

Abstract
Homer and others (2008. Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS Genetics 4, e1000167) recently showed that, given allele frequency data for a large number of single nucleotide polymorphisms in a sample together with corresponding population “reference” frequencies, by typing an individual's DNA sample at the same set of loci it can be inferred whether or not the individual was a member of the sample. This observation has been responsible for precautionary removal of large amounts of summary data from public access. This and further work on the problem has followed a frequentist approach. This paper sets out a Bayesian analysis of this problem which clarifies the role of the reference frequencies and allows incorporation of prior probabilities of the individual's membership in the sample.
Keywords: Bayesian analysis, Data confidentiality, Statistical genetics
1. INTRODUCTION
Homer and others (2008) have recently addressed the problem of inference of whether an individual has contributed to a mixture, given a large number of measurements on the individual and the mean measurements in the mixture. Their primary concern was with the forensic problem arising when one wishes to determine whether an individual contributes DNA to a pooled sample, but the problem also arises in assessing whether publication of allele frequencies for large numbers of single nucleotide polymorphisms (SNPs) in medical studies compromises undertakings given to subjects that they will not be identified. The publication of this paper raised sufficient concerns as to cause removal of large amounts of summary SNP data from publicly accessible web sites.
Homer and others took a frequentist, hypothesis testing approach to the problem. Briefly, and using their notation, their approach was as follows. Assume genotype is known from a “test” individual at SNPs j = 1,…, P and denote these by Yj∈(0,0.5,1), where 0 and 1 refer to the homozygous genotypes and 0.5 to the heterozygous genotype. In addition, it is assumed that allele relative frequencies are also available for a sample of size N and for a “reference population.” These were denoted Mj and Popj. For each SNP, define the difference
| (1.1) |
Under H0 (that the test subject does “not” belong to the sample), these differences are assumed to have zero mean. To test this hypothesis against the alternative hypothesis, H1 (the test subject “does” belong to the sample), Homer and others advocate a 1-sample, 1-sided t-test, initially treating the SNPs j = 1,…, P as independent observations.
This work appeared in a nonstatistical journal, and the statistical problem was stated somewhat informally. In particular, the precise role of the “reference population” in the inference is not very clear. Subsequently, Jacobs and others (2009) described a more formal likelihood-based hypothesis testing approach in which there is no reference but summary data are available for 2 samples, assumed to be drawn from the same population. A 2-sided test statistic was derived, which has zero expectation when the test sample belongs to neither sample, and a nonzero value otherwise, with sign determined by the sample to which it belongs.
Here, a Bayesian approach to the problem, as originally described by Homer and others (2008) is proposed. By deriving a Bayes factor for whether or not the test observation was in the sample, it becomes possible to incorporate additional prior information and to decide on an appropriate level of evidence to allow a decision to be made. The approach also clarifies the role of the “reference” frequencies and allows quantitative exploration of the effects of the use of nonrepresentative reference data.
2. A SIMPLE GAUSSIAN PROBLEM
It is convenient to start with a simple Gaussian problem which is closely analogous to that considered above. Consider a P-dimensional observation, x, assumed to be sampled at random from a multivariate normal distribution with mean vector μ and variance–covariance matrix IP. Also available is the mean of a sample of N such observations, 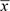 . We wish to determine whether the observation belonged to the sample (H1) or not (H0). Formally,
. We wish to determine whether the observation belonged to the sample (H1) or not (H0). Formally,
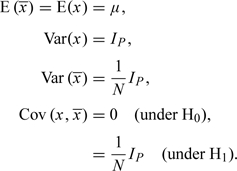 |
It seems natural to approach this problem by calculation of the Bayes factor, Pr(Data|H1)/Pr(Data|H0). This can then be multiplied by the prior odds (which would be expected to vary from one situation to another), yielding the posterior odds. However, the problem of the nuisance parameter, μ, must be addressed. Three different situations can be considered:
μ is known with certainty,
no information concerning μ is available, or
a preliminary estimate of μ is available from external data.
The problem considered by Homer and others is analogous to this simple problem and their use of a allele frequencies from a “reference population” is clearly analogous to provision of information about the means, μ, in the current formulation. However, it is not clear whether their reference population allele frequencies really do refer to population parameters or to estimates derived from external sources (although, clearly, only the latter would be available in practice).
When the value of μ is known, μ0 say, then some simple algebra yields
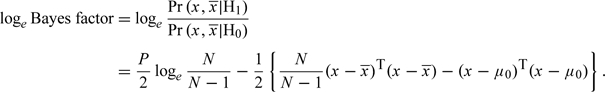 |
(2.1) |
Similarly to Homer and others, this contrasts the distance of x from with the distance of x from a “reference” value, μ0, although the distance metrics differ. For large N, the expectation of this is, to a close approximation, ±P/(2N), where the sign is positive under H1 and negative under H0. Thus, when the number of variables is even a relatively small multiple of the sample size, it will be possible to discriminate between the H0 and H1 with some confidence. Since modern gene chips deliver data on up to 10 00 000 SNPs and sample sizes rarely exceed a few thousand, this somewhat simplified analysis would suggest that, as suggested by Homer and others, it should be relatively easy to determine whether a DNA sample was one of those considered in a study for which we have summary data.
This conclusion, however, depends crucially on the availability of appropriate reference data. The situation of complete ignorance of μ can be modeled by integration with respect to μ over a uniform prior distribution. This yields
| (2.2) |
This depends only on the distance between x and . For large N, its expectation under H1 and H0 is approximately ±P/N2. Thus, in these circumstances, the number of variables needed to be measured to place an individual in a sample is proportional to N2 rather than to N.
The 2 scenarios discussed above represent extreme positions and real situations are likely to be intermediate between them. This can be represented by assuming an “informative prior” for μ:
| (2.3) |
This is the situation where the prior is derived entirely from a “representative reference sample” of size K, that is, a sample drawn from the same population as the study observations (both x and the observations contributing to ). Integration with respect to this prior then yields,
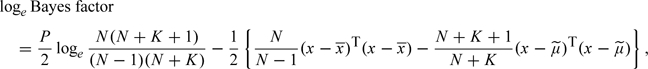 |
(2.4) |
where 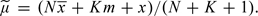 . Again, this expression has some similarity to the statistic of Homer and others (1.1), contrasting the distance of x from the sample mean, with its distance from a reference, now given by
. Again, this expression has some similarity to the statistic of Homer and others (1.1), contrasting the distance of x from the sample mean, with its distance from a reference, now given by 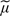 . However, whereas Homer and others used an L1 (Manhattan) distance metric, the current approach leads to the use of the L2, or Euclidean, metric.
. However, whereas Homer and others used an L1 (Manhattan) distance metric, the current approach leads to the use of the L2, or Euclidean, metric.
For large N and K, it is shown in the Appendix that the marginal expectations of the log Bayes factor (2.4) under H1 and H0 are given approximately by
| (2.5) |
Thus, the number of variables needed to place the test individual in the sample is proportional to the sample size N unless the amount of extraneous information (i.e. K) is small. As K approaches zero, the approximation eventually becomes dominated by the term in P/N2.
Table 1 shows the number of variables needing to be measured for the expectation of the log10 Bayes factor to be ±5 under H1 and H0.
Table 1.
Number of variables required to discriminate between hypotheses. The table shows, for sample sizes N = 100 and N = 1000, and with varying amounts of prior information concerning μ, the number of variables, P, needed for the expectation of log 10Bayes factor to be + 5 under H1 and – 5 under H0
| Prior knowledge of μ | Sample size |
|
| N = 100 | N = 1000 | |
| Known | 2300 | 23 000 |
| Ignorance | 115 000 | 11 500 000 |
| Informative prior | ||
| K = 200 | 3450 | 138 000 |
| K = 500 | 2760 | 69 100 |
| K = 5000 | 2350 | 27 600 |
3. NONREPRESENTATIVE REFERENCE SAMPLE
The last section concluded with a discussion of the more realistic situation in which we have partial prior knowledge of μ. We have assumed that this prior information is derived from a representative reference sample of size K drawn from the same population as the single observation, x, and the sample which may or may not include it, which has mean . If the mean of the representative reference sample is m, this yields the prior distribution (2.3). A simple model for nonrepresentativeness of the reference sample assumes that it is drawn from a reference population in which the variance is the same as that of the study population, that is, IP, but where the means in the study and reference populations differ by the unobserved vector
| (3.1) |
F is assumed to be a known constant which has an interpretation as an intraclass correlation coefficient. After integration over ∈, the prior distribution for μ is of the same form as (2.3), but with K replaced by
| (3.2) |
Consequently, the log Bayes factor is given by (2.4) with K replaced by K′. Note that
For example, when F = 0.005, K′ cannot exceed 199 no matter how large the reference sample.
If F is correctly specified, the expression (2.5) for the marginal expectation of the log Bayes factor will continue to hold when K is replaced by K′ as described above. However, misspecification of F will lead to poorly calibrated inference.
4. BINOMIAL OBSERVATIONS
In out-bred populations, autosomal SNP genotypes coded (0, 0.5, 1) as described in Section 1 can be treated as binomial variates on 2 trials. If the mean of the ith SNP genotype is μi, then its variance is μi(1 − μi)/2. A natural approach is to adopt a Gaussian approximation, but it is first necessary to relax the unrealistic and inflexible variance assumption of Section 3. This is achieved simply by replacing the identity matrix, IP, by a more general matrix, Σ. The results of Section 2 are changed only by replacement of inner products of the form vTv by quadratic forms vTΣ − 1v. Thus, in the case of partial prior knowledge of μ, the log Bayes factor (2.4) becomes
| (4.1) |
If the model for nonrepresentative prior is modified so that the differences between means of study and reference populations have variance proportional to Σ:
| (4.2) |
then, as before, K must be replaced by K′ as given by (3.2).
Assuming SNPs to be in linkage equilibrium, a Gaussian approximation for the log Bayes factor can be obtained by use of (4.1), approximating Σ by the diagonal matrix with elements 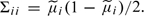 . With this variance assumption, the parameter F in the model for a nonrepresentative reference sample is equivalent to Wright's FST measure of divergence between the study and reference populations. The value F = 0.005 used as an illustration above is at the extreme of the values observed between different European populations (Heath and others, 2008). Thus, this would represent a relatively seriously nonrepresentative reference sample.
. With this variance assumption, the parameter F in the model for a nonrepresentative reference sample is equivalent to Wright's FST measure of divergence between the study and reference populations. The value F = 0.005 used as an illustration above is at the extreme of the values observed between different European populations (Heath and others, 2008). Thus, this would represent a relatively seriously nonrepresentative reference sample.
As has been pointed above, the log Bayes factor contrasts the L2 distances between x and with that between x and , whereas the statistic of Homer and others contrasts the corresponding L1 distances. However, whereas Homer and others weighted absolute distances for each SNP equally, the Bayes factor approach weights the squared distance for the ith SNP by 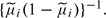 .
.
Figure 1 shows values of log10 Bayes factors for 1000 simulations of each of the combinations of (N,K,P) listed in the last part of Table 1 both when the observation was included in the sample (H1) and when it was not (H0). The numbers of SNPs were chosen to ensure that the expectation of the log Bayes factor was ±5 and the simulated values were distributed symmetrically around these expected values. However, the simulated values were quite variable, the extent of variability seemingly unrelated to either the sample size or the number of variables.
Fig. 1.

A simulation study. For each of the partial prior knowledge cases shown in Table 1, 1000 simulations were run for genotype (binomial) data under each of H1 (In sample) and H0 (Not in). The box plots show the distribution of values of the Gaussian approximation to log10(Bayes factor).
An exact treatment of the binomial case is also possible, at least in the case of independent variables. the likelihood contribution for the ith SNP is given by
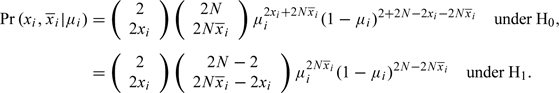 |
The conjugate prior for μi is a beta distribution. Parameterizing this to be equivalent to the information gained from observing an allele frequency mi in a reference sample of size K:
where B() is the beta function. Noting that 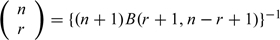 , it follows that the log Bayes factor is given by
, it follows that the log Bayes factor is given by
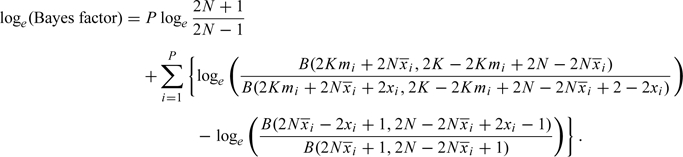 |
Figure 2 compares exact and approximate log Bayes factors for 20 simulations from each of H0 and H1 (N = 100,K = 200 and P = 3450). It can be seen that the Gaussian approximation performs very well in this relatively small problem.
Fig. 2.

Gaussian approximation log Bayes factors versus exact values. This figure shows the results of 20 simulations under each of H0 and H1 for binomial data with N = 100,K = 200 and P = 3450.
For large N and K, application of Stirling's approximation in the above expression yields the further approximation
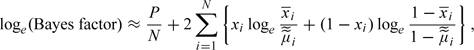 |
where 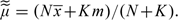 . This is similar in form to the test statistic proposed by Jacobs and others (2009).
. This is similar in form to the test statistic proposed by Jacobs and others (2009).
5. CORRELATED VARIABLES
Previous sections have assumed that the set of variables is independent. In the case of SNP genotypes, when only a relatively small number of SNPs are required, independence could be ensured by picking well-separated SNPs. However if, either because of large N or small K′, a large number of SNPs are required, the independence assumption is likely to break down because of “linkage disequilibrium” between SNPs which are physically close together on the genome. Homer and others address this problem without modifying their statistic by calculating an empirical estimate of its distribution. This is calculated by repeated sampling, with replacement, from a reasonably extensive set of individual-level data for the same set of SNPs. Jacobs and others discuss the problem of linkage disequilibrium only in very general terms but seem also to advocate use of an empirical distribution for an unmodified test statistic.
The current Bayesian approach requires estimates of the intercorrelation between SNPs and this, too, will generally require access to a set of individual-level data. Currently, the only widely available data set of this type is that from the International HapMap Project (The International HapMap Consortium, 2003) in which sample sizes are relatively small. However, larger sample sizes will become available, for example, from the 1000 Genomes Project.
Calculation of the Gaussian approximation to the Bayes factor can allow for correlation, as is clear from the general expression (4.1). Σ need not be diagonal and can be estimated from a suitable external data set. However, estimation of Σ − 1 is not a trivial problem since even the largest data sets likely to be available will have many fewer subjects than variables. It seems sensible to continue to estimate the diagonal elements of Σ from the study data. Thus, writing
where D is diagonal with 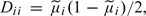 it is then necessary to estimate the inverse correlation matrix, R − 1 from the external data set. Note, however, that this matrix is likely to be extremely sparse and the nonzero elements will lie quite close to the diagonal.
it is then necessary to estimate the inverse correlation matrix, R − 1 from the external data set. Note, however, that this matrix is likely to be extremely sparse and the nonzero elements will lie quite close to the diagonal.
The problem of estimation of very large inverse correlation and partial correlation matrices has received some attention recently. The problems of instability of estimates and loss of rank are approached by the use of penalty functions. Schäfer and Strimmer (2005) proposed a shrinkage approach based on an L2 penalty function, but the use of L1 penalties leads to sparse solutions and has been preferred by most authors (Meinshausen and Bühlmann, 2006), (Yuan and Lin, 2007), (Friedman and others, 2008).
Returning to the Gaussian problem, a small simulation study was undertaken to test the sensitivity of the calculations to correlation between the variables and to assess the possibility of correcting the calculations using estimates of the inverse correlation matrix obtained from an additional sample. In this study, the variables formed a first order autoregressive (AR(1)) process with correlation 0.5 between neighboring variables. A slight adaptation of the method of Meinshausen and Bühlmann (2006) was used for estimation of the inverse correlation matrix, owing to its speed and its ease of implementation, taking account of the local nature of the dependencies. The method is based on the fact that the off-diagonol elements of the ith column of an inverse covariance matrix contain, with a change of sign, the regression coefficients of the ith variable on the remaining variables, multiplied by the reciprocal of the residual variance from this regression, which also provides the ith diagonal element of the inverse matrix. Meinshausen and Bühlmann (2006) estimated the inverse covariance matrix by estimating each of these regressions using the lasso. Here, the inverse correlation matrix was estimated in a similar manner, by a series of least angle regressions (LAR) (Efron and others, 2003), restricting the choice of variables to 10 on either side of each target (LAR is computationally faster than the lasso and gives very similar results). Since the initial estimate obtained in this way is not a symmetric matrix, in the calculations shown the final estimate was taken as the mean of the initial estimate and its transpose. However, using the initial nonsymmetric estimate gave almost identical results and is computationally more convenient. For this reason, the nonsymmetric estimate was used in the real data example discussed in Section 6.
Figure 3 shows the results of 100 simulations under H1 and under H0 of the case N = 100, K = 200 and P = 3450 for which the expected log10 Bayes factor is ±5. Figure 3(a) shows the log Bayes factor calculated by ignoring the correlation between variables against the correct values which use the known correlation structure. Although, as expected, the mean log Bayes factor remains approximately at ±5 (see Appendix), individual values can be seriously misestimated. Figure 3(b) shows the effect of estimating the correlation structure, as described above, using an additional sample of size 200, while in Figures 3(c) and (d), the additional sample size is 500 and 1000, respectively. The LAR calculations were carried out in the R lars package, allowing up to 10 steps and picking the solution with the smallest Cp statistic. This performed well although, when the sample size for estimation of the partial correlations was small, there was a tendency for Bayes factors to be biased downward, particularly in the H0 simulations. This is presumably a consequence of the bias in the estimates of the partial correlations, which will be more pronounced at smaller sample sizes.
Fig. 3.

Effect of correlation between variables. This figure shows the results of 100 simulations under each of H0 and H1 for binomial data with N = 100,K = 200 and P = 3450 when the variables are intercorrelated according to an AR(1) model with lag 1 correlations 0.5. (a) No correction for correlation. (b) Correction using estimated inverse correlation matrix from a sample of size 200. (c) Correction using a sample of size 500. (d) Correction using a sample of size 1000.
6. A REAL EXAMPLE
The data from this example were drawn from the Wellcome Trust Case Control Consortium study (Wellcome Trust Case Control Consortium, 2007). The sample allele frequencies, concern the N = 145 subjects from the British Birth Cohort (BBC), who were resident in Scotland, and the reference sample consisted of the K = 1455 National Blood Service controls drawn from throughout Great Britain. This reference sample provides both a prior estimate, m, of the population allele frequencies and an estimate of the inverse correlation matrix between SNPs. Only data for the P = 4743 SNPs on chromosome 20 which have call rates of at least 97.5% and minor allele frequencies of at least 10% were used. For these values of N, K, and P, the expected values of the log10 Bayes factor are approximately ±6.5 so that this chromosome 20 data should be adequate, most of the time, to determine whether an individual observation, x, was or was not drawn from the 145 Scottish subjects. To test this, 50 subjects were drawn at random from the Scottish BBC subjects and a further 50 were drawn the BBC subjects who were resident in the rest of Great Britain.
The distribution of log10 Bayes factors for the 2 groups of subjects are shown in Figure 4(a). Mean log10 Bayes factors were +5.91 and − 6.21 in the 2 groups of subjects, which agrees reasonably closely with expectation. This suggests that the reference sample was not seriously nonrepresentative, despite the fact that it was drawn from all over Great Britain rather than from Scotland. A pessimistic scenario was also investigated, taking FST = 0.003, which corresponds with the value found by Heath and others (2008) for the FST between Russia and the United Kingdom. This has the effect of rendering the effective reference sample size to be only 270.5 and the expected log10 Bayes factors to be ±4.6. Figure 4(b) compares the 2 sets of Bayes factors. The allowance for a seriously unrepresentative prior has, as expected, made it rather more difficult to identify individuals as belonging to the summarized sample. The mean log10 Bayes factors in the 2 groups, at + 3.03 and − 5.62, were some way from expectation indicating that this prior was not well specified.
Fig. 4.

A real example using chromosome 20 data drawn from the Wellcome Trust Case Control Consortium (N = 145, P = 4743 and K = 1455). The ordinate in (b) is calculated under the assumption of a nonrepresentative reference sample (FST = 0.003), while that in (c) ignores linkage disequilibrium. Otherwise Bayes factors are calculated on the assumption that the reference sample is representative and allowing for linkage disequilibrium between SNPs.
Finally, Figure 4(c) shows the effect of ignoring linkage disequilibrium. As before, the mean values of the log10 Bayes factors was not seriously affected although individual values could be changed substantially.
7. DISCUSSION
A question often asked is whether the difficulty identified by Homer and others extends to widespread dissemination of other study results, in particular to publication of test statistics for case/control comparisons. That it does, at least in some circumstances can be demonstrated by a simple extension of the Gaussian problem set out in Section 2. Suppose that vectors of means for 2 samples of size N1 and N2 have been compared, using a vector of z-tests, 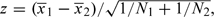 , and also suppose that we have a test observation, x. Given z and an estimate of the population mean μ, can we determine whether x was part of Sample 1, Sample 2, or neither?
, and also suppose that we have a test observation, x. Given z and an estimate of the population mean μ, can we determine whether x was part of Sample 1, Sample 2, or neither?
The methods described here can readily be adapted to this problem. If x is in neither sample, (x − μ) is uncorrelated with z. However, if it is in either sample,
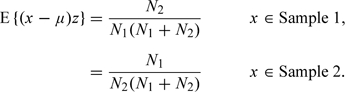 |
Thus, 2 Bayes factors can be calculated using similar arguments to those developed above. Note, however, that the chi-squared tests, z2, could not be used in this way since the loss of sign destroys the correlations which provide the information for discrimination between hypotheses. This also applies to p-values calculated from the tests.
It could be argued that scenarios in which an individual might be identified in this manner are somewhat improbable—particularly when so many SNPs would be needed that linkage disequilibrium could not be ignored (so that any potential invader of privacy would also require access to an individual-level data set from which to estimate the linkage disequilibrium structure). However, this is usually not relevant; in agreeing to take part in a study, subjects will have been assured that data which would allow them to be identified would not be made public. It is now clear that publication of summary data could, in some circumstances, violate this assurance.
Homer and others also considered the case in which raw allele intensity measures are available for an individual and for a pooled DNA sample. The problem is to determine whether or not the individual contributed to the pool. The details of their treatment of allele signal intensities from high-density SNP genotyping chips are beyond the scope of this paper but result in a continuous measure for each SNP which they are able to assume to be approximately Gaussian; these are essentially the signals which are converted by genotype scoring algorithms into a 3-level discrete genotype code. The methods described here are potentially applicable to such intensity data, although additional work might be necessary to ensure outlier resistance. However, as pointed out by Homer and others, N is replaced by the reciprocal of the proportion of the DNA in the pool which could derive from the individual of interest and, particularly in forensic applications, this will usually be unknown. In such cases, it will be necessary to extend the Bayesian treatment to incorporate a prior dstribution for N.
FUNDING
Wellcome Trust Principal Research Fellowship to D.C. Funding for the WTCCC project was provided by the Wellcome Trust under award 076113 and 085475.
Acknowledgments
The example of Section 6 makes use of data generated by the Wellcome Trust Case Control Consortium (WTCCC). A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. I should like to thank colleagues and the referees for helpful comments on an earlier draft. Conflict of Interest: None declared.
APPENDIX: THE EXPECTATION OF THE LOG BAYES FACTOR
The distributions of x and under H0 and H1, conditional upon the vector of means μ, have been set out in Section 2. Further, the prior distribution of μ has been assumed to be N(m,IP/K). In this section, the expectation of the log Bayes factor (2.4) over the joint distribution of x, , and μ is derived.
It is easily shown that the elements of (x − ) have zero mean and variances (N + 1)/N under H0 and (N − 1)/N under H1. We can express (x − ) as
It follows that the elements of (x − ) also have zero mean and variances (N + K)/(N + K + 1) under H0 and (N + K)(N + K − 1)/(N + K + 1)2 under H1. From these results, it follows that
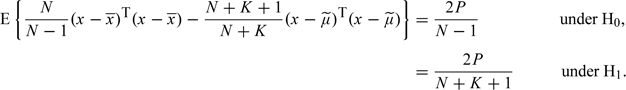 |
For large N, K these become, approximately, 2P/N and 2P/(N + K), respectively, and
The approximate expectation (2.5) follows directly from these results.
Note that this argument makes it clear that the expectation of (2.4) is unaffected by any correlation between elements within (x − ) and between elements within (x − ) although, in these circumstances, the correct log Bayes factors are given by (4.1). This explains the results of Figure 3(a). A minor extension of the argument outlined above shows that the expectation of the correct log Bayes factor (4.1) is also approximated by (2.5).
References
- Efron B, Hastie T, Johnstone I, Tibshirani R. Least angle regression. Annals of Statistics. 2003;32:407–499. [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics. 2008;9:432–441. doi: 10.1093/biostatistics/kxm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath SC, Gut IG, Brennan P, McKay JD, Bencko V, Fabianova E, Foretova L, Georges M, Janout V, Kabesch M. Investigation of the fine structure of European populations with applications to disease association studies. European Journal of Human Genetics. 2008;16:1413–1429. doi: 10.1038/ejhg.2008.210. and others. [DOI] [PubMed] [Google Scholar]
- Homer N, Szelinger S, Redman M, Duggan D, Tembe W, Muehling J, Pearson JV, Stephan DA, Nelson SF, Craig DW. Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS Genetics. 2008 doi: 10.1371/journal.pgen.1000167. 4, e1000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KB, Yeager M, Wacholder S, Craig D, Kraft P, Hunter DJ, Paschal J, Manolio TA, Tucker M, Hoover RN. A new statistic and its power to infer membership in a genome-wide association study using genotype frequencies. Nature Genetics. 2009;41:1253–1257. doi: 10.1038/ng.455. and others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinshausen N, Bühlmann P. High dimensional graphs and variable selection with the lasso. Annals of Statistics. 2006;34:1436–1462. [Google Scholar]
- Schäfer J, Strimmer K. A shrinkage approach to large-scale covariance matrix estimation and impications for functional genomics. Statistical Applications in Genetics and Molecular Biology. 2005 doi: 10.2202/1544-6115.1175. 4, Article 32. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. The International HapMap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Lin Y. Model selection and estimation in the gaussian graphical model. Biometrika. 2007;94:19–35. [Google Scholar]