNA preparations, using the RNeasy Qiagen Mini kit, were reverse transcribed and PCR amplified using HIF-1α primers.
An induction of HIF-1α RNA levels in response to hypoxia was seen in the control group (Figure 3B). Morphine alone showed significant suppression of hypoxia-induced HIF-1α expression. Stimulation of cells with LPS further induced HIF-1α message levels above hypoxic levels (control). However, morphine pretreatment significantly inhibited LPS-induced HIF-1 alpha mRNA levels.
Figure 3B. Morphine Suppresses O2-dependent HIF-1α RNA Expression.

Mouse macrophage RAW-264 cells were pretreated with 1 μM morphine 16 hours prior to administration of 1 μM LPS. Cells were subjected to a hypoxic state (95% N2, 5%CO2) for 6 hours. RNA preparations, using the RNeasy Qiagen Mini kit, were reverse transcribed and PCR amplified using HIF-1α primers. Inductions of HIF-1α RNA levels were present under hypoxia (control) and LPS stimuli. Morphine significantly suppressed both hypoxia- and LPS-induced HIF-1α expression.
To further determine if morphine modulates transcriptional activation of VEGF, the full length VEGF promoter was transfected into RAW cells and pretreated with morphine for 12 hours before the exposing the cells to either LPS, hypoxia or both LPS and hypoxia. Our results show that morphine treatment significantly inhibited hypoxia-induced promoter activation. The 1.5 kb full-length promoter includes the hypoxia responsive element (HRE) to which hypoxia inducible factor 1 alpha binds.
Taken together, our results demonstrate that chronic morphine treatment suppresses both hypoxia- and LPS-mediated transcriptional induction of HIF-1α. Since morphine has been shown to suppress these effects on pro-angiogenic factors in both primary and immortal macrophages in vitro, we set out to assess what effect morphine would have on the migration patterns of macrophages and neutrophils using an in vivo wound model.
Neutrophil and macrophage infiltration into PVA sponge implants following chronic morphine treatment
Cells sequestered into LPS loaded PVA sponges underwent flow cytometry analysis on Day 1 and 7 post implantation (Figure 4). Neutrophils were identified using the surface marker LYG-6/Gr-1. Sponge extracts taken from mice treated with LPS (placebo+LPS, black bar) or morphine+LPS (white) showed a significant differences in neutrophil migration patterns in response to chronic morphine administration at day 1. Interestingly, in the morphine treatment group neutrophil numbers at day 7 was similar to placebo treatment.
Figure 4. Recruitment patterns of neutrophils into PVA sponged loaded with LPS.

Following removal of PVA sponges, cells extracted were counted and labeled with the neutrophil markers LY6G/GR1 (n=4). FACs analysis revealed a significant decrease in neutrophil migration at day 1 in morphine treated animals but no differences in neutrophil migration amongst placebo+LPS and morphine+LPS treated groups seven days post injury.
Macrophage recruitment to LPS loaded PVA sponges were evaluated at day 4 and 7. Monocyte recruitment peaked at day 4 in the placebo group. In the morphine treated animals a decreased recruitment was observed at day 1 (data not shown) and 4 but at day 7 monocytes numbers were greater than placebo animals (Figure 5). These data indicate that LPS-modulation inflicts a lasting detrimental effect on the migratory pattern of macrophages instrumental for wound repair processes during healing and morphine treatment further exacerbates the migration.
Figure 5. Recruitment patterns of monocytes into PVA sponged loaded with LPS.

Following removal of PVA sponges, cells extracted were counted and labeled with the macrophage markers F4/80(n=4). FACs analysis revealed a significant decrease in monocyte migration in the morphine treated group at day 4 but at day 7 a sustained recruitment was observed.
Next, we explored whether or not morphine modulates hypoxia induced macrophage chemoattractant MCP-1 protein levels.
Effects of secreted protein levels for monocyte chemo-attractant MCP-1 following morphine treatment
Peritoneal monocytes/macrophages were treated with morphine and saline and LPS was added to the culture. Cells were then subjected to hypoxia and MCP- levels measured after 24 hours. Culture supernatants underwent MCP-1 Quantikine ELISA detection analysis (Figure 6). A significant level of MCP-1 was seen in response to LPS + hypoxia. Morphine inhibited both hypoxia and LPS+ hypoxia induced MCP 1 expression.
Figure 6. MCP-1 expression levels in response to morphine.

The potent macrophage chemoattractants MCP-1 were analyzed using ELISA. Both LPS and hypoxia treatment alone resulted in a significant increase in MCP-1 levels. The expression was significantly greater when both insults were presented together. Morphine treatment resulted in a significant inhibition of both Hypoxia and LPS induced MCP-1 synthesis.
Discussion
Although chronic opioid use lead to severe immune compromise but the underlying mechanisms why heroin-addicted patients present with delayed wound healing and wound complications is not known. The proposed studies attempt to address the mechanisms by which chronic morphine modulate the wound healing process. Furthermore, since chronic morphine increases the risk for opportunistic infections, investigating the mechanism by which would healing occur in the context of infection and modulate the healing process will allow for the development of therapeutic strategies that will decrease wound complications and facilitate the healing process.
Angiogenesis is critical for supplying blood to areas requiring oxygen and nutrients/glucose for cell/tissue survival. The literature describing the effects of morphine on angiogenic events has been extensively studied for the purposes of understanding tumorgenesis. In the tumor model, suppression of angiogenesis would prove beneficial in starving these tumors of nutrients and oxygen necessary for growth. However, during wound repair, suppression of angiogenesis would prove detrimental. Promotion of angiogenesis prematurely could result in dissemination of unresolved pathogens. Delayed angiogenic events could also pose problems by delaying the delivery of oxygen, nutrients and cells required for wound repair. Because both arms of wound healing are orchestrated by the same types of cells, particularly leukocytes, we set out to investigate the effects chronic morphine play in disrupting this delicate balance between pathogen resolution and repair following injury.
The body of work in this chapter employed a matrigel model to assess chronic morphine effects on the second arm of wound healing; wound angiogenesis. Several studies have implicated macrophages in acting as a vital producer and secretor of several identified pro-angiogenic factors including HIF-1α and VEGF[14–18].
Our studies suggest that morphine plays both a direct and indirect role in suppressing angiogenesis during the wound repair process and that this suppression involves macrophages. Morphine's immunosuppressive effects on the migration and function of leukocytes during the initial innate response indirectly affects later repair outcomes. The morphine-mediated decrease in leukocyte infiltration instrumental for bacterial clearance results in inadequate bacterial resolution and sustained elevation of pro-inflammatory factors. Inadequate resolution and elevated inflammatory factors fail to trigger signals required for switching the pro-inflammatory arm off and the pro-angiogenic arm on. These events yield a net effect of delayed wound healing.
We have shown morphine to elicit a direct effect on the wound repair process by suppressing the formation of new blood vessels/angiogenesis in matrigel plugs containing an angiogenic inducer, LPS. New blood vessel nodes, length and branching throughout the matrigel were all significantly decreased following morphine treatment.
In recent years, studies have helped the scientific community appreciate the magnitude by which the transcription factor HIF-1α plays in regulating angiogenesis[19–22]. We have shown that morphine suppresses hypoxia- and LPS-induced HIF-1α transcription and protein stability. Nuclear fractions of hypoxia- and LPS-induced HIF-1α showed a decrease in protein levels indicating that less HIF-1α translocates into the nucleus thus leading to a reduction in proangiogenic factor transcription and hence, decreased signals necessary for initiating angiogenesis. Transcription of VEGF is one of the identified potent angiogenic factors mediated by HIF-1α.
When macrophages were chronically treated with morphine, VEGF expression levels were decreased even in the presence of the stimulus LPS. This suppression of VEGF protein levels occurred in a dose-dependent manner.
In addition to a decrease in LPS and hypoxia induced VEGF, we also show morphine treatment results in a significant decrease in macrophage recruitment, a key cell that has been implicated for the initiation of angiogenesis. By day seven, although no significant differences were seen in neutrophil recruitment patterns into PVA sponges following chronic morphine treatment, macrophage populations were considerably lower than expected. Typically, a second robust wave of infiltrating macrophages ensue by seven days post wounding to provide signals essential in the angiogenic and wound repair processes[11]. Macrophages play a vital role not only in producing and secreting potent pro-inflammatory factors, but also in their secretion of potent pro-angiogenic factors. With lower macrophage influx, less pro-angiogenic factors, including the transcription factor HIF-1α and growth factor VEGF, are produced and secreted by these cells. In the absence of these factors, the second arm of healing will not proceed resulting in a significantly delayed wound repair.
Taken together, our studies show that morphine regulates wound angiogenesis by inhibiting hypoxia and LPS induced VEGF secretion by modulating HIF-1 alpha induction and nuclear translocation. These studies shed light on some of the multifaceted levels morphine plays in delaying wound healing, yet further studies are needed to tease out the complex mechanisms by which morphine regulates wound repair.
Figure 3C. Effect of morphine on hypoxia-induced promoter activation in RAW cells.

A full-length promoter (−1176 to +336) of the human VEGF gene was used to transfect Mouse macrophage RAW-264. Promoter induction was measured by firefly luciferase activity normalized to that of renilla luciferase. Fold induction of each group over respective control was expressed as relative luciferase activity.
Acknowledgement
This work was supported by grants R01 DA12104, R01 DA022935, K02 DA015349 and P50 DA11806 (S. Roy) from The National Institutes of Health and funds from the Minneapolis Veterans Affairs Medical Center (RA.Barke).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia-inducible factor (HIF-1) alpha: Its protein stability and biological functions. Experimental & Molecular Medicine. 2004;36(1):1–12. doi: 10.1038/emm.2004.1. [DOI] [PubMed] [Google Scholar]
- 2.Lynch SE, Colvin RB, Antoniades HN. Growth factors in wound healing. single and synergistic effects on partial thickness porcine skin wounds. The Journal of Clinical Investigation. 1989;84(2):640–646. doi: 10.1172/JCI114210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates DO, Jones RO. The role of vascular endothelial growth factor in wound healing. The International Journal of Lower Extremity Wounds. 2003;2(2):107–120. doi: 10.1177/1534734603256626. [DOI] [PubMed] [Google Scholar]
- 4.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacological Reviews. 2004;56(4):549–580. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 5.Motani A, Forster L, Tull S, Anggard EE, Ferns GA. Insulin-like growth factor-I modulates monocyte adhesion to EAhy 926 endothelial cells. International Journal of Experimental Pathology. 1996;77(1):31–35. doi: 10.1046/j.1365-2613.1996.960098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roesel JF, Nanney LB. Assessment of differential cytokine effects on angiogenesis using an in vivo model of cutaneous wound repair. The Journal of Surgical Research. 1995;58(5):449–459. doi: 10.1006/jsre.1995.1071. [DOI] [PubMed] [Google Scholar]
- 7.Roy H, Bhardwaj S, Yla-Herttuala S. Biology of vascular endothelial growth factors. FEBS Letters. 2006;580(12):2879–2887. doi: 10.1016/j.febslet.2006.03.087. [DOI] [PubMed] [Google Scholar]
- 8.Roy S, Khanna S, Sen CK. Redox regulation of the VEGF signaling path and tissue vascularization: Hydrogen peroxide, the common link between physical exercise and cutaneous wound healing. Free Radical Biology & Medicine. 2008;44(2):180–192. doi: 10.1016/j.freeradbiomed.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 9.Somanath PR, Chen J, Byzova TV. Akt1 is necessary for the vascular maturation and angiogenesis during cutaneous wound healing. Angiogenesis. 2008 doi: 10.1007/s10456-008-9111-7. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toda M, Suzuki T, Hosono K, Kurihara Y, Kurihara H, Hayashi I, et al. Roles of calcitonin gene-related peptide in facilitation of wound healing and angiogenesis. Biomedicine & Pharmacotherapy Biomedecine & Pharmacotherapie. 2008 doi: 10.1016/j.biopha.2008.02.003. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 11.Park JE, Barbul A. Understanding the role of immune regulation in wound healing. American Journal of Surgery. 2004;187(5A):11S–16S. doi: 10.1016/S0002-9610(03)00296-4. [DOI] [PubMed] [Google Scholar]
- 12.Martin JL, Koodie L, Krishnan AG, Charboneau R, Barke RA, Roy S. Chronic morphine administration delays wound healing by inhibiting immune cell recruitment to the wound site. Am J Pathol. 2010 Feb;176(2):786–99. doi: 10.2353/ajpath.2010.090457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balasubramanian S, Ramakrishnan S, Charboneau R, Wang J, Barke RA, Roy S. Morphine sulfate inhibits hypoxia-induced vascular endothelial growth factor expression in endothelial cells and cardiac myocytes. J Mol Cell Cardiol. 2001 Dec;33(12):2179–87. doi: 10.1006/jmcc.2001.1480. [DOI] [PubMed] [Google Scholar]
- 14.Blouin CC, Page EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1alpha. Blood. 2004;103(3):1124–1130. doi: 10.1182/blood-2003-07-2427. [DOI] [PubMed] [Google Scholar]
- 15.Komatsu DE, Hadjiargyrou M. Activation of the transcription factor HIF-1 and its target genes, VEGF, HO-1, iNOS, during fracture repair. Bone. 2004;34(4):680–688. doi: 10.1016/j.bone.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 16.Sen CK, Khanna S, Babior BM, Hunt TK, Ellison EC, Roy S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. The Journal of Biological Chemistry. 2002;277(36):33284–33290. doi: 10.1074/jbc.M203391200. [DOI] [PubMed] [Google Scholar]
- 17.Varney ML, Olsen KJ, Mosley RL, Singh RK. Paracrine regulation of vascular endothelial growth factor--a expression during macrophage-melanoma cell interaction: Role of monocyte chemotactic protein-1 and macrophage colony-stimulating factor. Journal of Interferon & Cytokine Research: The Official Journal of the International Society for Interferon and Cytokine Research. 2005;25(11):674–683. doi: 10.1089/jir.2005.25.674. [DOI] [PubMed] [Google Scholar]
- 18.Vink A, Schoneveld AH, Lamers D, Houben AJ, van der Groep P, van Diest PJ, et al. HIF-1 alpha expression is associated with an atheromatous inflammatory plaque phenotype and upregulated in activated macrophages. Atherosclerosis. 2007;195(2):e69–75. doi: 10.1016/j.atherosclerosis.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 19.Hickey MM, Simon MC. Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Current Topics in Developmental Biology. 2006;76:217–257. doi: 10.1016/S0070-2153(06)76007-0. [DOI] [PubMed] [Google Scholar]
- 20.Lin SK, Shun CT, Kok SH, Wang CC, Hsiao TY, Liu CM. Hypoxia-stimulated vascular endothelial growth factor production in human nasal polyp fibroblasts: Effect of epigallocatechin-3-gallate on hypoxia-inducible factor-1 alpha synthesis. Archives of Otolaryngology--Head & Neck Surgery. 2008;134(5):522–527. doi: 10.1001/archotol.134.5.522. [DOI] [PubMed] [Google Scholar]
- 21.Ruas JL, Lendahl U, Poellinger L. Modulation of vascular gene expression by hypoxia. Current Opinion in Lipidology. 2007;18(5):508–514. doi: 10.1097/MOL.0b013e3282efe49d. [DOI] [PubMed] [Google Scholar]
- 22.Wu S, Nishiyama N, Kano MR, Morishita Y, Miyazono K, Itaka K, et al. Enhancement of angiogenesis through stabilization of hypoxia-inducible factor-1 by silencing prolyl hydroxylase domain-2 gene. Molecular Therapy: The Journal of the American Society of Gene Therapy. 2008 doi: 10.1038/mt.2008.90. Epub ahead of print. [DOI] [PubMed] [Google Scholar]