

SUMMARY
Aggregates of the hyperphosphorylated microtubule associated protein tau (MAPT) are an invariant neuropathological feature of tauopathies. Here we show that microglial neuroinflammation promotes MAPT phosphorylation and aggregation. First, lipopolysaccharide-induced microglial activation promotes hyperphosphorylation of endogenous mouse MAPT in non-transgenic mice that is further enhanced in mice lacking the microglial-specific fractalkine receptor (CX3CR1) and is dependent upon functional toll-like receptor 4 and interleukin 1 (IL1) receptors. Second, humanized MAPT transgenic mice lacking CX3CR1 exhibited enhanced MAPT phosphorylation and aggregation as well as behavioral impairments that correlated with increased levels of active p38 MAPK. Third, in vitro experiments demonstrate that microglial activation elevates the level of active p38 MAPK and enhances MAPT hyperphosphorylation within neurons that can be blocked by administration of an interleukin 1 receptor antagonist and a specific p38 MAPK inhibitor. Taken together, our results suggest that CX3CR1 and IL1/p38 MAPK may serve as novel therapeutic targets for human tauopathies.
INTRODUCTION
Increasing evidence suggests that neuroinflammation is a common feature of tauopathies. First, activated microglia are found in the postmortem brain tissues of various human tauopathies including Alzheimer’s disease (AD), Frontotemporal Dementia (FTD), Progressive Supranuclear Palsy and Corticobasal Degeneration (Gebicke-Haerter, 2001; Gerhard et al., 2006; Ishizawa and Dickson, 2001). Second, induction of systemic inflammation via administration of the Toll-like receptor 4 (TLR4) ligand, lipopolysaccharide (LPS) significantly induced MAPT hyperphosphorylation in a triple transgenic mouse model of AD (Kitazawa et al., 2005). Third, immunosuppressant drug FK506 attenuated microglial activation and extended the lifespan of P301S transgenic mouse model of FTD (Yoshiyama et al., 2007). Finally, a growing number of studies suggest that pro-inflammatory cytokines, such as interleukin 1 (IL1), interleukin 6 and nitric oxide released from astrocytes can accelerate MAPT pathology and formation of neurofibrillary tangles (NFTs) in vitro (Li et al., 2003; Quintanilla et al., 2004; Saez et al., 2004). While these findings suggest a correlative link between neuroinflammation and tauopathies, there is little mechanistic evidence that altered microglial activation plays a pathogenic role in the formation of MAPT pathologies.
Recent experimental evidence has directly implicated microglia in the pathogenesis of a variety of neurodegenerative diseases, including Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and a mouse model of systemic inflammation. One signaling pathway through which neurons and microglia communicate and has been demonstrated to play an important role in neuroinflammation and neuroprotection is fractalkine (CX3CL1) and its cognate receptor (CX3CR1), a unique, one-to-one ligand-receptor chemokine pair. Notably, CX3CL1 is highly expressed in neurons while CX3CR1 is exclusively expressed in microglia within the CNS (Harrison et al., 1998). Furthermore, exogenously added CX3CL1 is neuroprotective in models of in vitro neuroinflammation (Meucci et al., 1998; Mizuno et al., 2003). In addition, a genetic variant with reduced levels of CX3CR1 has been associated with age-related macular degeneration in humans (Combadiere et al., 2007). Notably, disruption of CX3CL1-CX3CR1 signaling by deletion of the Cx3cr1 gene induces neurotoxicity in mouse models of systemic inflammation, PD and ALS (Cardona et al., 2006), although is protective against neuronal loss in a mouse model of focal cerebral ischemia (Denes et al., 2008) and in the triple transgenic (3xTg) mouse model of AD (Fuhrmann et al., 2010). However, the CX3CR1 deficient 3xTg animals were examined at an age prior to the development of either extracellular Aβ deposition or intracellular MAPT aggregation that defines AD and thus the nature of the signal that leads to the neurotoxicity in this model that is protected by CX3CR1 deficiency is unclear. Finally, we recently observed that CX3CR1 deficiency leads to reduced Aβ deposition in two different transgenic mouse models of AD, potentially through enhanced uptake of fibrillar Aβ by CX3CR1 deficient microglia (Lee et al., 2010). Together, these studies demonstrate that altered microglial-signaling through CX3CR1 plays a direct role in neurodegeneration and/or neuroprotection depending upon the CNS insult.
In the current studies, we evaluated the effects of either LPS administration and/or CX3CR1 deficiency on MAPT hyperphosphorylation, and aggregation in both non-transgenic mice and in a humanized mouse model of tauopathy (hTau). LPS administration induced hyperphosphorylation of both endogenous and transgene-derived MAPT that was dependent upon LPS dose and CX3CR1 deficiency. Furthermore, introduction of CX3CR1 deficiency into hTau mice resulted in altered microglial activation, enhanced MAPT phosphorylation and aggregation, as well as behavioral abnormalities. Finally, we provide mechanistic data suggesting that the pathways responsible for these effects include microglial-derived interleukin 1 (IL1) and neuronal p38 mitogen activated protein kinase (MAPK). These results suggest a direct link between microglial activation and MAPT phosphorylation and aggregation within neurons.
RESULTS
Enhanced MAPT phosphorylation after systemic LPS administration
To study the relationship between microglial activation and phosphorylation of endogenous mouse MAPT, two-month old C57BL/6 mice were exposed to an inflammatory stress via peripheral administration of either low (1 mg/kg; intraperitoneal) or high (10 mg/kg; i.p) dose of LPS and the brain extracts were prepared for Western blot analysis (Figure 1A). With increasing concentrations of LPS, there were elevated levels of mouse MAPT phosphorylated at both ser202 (AT8) and thr231 (AT180) in brain extracts when compared to the vehicle injected group (Figure 1B–D). In addition, there were increases in other phosphorylated MAPT epitopes, including ser396/ser404 (PHF1, data not shown).
Figure 1. Enhancement of microglial activation via injection of LPS or genetic deletion of Cx3cr1 promotes MAPT hyperphosphorylation.
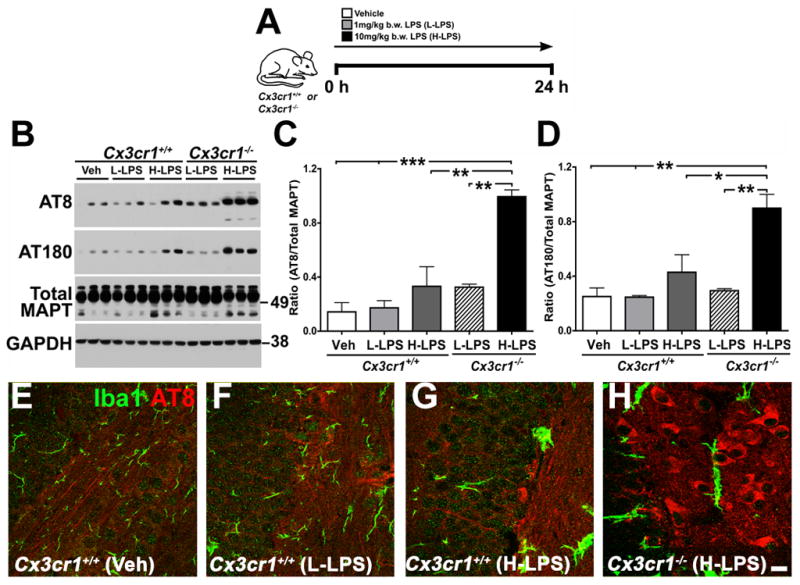
(A) Schematic diagram showing that two-month-old Cx3cr1+/+ or Cx3cr1−/− mice were injected with a single dose of L-LPS (1.0 mg/kg b.w; i.p.) or H-LPS (10.0 mg/kg b.w; i.p) or Vehicle (Veh), sacrificed after 24 h and brains processed either for biochemical or immunohistochemical analysis. (B) Western immunoblot of hippocampal extracts shows LPS induces dose-dependent increases in AT8 (ser202) and AT180 (thr231) site phosphorylation of endogenous mouse MAPT in Cx3cr1+/+ mice, which was further elevated in Cx3cr1−/− mice injected with L-LPS or H-LPS. Total MAPT and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels were similar across all samples. (C–D) Quantification of Western blots revealed a statistically significant (n=4 for Cx3cr1+/+ with Veh; n=5 for Cx3cr1+/+ with L-LPS; n=4 for Cx3cr1−/− with L-LPS; n=3 for Cx3cr1+/+ with H-LPS and n=3 for Cx3cr1−/− with H-LPS; mean±s.e.m of integrated density value – IDV ratio; *p<0.05; **p<0.01; ***p<0.001; one-way ANOVA with Tukey multiple comparison posthoc test) increase in the AT8/Total MAPT and AT180/Total MAPT with H-LPS treatment of wild-type mice and an even greater increase in LPS injected Cx3cr1−/− mice. (E–H) Double immunofluorescence revealing Iba1+ morphological activation of microglia (green) and AT8 + neurons (red) in wild-type mice injected with LPS that was further enhanced in LPS injected Cx3cr1−/− mice. Scale bar 10 μm. See also Figure S1.
To examine the effects of further enhancing microglia activation on MAPT phosphorylation, we utilized Cx3cr1−/− mice. Previous studies demonstrated that loss of Cx3cr1 promotes microglial activation and neurodegeneration in mouse models of PD, ALS, as well as in systemic inflammation induced by LPS (Cardona et al., 2006). Notably, LPS administration to Cx3cr1−/− mice resulted in a dramatic, dose-dependent elevation in phosphorylation of endogenous mouse MAPT at both the AT8 and AT180 (Figure 1B–D), as well as at PHF1 (data not shown), sites when compared to non-transgenic controls and vehicle injected Cx3cr1−/− mice (data not shown). To confirm the biochemical data, fluorescent immunohistochemistry was performed on mouse brain sections revealing the presence of AT8+ cells within the dentate gyrus of LPS administered non-transgenic mice when compared to vehicle injected controls (Figure 1E–G) that was further enhanced in the Cx3cr1−/− mice (Figure 1H). These findings demonstrate that LPS induces elevated endogenous MAPT phosphorylation that is dependent upon LPS dose and the presence/absence of CX3CR1.
Alterations in microglial activation in hTau mice
To examine whether altered microglial activation was observed in a mouse model of tauopathy, transgenic mice in which the wild-type human MAPT gene replaced the endogenous mouse Mapt gene (hTau mice) were examined for the age-related appearance of MAPT pathology and microglial activation. hTau mice first develop hyperphosphorylated MAPT at 3 months of age, MAPT aggregates at 9 months and neuronal loss by 15 months of age (Andorfer et al., 2003). Notably, by about 12 months of age, hTau mice exhibited Iba1+ microglia in the hippocampus with shorter processes and rounder cell bodies consistent with microglial activation (Figure S1B) that was even more pronounced in 18-month-old hTau mice (Figure S1C). By contrast, non-transgenic mice at 18 months of age exhibited Iba1+ microglia that were more ramified in appearance, consistent with a quiescent phenotype (Figure S1A).
To support the immunohistochemistry results, the levels of various cytokines and chemokines were also examined in the hTau mice by ELISA. Notably, by as early as six months of age, the levels of cleaved CX3CL1 were substantially elevated in hippocampal extracts from hTau mice when compared to non-transgenic controls (Figure S1H), whereas the levels of other cytokines (TNFα, INFγ, IL1β, IL6) remained unchanged (data not shown). Furthermore, analysis of the levels of additional chemokine/cytokine transcripts via quantitative real-time PCR revealed elevated levels of both NOS2 and MPC1/CCL2 transcripts in hemibrains of six-month old hTau mice when compared to age-matched controls (Figure S1I–J). Taken together, these results suggest that altered age-related alterations in neuroinflammation are observed in hTau mice at early stages of disease progression, including the production of CX3CL1, the ligand for the microglial receptor, CX3CR1.
Enhanced MAPT phosphorylation in LPS injected hTau mice
To test whether further enhancing microglial activation in the hTau mice could promote MAPT hyperphosphorylation, two-month-old hTau mice (prior to the first appearance of hyperphosphorylated MAPT in this model) were peripherally administered with a low-dose (1 mg/kg; i.p) of LPS. LPS induced MAPT hyperphosphorylation at the phospho-thr231 (TG3) and AT8 epitopes in the hippocampus of hTau mice (Figure S1E, S1G) when compared to vehicle injected hTau mice (Figure S1D, S1F). Considering that the somato-dendritic accumulation of AT8+ MAPT does not occur until three months in the hippocampus of hTau mice (Andorfer et al., 2003), these results suggest that induction of TLR4-mediated systemic inflammation via LPS induces MAPT hyperphosphorylation in pre-pathological hTau mice.
Worsened disease in hTau-Cx3cr1−/− transgenic mice
LPS exerts its effects through TLR4 signaling in astrocytes, endothelial cells, polydendrocytes and microglia. To specifically examine the role of microglial signaling in modulating MAPT pathologies, hTau mice were mated with mice lacking the microglial receptor, CX3CR1. Given our findings that CX3CR1 deficiency promotes phosphorylation of MAPT following peripheral LPS administration in non-transgenic mice (Figure 1) and that six-month-old hTau mice exhibit elevated levels of cleaved CX3CL1 (Figure S1H), we postulated that a deficiency in CX3CR1 would promote MAPT pathologies in the hTau mice.
Six-month-old hTau-Cx3cr1−/− mice exhibited significantly increased MAPT phosphorylation on AT8, AT180 and PHF1 sites when compared to age-matched hTau mice (Figure 2A–2D), while detergent soluble total MAPT levels remained unchanged (Figure 2E). Consistent with the biochemical studies, immunohistochemical analysis revealed a marked increase in the total number of AT8+ (Figure 2F–M), CP13+ and AT180+ (Figure S2A–B) neurons in the dentate gyrus and CA3 regions of hippocampus of hTau-Cx3cr1−/− mice compared to hTau mice or non-transgenic controls. The enhancement of MAPT phosphorylation in the hTau-Cx3cr1−/− mice required both the hTau transgene and loss of CX3CR1 signaling, as neither 6-month-old Cx3cr1−/− (Figure 2F–M and Figure S2J) nor Cx3cl1−/− mice (data not shown) exhibited alterations in AT8, AT180 or PHF1 epitopes when compared to non-transgenic controls.
Figure 2. CX3CR1 deficiency induces MAPT hyperphosphorylation and aggregation as well as p38 MAPK activation in six-month-old hTau mice.

(A) Western blot analysis of hippocampal lysates revealed an increase in MAPT phosphorylation at AT8, AT180 and PHF1 in hTau-Cx3cr1−/− mice, but no alterations in total MAPT levels (antibody Tau5); GAPDH was loading control. (B–E) Quantification of western blots for AT8, PHF1, AT180 and total MAPT revealed a statistically significant (n=5 for hTau-Cx3cr1+/+; n=5 for hTau-Cx3cr1−/−; mean±s.e.m of IDV; *p<0.05; unpaired t test) increase in AT8 (B), AT180 (D) and PHF1 (C), but not detergent-soluble total MAPT (E) in hTau-Cx3cr1−/− mice compared to hTau-Cx3cr1+/+ mice. (F–M) Immunohistochemistry revealed an increase in the number of AT8 positive neurons in the dentate gyrus (DG) (I) and CA3 (M) in hTau-Cx3cr1−/− mice compared to hTau-Cx3cr1+/+ mice (H, L), Cx3cr1−/− (G, K) or non-transgenic (Non-Tg) controls (F, J), Scale bar 20 μm. (N) Sarkosyl insoluble and AT8 + MAPT was higher in the hippocampi of hTau-Cx3cr1−/− mice than hTau-Cx3cr1+/+ mice. (O) Numerous Gallyas silver positive CA3 neurons were detected in the hippocampus of hTau-Cx3cr1−/− mice but not hTau-Cx3cr1+/+ mice. Scale bar 10 μm. (P–Q) Six-month-old Cx3cr1−/−, hTau-Cx3cr1+/+ and hTau-Cx3cr1−/− mice (n=8 per age group) were subjected to Y-maze test. While hTau-Cx3cr1−/− mice appeared similar to Cx3cr1−/− and hTau-Cx3cr1+/+ mice motor abilities, including the number of arms entered in the Y-maze (P), hTau-Cx3cr1−/− mice exhibited a significant deficit (*p<0.05; one-way ANOVA with Dunnett’s multiple comparison test at 99% confidence interval) in the percent spontaneous alternation when compared to control group (Q), indicating deficiencies in spatial working memory. See also Figure S2.
MAPT hyperphosphorylation is thought to be the initial step in the formation of MAPT aggregates and NFTs in the human AD brain (Greenberg and Davies, 1990). Typically, when MAPT undergoes self-aggregation, it becomes detergent insoluble in Sarkosyl (N-laurylsarcosine). To examine the effects of CX3CR1 deficiency on MAPT aggregation in the hTau mice, 1% Sarkosyl hippocampal extracts were prepared from six-month-old hTau-Cx3cr1+/+ and hTau-Cx3cr1−/− mice (Greenberg and Davies, 1990; Mocanu et al., 2008) to generate both Sarkosyl-soluble and -insoluble MAPT (“pretangles”) fractions (Mocanu et al., 2008). Western blot analysis of these fractions revealed elevated levels of Sarkosyl insoluble MAPT in hTau-Cx3cr1−/− mice when compared to hTau mice (Figure 2N) and that MAPT in the insoluble fraction was hyperphosphorylated at the AT8 site (Figure 2N).
To confirm the presence and examine the localization of insoluble MAPT in the brains of in hTau-Cx3cr1−/− mice, Gallyas silver staining, a standard method for the detection of MAPT pathology (Braak and Braak, 1991), was performed. Strikingly, numerous Gallyas positive neurons in the CA3 region of the hippocampus as well as Gallyas-positive dystrophic neurites were identified in the six-month-old hTau-Cx3cr1−/− mice but not in age-matched hTau mice (Figure 2O), non-transgenic or Cx3cr1−/− controls (data not shown). Furthermore, since Gallyas can stain other filamentous proteins including neurofilaments, immunohistochemistry for phosphorylation-dependent (SMI31 antibody) and phosphorylation-independent (SMI32 antibody) neurofilament was performed revealing the lack of detectable neurofilament aggregates in hTau-Cx3cr1−/− mice (data not shown). Given that MAPT aggregates in the hTau mice do not appear until 9 months of age (Andorfer et al., 2003), our data demonstrates that pathologic MAPT aggregation is accelerated in the hTau-Cx3cr1−/− mice.
hTau mice have been reported to exhibit behavioral impairments at 12 months of age, three months after the first appearance of MAPT aggregates. To examine whether CX3CR1 deficiency resulted in both altered behavior as well as brain pathology at an earlier time point, 6-month-old hTau-Cx3cr1−/−, hTau-Cx3cr1+/+, Cx3cr1−/− and non-transgenic mice were tested in the Y-maze. While the animals were similar in measures of arms entered (Figure 2P), a measure of general activity, the spontaneous alternation ratio, which is a measure of hippocampal-dependent spatial working memory, was significantly reduced in the hTau-Cx3cr1−/− mice when compared to hTau-Cx3cr1+/+ and Cx3cr1−/− controls (Figure 2Q). Other measures of motor abilities of the different genotypes, including open field and rotarod were not significantly different (data not shown). Taken together, these results demonstrate that CX3CR1 deficiency promotes MAPT phosphorylation and aggregation as well as behavioral abnormalities in working memory in the hTau mice.
Enhanced microglial activation in hTau-Cx3cr1−/− transgenic mice
To examine whether CX3CR1 deficiency leads to altered microglial activation in the hTau mice, brain sections were stained with a microglia-specific marker, Iba1 as well as CD68, a phagocytic marker induced upon microglial activation. Notably, CX3CR1 deficiency was associated with Iba1 positive cells that were less ramified with shorter processes and rounder cell bodies in hTau-Cx3cr1−/− mice when compared to hTau, non-transgenic or Cx3cr1−/− mice (Figure 3A–H). Quantification of Iba1 positive cells using fractal dimension analysis (Soltys et al., 2001) revealed a statistically significant reduction in fractal dimension in the hTau-Cx3cr1−/− mice (Figure 3I), consistent with enhanced microglial activation. Furthermore, CD68 staining was noticeably elevated in the hTau-Cx3cr1−/− mice when compared to hTau, non-transgenic and Cx3cr1−/− controls (Figure 3J–Q). This visual impression was confirmed upon quantification of the densities of CD68 staining (Figure 3R). In addition, both CD11b and iNOS staining were enhanced in hTau-Cx3cr1−/− mice (Figure S3A–3B).
Figure 3. CX3CR1 deficiency results in enhanced microglial activation hTau mice.

(A–H) Iba1+ microglia in the DG (A–D) and CA3 (E–H) of six-month-old hTau-Cx3cr1−/− mice displayed activated phenotype compared to hTau-Cx3cr1+/+, Cx3cr1−/− and non-transgenic (Non-Tg) controls. (I) Morphometric analysis of Iba1+ microglia from hTau-Cx3cr1−/− mice displayed significantly (n=3 animals per group; mean±s.e.m; *p<0.05 for hTau-Cx3cr1−/− compared to control groups; one-way ANOVA followed by Dunnett’s posthoc test) lower fractal dimension value compared to hTau-Cx3cr1+/+, Cx3cr1−/− and non-tg controls. (J–Q) Increase in the number of CD68 (phagocytic marker) positive cells in the DG (J–M) and CA3 (N–Q) of hTau-Cx3cr1−/− mice compared to hTau-Cx3cr1+/+, Cx3cr1−/− or non-tg controls. (R) Quantification of the CD68+ density revealed a significant increase (n=3 mice per group; mean ± s.e.m; ***p<0.0001; one-way ANOVA followed by Tukey posthoc test) in hTau-Cx3cr1+/+ mice when compared to hTau-Cx3cr1+/+, Cx3cr1−/− or non-tg controls. Scale bar 10 μm. (S–T) Quantitative real-time PCR analysis for CD45 and CD68 transcripts revealed a significant increase (normalized to non-transgenics; n=3 mice per group; mean±s.d; *p<0.05, **p<0.01; one-way ANOVA followed by Tukey posthoc test) in both CD45 and CD68 in the hemibrains of six-month-old hTau-Cx3cr1−/− mice compared to age-matched controls. See also Figure S3.
To confirm the immunohistochemical findings, the levels of various transcripts associated with microglial inflammation were next examined, revealing an increase in the levels of both CD68 and CD45 (Figure 3S–T) in hTau-Cx3cr1−/− mice when compared to controls. As noted above, the six-month old hTau mice displayed elevated levels of NOS2 and MCP1/CCL2 transcripts (Figure S1I–J) compared to non-transgenic controls that remained elevated in hTau-Cx3cr1−/− mice (Figure S3C–D). These results demonstrate that removal of CX3CR1 results in both enhanced MAPT phosphorylation, aggregation and microglial activation in hTau mice. Because CX3CR1 is expressed exclusively by microglia in the CNS, these results suggest that alterations in microglial activation are responsible for the observed alterations in MAPT phosphorylation and aggregation.
Elevated p38 MAPK signaling in hTau-Cx3cr1−/− transgenic mice
To identify the downstream kinase(s) and/or phosphatase pathways responsible for the elevated MAPT phosphorylation in the hTau-Cx3cr1−/− mice, the levels of p35 and p25 (regulators of the Cdk5) (Lew et al., 1994; Tsai et al., 1994), glycogen synthase kinase 3β (GSK3β) phosphorylated at serine-9 (Sutherland et al., 1993), p38 MAPK phosphorylated at thr180/tyr182 (Goedert et al., 1997; Lee et al., 1994) and protein phosphatase 2A (PP2A) were measured in hippocampal extracts from six-month-old hTau-Cx3cr1−/− mice and hTau controls. While the levels of p35/p25, phospho-ser9 GSK3β and PP2A remained unchanged (Figure S2C–F), the levels of phospho-p38 MAPK were significantly elevated in the hippocampus of 6-month-old hTau-Cx3cr1−/− mice when compared to age-matched hTau controls (Figure S2G and 2H). Previous studies on the hTau mice demonstrated an elevation in phospho-p38 MAPK at 10 months of age (Kelleher et al., 2007). Finally, activated transcription factor 2 (ATF2), which is phosphorylated at thr71 by active p38 MAPK, was also significantly increased in 6-month-old hTau-Cx3cr1−/− mice when compared to hTau mice alone (Figure S2G, S2I), suggesting that the levels of both active p38 MAPK and one of its downstream targets were elevated and could be responsible for the enhanced phosphorylation and aggregation of MAPT.
p38 MAPK Promotes MAPT Phosphorylation Upon Exposure to Microglial Factors
We postulated that the enhancement of MAPT phosphorylation and aggregation as well as p38 MAPK activation in CX3CR1 deficient hTau mice was due to altered communication between microglia and neurons. To examine this possibility in greater detail, wild-type and CX3CR1 deficient microglia were co-cultured in the bottom compartment of transwell culture dishes with wild-type primary cortical neurons cultured in the top compartment for 24 hours prior to evaluation of neuronal cell lysates for alterations in MAPT phosphorylation (Figure 4A). Co-cultures of neurons with wild-type microglia significantly induced MAPT phosphorylation at the AT8 site compared to neurons alone, while co-culture of CX3CR1 deficient microglia with neurons further increased the levels of AT8 MAPT epitopes (Figure 4C, 4F). Furthermore, the levels of phosphorylated p38 MAPK was increased in neuronal lysates co-cultured with either wild-type or CX3CR1 deficient microglia (Figure 4C, 4I). These data suggest that a soluble factor(s) released from microglia induces MAPT phosphorylation in neurons and lack of CX3CR1 in microglia enhances this phenotype.
Figure 4. Microglial-derived soluble factors induce MAPT phosphorylation in neurons via p38 MAPK pathway.

(A–B) Schematics displaying details of microglia–neuron co-culture experiment (A) and microglia conditioned media (CM) treatment on primary neurons (B). Non-transgenic controls (Cx3cr1+/+) or Cx3cr1−/− microglia were co-cultured with 21 DIV primary cortical neurons derived from non-transgenic (Cx3cr1+/+) embryos for 24 h and the neuronal lysates were processed for biochemical analysis. For the CM experiments, 21 DIV Cx3cr1+/+ neurons were treated with 25% ( + ) Cx3cr1−/− microglial CM for 24 h prior to biochemical analysis of neuronal lysates. CM from empty wells ( − ) served as a negative control. (C, F, I) Neurons co-cultured with Cx3cr1+/+ microglia displayed significant induction of AT8 site MAPT phosphorylation (C, F) and p38 MAPK activation (C, I) compared to neurons co-cultured with no-microglia. AT8 site phosphorylation was further enhanced when the neurons were co-cultured with Cx3cr1−/− microglia (n=3 independent cultures; mean±s.e.m of IDV ratios; *p<0.05, **p<0.01; one-way ANOVA followed by Tukey posthoc test). (D, E, G, H) Cx3cr1−/− microglial CM significantly induced neuronal AT8 and PHF1 site MAPT phosphorylation and p38 MAPK (phospho-thr180/tyr182) activation. Heat inactivation of microglial CM (microwaved until boiling and cooled) prior to neuronal treatment blocked neuronal MAPT phosphorylation and p38 MAPK activation. Pre-treatment (30 min) of primary neurons with SB203580, a p38 MAPK inhibitor, also significantly blocked the effects of microglial CM (n=3 independent cultures; mean±s.e.m of IDV ratios; *p<0.05, **p<0.01; one-way ANOVA-Dunnett’s posthoc test for AT8 and Tukey posthoc test for PHF1 and phospho-p38 MAPK). (J, K) Levels of activated-ATF2 (phospho-thr71) were elevated in microglia CM treated neuronal lysates and was blocked by SB203580 pre-treatment. All experiments were performed in duplicates in three independent cultures.
To confirm and extend these results, primary microglia were prepared from CX3CR1 deficient mice, conditioned media (CM) obtained and transferred to primary cortical neurons at a final concentration of 25% (Figure 4B). Notably, exposure of primary neurons to CM from Cx3cr1−/− microglia resulted in enhanced levels of MAPT phosphorylation at both the AT8 and PHF1 sites (Figure 4D, 4E, 4G). Finally, consistent with our in vivo as well as co-culture findings, the levels of activated p38 MAPK were also increased in neurons exposed to CM from the CX3CR1 deficient microglia (Figure 4D, 4H). The effects of the microglial CM could be blocked by heat inactivation (Figure 4D–4H), suggesting the factor(s) responsible was likely proteinaceous. Significantly, the effects of CX3CR1 deficient microglial CM on both p38 MAPK activation and MAPT phosphorylation could be blocked by 30 min pre-incubating neurons with a specific p38 MAPK inhibitor, SB203580 (Cuenda et al., 1995) (Figure 4D–4H), indicating that the enhancement of MAPT phosphorylation occurred via a p38 MAPK dependent pathway. Consistent with this, phosphorylation of ATF2 at thr71 was also elevated in neurons treated with microglial CM and blocked by treatment with SB203580 (Figure 4J–K). Taken together, our in vitro and in vivo studies suggest that a microglia-derived soluble factor(s) induces a p38 MAPK dependent increase in neuronal MAPT phosphorylation that is further enhanced in CX3CR1 deficient microglia.
Induction of MAPT phosphorylation dependent upon TLR4 and IL1 signaling
Several previous in vitro studies demonstrated that microglial-derived interleukin 1β (IL1β) could induce MAPT phosphorylation in neurons through a p38 MAPK dependent pathway (Hartzler et al., 2002; Li et al., 2003; Sheng et al., 2001). In addition, previous findings from our laboratory demonstrated that the neurotoxic effects of LPS stimulated CX3CR1 deficient microglia was mediated in part by IL1β (Cardona et al., 2006). To determine if the MAPT phosphorylation observed upon LPS administration is dependent upon TLR4 and IL1 signaling, we performed an additional in vivo LPS experiment. H-LPS (10 mg/kg) was administered to wild-type, Tlr4−/− and interleukin 1 receptor type I knockout (Il1r1−/−) mice and endogenous MAPT phosphorylation analyzed by Western blot analysis. While H-LPS induces AT8 and AT180 site phosphorylation of MAPT in Cx3cr1+/+ and Cx3cr1−/− mice, these effects were completely blocked in both Tlr4−/− (Figure 5A–B) and Il1r1−/− mice (Figure 5C–D). The block in MAPT phosphorylation in the Tlr4−/− mice demonstrates that the effects of LPS act through the canonical TLR4 signaling pathway within the brain, while the block of MAPT phosphorylation in the Il1r1−/− mice suggests that one of the cytokines responsible for induction of MAPT phosphorylation is IL1. To examine whether microglial derived IL1 can directly signal to neurons, induce activation of p38 MAPK and promote MAPT phosphorylation, we turned to the in vitro culture system (Figure 4B) in which CM from CX3CR1 deficient microglia was exposed to primary cortical neurons. Notably, the effects of microglial conditioned media on activation of p38 MAPK and neuronal MAPT phosphorylation was attenuated by pre-treatment of the neurons with Kineret, an IL1R antagonist (Figure 5E–G). In summary, our data suggests microglially derived IL1 can promote p38 MAPK activation and lead to enhanced MAPT phosphorylation within neurons.
Figure 5. IL1R1 and TLR4 deficiency blocks LPS-induced MAPT phosphorylation.

Two-month-old Cx3cr1+/+, Cx3cr1−/−, Tlr4−/− (A, B) and Il1r1−/− (C, D) mice were injected with either a single dose of vehicle or high dose LPS (10 mg/kg). (A, C) Western immunoblot of hippocampal extracts shows LPS induces dose-dependent increases in AT8 and AT180 site phosphorylation of endogenous mouse MAPT in Cx3cr1+/+ mice, that was further elevated in Cx3cr1−/− mice, but blocked in Tlr4−/− (A) and Il1r1−/− (C) mice to levels similar to that observed in vehicle injected Cx3cr1+/+ mice. Total MAPT and GAPDH levels were similar across all samples. (B, D) Quantification of Western blots revealed a statistically significant decrease in the ratios of AT8/GAPDH and AT180/GAPDH (data not shown) in the IL1R1−/− (n=3) and TLR4−/− (n=3) mice injected with H-LPS when compared to H-LPS injected treated Cx3cr1+/+ mice (n=4; mean ± s.e.m of integrated density value – IDV ratio; * p<0.05; unpaired t test). (E–G) IL1 Receptor Antagonist (IL1-RA/Kineret) Attenuates Microglial-Induced Neuronal MAPT Phosphorylation. (E) 21 DIV primary cortical neurons were pretreated (15 min) with vehicle (−) or 50 ng/ml IL1-RA (Kineret) prior to incubation with conditioned media (CM) from Cx3cr1−/− microglia. 24 h after the incubation, the neuronal lysates were probed for AT8, total MAPT (Tau5), phospho-p38 MAPK, total p38 MAPK and GAPDH. Note reduction in AT8 and phospho-p38 MAPK immunoreactive bands in IL1-RA treated neurons. (F and G) Quantification of the AT8/Total MAPT and phospho-p38 MAPK/total p38 MAPK ratios revealed more than four-fold reduction in the AT8 site phosphorylation (F) and statistically significant (*p<0.05; mean±s.e.m; n=3; unpaired t test) reduction in phospho-p38 MAPK levels (G) as a result of Kineret pretreatment. All experiments were performed in duplicates in three independent cultures.
DISCUSSION
In the current studies, we demonstrated that microglial-mediated neuroinflammation induces MAPT phosphorylation in several different models: a single LPS injection model of modest systemic inflammation, the hTau mouse model of tauopathy mated to Cx3cr1−/− mice and an in vitro microglial/neuronal culture system.
While other studies have linked microglial activation to MAPT phosphorylation, a majority of these studies were performed in vitro or were correlational in nature in postmortem tissue. The only two previous in vivo studies to address the relationship between neuroinflammation and MAPT phosphorylation and aggregation was a study examining the effects of LPS administration in the 3xTg mouse model of AD and a separate study on the effects of the immunosuppressant drug FK506 in P301S transgenic mice. However, the 3xTg mice contains mutant APP, PSEN1 as well as MAPT transgenes, making it difficult to assess the direct contribution of LPS to MAPT pathology, as microglial activation is also observed in response to the aggressive beta-amyloid (Aβ) deposition observed in this model. In addition the effects of LPS can act through numerous cell types within the brain. By contrast, the P301S study provided the first evidence that microglial neuroinflammation precedes MAPT pathology (in the absence of Aβ pathology) and that intervention with FK506 could ameliorate MAPT pathology and increase lifespan (Yoshiyama et al., 2007). While these studies suggested a links between microglial neuroinflammation and MAPT pathology, the current study documents the effects of a specific microglial receptor in modulating MAPT pathology and provides mechanistic insights into the downstream signaling molecules.
The present study demonstrates that a single dose of LPS, administered peripherally, is sufficient to induce MAPT hyperphosphorylation. LPS is an endotoxin that is part of the outer wall of Gram negative bacteria and can induce systemic inflammation via engagement of TLR4 (Lien et al., 2000). LPS has also been utilized as model to examine the role neuroinflammation via either peripheral administration in systemic inflammation or direct administration into the brain (Hauss-Wegrzyniak et al., 1998). Depending upon the dose of LPS, the route of administration, the time frame and the model examined, LPS can induce either neurodegeneration or neuroprotection with both short term (hours to days) and long term (months to years) effects. The LPS paradigm we have employed to examine the effects of induction of systemic inflammation has been widely utilized in numerous animal models of neurodegeneration (Cunningham et al., 2009; Cunningham et al., 2005; Masocha, 2009; Qin et al., 2007). Notably, several recent studies have suggested that the acute effects of LPS can have long lasting effects (months to years) within the CNS (Masocha, 2009; Qin et al., 2007). Our studies demonstrate a graded response to LPS with increased MAPT phosphorylation that is dependent upon the dose of LPS and deficiency of CX3CR1. Furthermore, either TLR4 or IL1R1 deficiency blocked the enhanced MAPT phosphorylation observed upon LPS administration, demonstrating that LPS-induced MAPT phosphorylation is acting through a canonical inflammatory pathway mediated in part by IL1. In addition, in vitro studies demonstrated that blocking IL1 signaling with an IL1 receptor antagonist reduced both p38 MAPK activation as well as MAPT phosphorylation within neurons. Future studies will be required to examine the relative contribution of IL1α versus IL1β and to determine whether the enhanced MAPT phosphorylation and aggregation observed in CX3CR1 deficient hTau mice is also dependent upon IL1-p38 MAPK signaling.
Previous studies demonstrated that hTau mice develop age-related MAPT pathology (Andorfer et al., 2003) and neurodegeneration (Andorfer et al., 2005). In the current studies, we document that hTau mice develop age-related alterations in microglia activation at 12 to 18 months of age. This is consistent with several other recent observations in the hTau mice (Kelleher et al., 2007; Noble et al., 2009) as well as other mouse model of tauopathies (Kitazawa et al., 2005; Yoshiyama et al., 2007), suggesting that robust microglia activation occurs upon development of extensive MAPT pathology. In addition, our studies suggest that at six months of age, an age at which there is minimal development of MAPT aggregates, there are already alterations in expression of several cytokines and chemokines, including CCL2, NOS2 and most notably CX3CL1. CX3CL1 is produced by neurons in the CNS and signals directly to the CX3CR1 receptor, which is exclusively produced by microglia. Finally, consistent with the findings in the 3xTg mouse model, LPS administration to hTau mice induced enhanced MAPT phosphorylation (Kitazawa et al., 2005).
CX3CL1 and its cognate receptor, CX3CR1, play an important role in neuroinflammation via paracrine signaling between neurons and microglia (Figure 6). Previous studies from our laboratory (Cardona et al., 2006) as well as others (Jung et al., 2000) demonstrated that CX3CR1 deficiency alone does not lead to acute microglial activation or neurodegeneration. However, in mouse models of systemic inflammation, PD and ALS, CX3CR1 deficiency leads to enhanced neurodegeneration (Cardona et al., 2006), while conversely in mouse models of stroke, CX3CR1 deficiency lead to neuroprotection (Denes et al., 2008).
Figure 6. Model of fractalkine (CX3CL1/CX3CR1) signaling in neurodegenerative tauopathies.
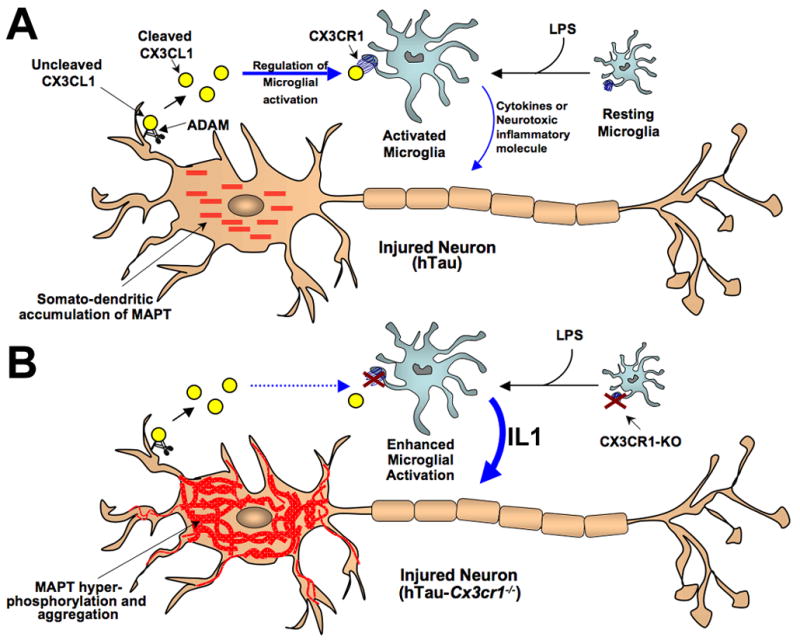
(A) In the CNS, the chemokine fractalkine (CX3CL1) is expressed by neurons and cleaved by the A Disintegrin And Metalloproteases (ADAM10 and ADAM17) to release cleaved soluble CX3CL1, which binds to the G-coupled transmembrane receptor (CX3CR1) present on microglia and regulates microglial activity. Lipopolysaccharide (LPS) induces microglial activation, while neuronal stress (such as overexpression and somato-dendritic accumulation of human MAPT in case of hTau) results in enhanced release of CX3CL1. An interaction between fractalkine (CX3CL1) and its receptor (CX3CR1) downregulates microglial activity and dampens the toxic effects of activated microglia. (B) Lack of CX3CR1 (Cx3cr1−/−) in microglia disrupts CX3CL1-CX3CR1 communication (dashed blue arrow), dysregulates microglial activity induced by LPS or neuronal stress (human MAPT overexpression in hTau mice) and results in enhanced release of soluble factors (including IL1) from activated microglia, can induce active neuronal p38 MAPK and lead to enhanced phosphorylation and aggregation of MAPT. Notably, this molecular mechanism would lead to a feed-forward induction of MAPT phosphorylation/aggregation and microglial activation.
Notably, a recent study by Fuhrmann and colleagues suggested that CX3CR1 deficiency could prevent neuronal loss in the 3xTg mouse model of AD (Fuhrmann et al., 2010). A 1.8% loss of neurons in cortical layer II per month observed in the 3xTg mouse model detected by two photon microscopy was prevented in the CX3CR1 deficient 3xTg animals. While this study suggested that CX3CR1 deficiency could modulate phenotypes in an AD mouse model, there are a number of important differences between the two studies, their endpoints and their conclusions.
First, the 3xTg mice exhibits both Aβ and MAPT pathologies due to the inclusion of three different mutant human transgenes (APP, PSEN1 and MAPT) that causes AD and/or FTD. Because of this transgenic strategy, it remains difficult to attribute the effects of CX3CR1 deficiency to specific alterations in Aβ or MAPT pathologies. Notably, we have recently completed a separate study examining the effects of CX3CR1 deficiency on Aβ pathologies in two different mouse models of AD and demonstrate that blocking CX3CL1-CX3CR1 signaling actually reduces Aβ pathologies via a mechanism involving alterations in microglial phagocytosis of extracellular Aβ aggregates (Lee et al., 2010). Thus, taken together, our results suggest that CX3CR1 deficiency has opposing effects on the two primary pathologies of AD, extracellular Aβ plaques and intracellular MAPT aggregates, information that could not be discerned in the Fuhrmann et al. studies.
Second, Fuhrman et al. examined CX3CR1 deficient 3xTg animals at 4–6 months of age, which is just prior to substantial extracellular Aβ deposits exhibited in this model and at least 6–8 months prior to the first appearance of alterations in MAPT phosphorylation and aggregation (Oddo et al., 2003). Notably, the authors also report that there are no observable alterations in MAPT phosphorylation in 4–6 month-old CX3CR1 deficient 3xTg mice. In addition, due to restrictions in live imaging within the brain by two photon microscopy, the neuronal loss detected by Fuhrman et al., was observed in the outer layers of the cortex and not within the hippocampus, where the initial alterations in MAPT phosphorylation and aggregation were observed in the hTau mouse model and have also been reported in the 3xTg mouse model. Because of this, the Fuhrman et al. study focuses on the abundant intracellular Aβ present at this age within this cortical neuronal population in the 3xTg mice as a potential neurotoxic trigger for the neuronal loss that can be blocked by CX3CR1 deficiency. How these results translate to effects on extracellular Aβ deposition, similar to that published by Lee et al., and intracellular MAPT aggregates, as presented here, remain to be determined.
Third, most of the transgenic mouse models of AD, including the 3xTg mice do not exhibit robust age-related neurodegeneration (reviewed in McGowan et al., 2006), as assessed by other post-mortem methods utilized to detect neuronal cell loss. Thus, it remains to be determined how the neuronal loss observed by Fuhrmann et al. via two photon microscopy relates to the long-term Aβ toxicity observed in the AD mouse models.
Fourth, our studies suggest that CX3CR1 deficiency has opposing effects on Aβ and MAPT pathologies (Lee et al., 2010). Given increasing evidence that Aβ pathologies precede MAPT pathologies by as much as 10 years in humans (Perrin et al., 2009) our studies strongly suggest that blocking CX3CL1/CX3CR1 signaling has opposing effects at different stages of disease progression. Thus, while CX3CR1 deficiency at early stages of intracellular Aβ pathology (Fuhrmann et al., 2010) and extracellular Aβ pathology (Lee et al., 2010) may be protective, the effects of CX3CR1 deficiency are deleterious as the disease progresses towards development of enhanced microglia-mediated neuroinflammation and/or MAPT aggregation. Future studies will be required to assess the validity of this hypothesis in additional disease relevant animal models, as well as assess the effects on microglial phenotypes at different stages of disease progression.
Finally, the present study provides mechanistic links between CX3CR1 and specific AD pathologies and signaling pathways. CX3CR1 deficiency has specific effects on MAPT phosphorylation and aggregation, microglial activation and behavior in the hTau mouse model. In addition, these studies link CX3CR1 deficiency to the release of microglia factors (including IL1) that subsequently induces p38 MAPK and MAPT phosphorylation within neurons.
Interestingly, our previous studies demonstrated that administration of LPS to Cx3cr1−/− mice induces neurodegeneration that is also dependent upon IL1 signaling (Cardona et al., 2006), suggesting that IL1 may represent as an important signaling molecule between microglia and neurons. Notably, the relevance of CX3CL1-CX3CR1 signaling to human neurodegeneration has been highlighted by the identification of V249I and T280M polymorphisms in CX3CR1 that are associated with human age-related macular degeneration (Combadiere et al., 2007). The current studies extends these observations and provides evidence that CX3CL1-CX3CR1 signaling plays a role in the development of tauopathies, as CX3CR1 deficient hTau mice exhibit enhanced microglial activation, pronounced MAPT hyperphosphorylation and pathology as well as memory disturbances (Figure 6). These results also suggest that CX3CL1-CX3CR1 signaling may provide a novel therapeutic target for tauopathies (Figure 6).
Several different studies have provided evidence that p38 MAPK is one of the MAPT kinases that links neuroinflammation and MAPT phosphorylation in tauopathies (Hartzler et al., 2002; Hensley et al., 1999; Sun et al., 2003). In the present studies, we observed increased levels of activated p38 MAPK and ATF2 in the brains of CX3CR1 deficient hTau mice, but not other established MAPT kinases. Furthermore, our in vitro studies demonstrated that soluble factors released by microglia (and enhanced in CX3CR1 deficient microglia) induces MAPT phosphorylation in cultured neurons that is dependent upon p38 MAPK activity and can be blocked by an IL1 receptor antagonist.
In conclusion, this study demonstrates that altered microglia activation plays a direct role in modulating the hyperphosphorylation and aggregation of MAPT within neurons and suggests potential strategies for therapeutic intervention in tauopathies.
EXPERIMENTAL PROCEDURES
Animals
hTau (Andorfer et al., 2003), Il1r1−/−, Tlr4−/− and Cx3cr1−/− (Jung et al., 2000) mice were in C57BL/6J genetic background and were obtained from the Jackson Laboratory and Dan Littman (HHMI, New York University School of Medicine). Experimental protocols were performed in accordance with US National Institutes of Health guidelines on animal care and were approved by the Cleveland Clinic Institutional Animal Care and Use Committee.
Antibodies
MAPT antibodies: Mouse monoclonal antibodies - AT8, AT180 (Pierce) and Tau5 (Invitrogen); PHF-1, CP13, TG3 (provided by Peter Davies, Albert Einstein College of Medicine). Inflammatory markers: Rabbit polyclonal antibody against ionized calcium binding adaptor molecule 1 (Iba1) (Wako); rat monoclonal antibody against CD68 (Serotec). Mouse monoclonal antibodies against CD11b/Mac-1 (Chemicon/Millipore) and iNOS (Transduction Laboratories). Other antibodies: Mouse monoclonal antibody against GAPDH; rabbit polyclonal antibodies against activated ATF-2 (CRE-BP1) phosphorylated at thr71, total p38 MAPK and activated p38 MAPK (phospho-thr180/tyr182) were from Cell Signaling; mouse monoclonal antibody against Cdk5 regulators p35/p25 (05-380; Millipore), rabbit monoclonal antibody against GSK3b-phospho-ser9 (04-1075; Millipore); rabbit monoclonal antibody to PP2A C-subunit (52F8; 2259; Cell Signaling).
LPS administration
Vehicle (Veh, Hank’s Balanced Saline Solution) or lipopolysaccharide (LPS; Sigma-Aldrich, USA) at two concentrations (1 mg/kg or 10 mg/kg body weight L-LPS or H-LPS respectively; intraperitoneal; single dose) was injected into two-month-old non-transgenic C57BL/6, Cx3cr1−/−, Il1r1−/− and Tlr4−/− mice. 2 month old hTau mice received LPS at 1 mg/kg b.w. The doses of LPS selected for the presented study is based on a study, which reported sub-lethal (<5 mg/kg b.w) and lethal (>5 mg/kg b.w) LPS doses and that the latter dose produced neurodegeneration and chronic microglial activation, which could be observed 10 months post-injection (Qin et al., 2007). After 24 h, the mice were sacrificed and the hippocampi from left hemisphere were processed for biochemical analysis and the entire right hemisphere was drop-fixed in 4% paraformaldehyde and processed for neuropathological analysis.
SDS-PAGE and Western Immunoblotting
Hippocampal lysates were prepared by homogenizing in Tissue-Protein Extraction Reagent (T-PER, 78510; Pierce) with protease (P8340 Sigma-Aldrich) and phosphatase (P5726; Sigma-Aldrich) inhibitor cocktails. Protein (20 μg) was resolved in 4–12 % Bis-Tris Novex NuPage gels (Invitrogen) and transferred to PVDF membrane, blocked (in 5% milk) and incubated overnight in primary antibodies (AT8 1:5000; AT180 1:5000; PHF-1 1:10,000; Tau5 1:10,000; GAPDH 1:20,000; phospho-p38 MAPK, total p38 MAPK, phospho-ATF-2 were used at 1:1000) following by respective secondary antibodies. Membranes were developed using ECL reagent (NEL101001EA; Perkin Elmer) and immunoreactive bands were quantified in AlphaEaseFC™ Software (Alpha Innotech Corporation).
Immunohistochemistry
Free-floating sections were processed for standard immunohistochemistry or immunofluorescence staining. Primary antibodies were used are at following dilution; AT8, AT180, CD68, at 1:200; TG3 and CP13 1:100; Iba1 1:500; incubated overnight at 4°C. Secondary antibodies (1:500) conjugated to either biotin (for immunohistochemistry from Vector Laboratories) or Alexa Fluor® dyes (for immunofluorescence from Invitrogen) were used. Sections then were either mounted in DAPI Hardset Reagent (for immunofluorescence) or incubated with Avidin:Biotinylated enzyme Complex (ABC reagent, Vector Laboratories; for immunohistochemistry) reagent for 1 h at RT. The immunoreactive signals were revealed by developing sections in SigmaFast with (for Iba1, CD68, CD11b and iNOS) or without (for all MAPT antibodies) the metal enhancer.
Morphometric analysis of microglial morphology and CD68 density
Fractal Dimension analysis to determine the activation state of microglia was performed on Iba1-stained images acquired at x20 objective using Image-Pro Plus software as described before (Soltys et al., 2001; Varvel et al., 2009). To quantify the density of CD68 immunoreactive cells/structures, we referred earlier methods (Jimenez-Andrade et al., 2006) with slight modification. Briefly, CD68 stained images were background subtracted, normalized (segmentation and threshold setting) and the immunoreactive densities were determined using a specific Image-Pro Plus macro (“Density”) and expressed as mean± s.e.m per each group. For both Iba1 and CD68 quantification three sections per animal; three 8 bit RGB digital images at the dentate gyrus and CA3 regions per animal; n=3 animals)
Primary cortical neuronal cultures
Neuronal cultures were prepared from E16.5 C57BL/6J wild-type mouse embryos as described before (Bhaskar et al., 2009). Specifically, primary cortical neurons were seeded on poly-L-lysine coated coverslips at a density of 1.6×105 cells/well in 6-well plate (for co-culture experiments). Alternatively, cells were also plated at 1.6×105 cells/well directly onto poly-L-lysine coated 6-well plate without coverslips (for conditioned media experiment). All cultures were grown for 21 days in vitro (DIV) at 37°C in humidified 5% CO2/95% air prior to any treatment.
Primary microglial culture
Microglial cultures were prepared from post-natal day 3 (P3) pups from Cx3cr1+/+ or Cx3cr1−/− genotypes as described before (Saura et al., 2003). Specifically, mixed glial cells were seeded at a density of 1.0×105–1.2×105 cells/cm2 to obtain approximately 6×104–8×105 microglial cells/cm2 after removal of astrocyte layer at 14th day. Furthermore, the DMEM-F12 media was replaced with Neurobasal media (with no B27 supplements) 24 h prior to the co-culture or conditioned media experiment to match the culture media with that of primary neurons.
Neuron-microglia co-cultures
Primary neuronal and microglial cultures were prepared as described above. 21 DIV primary cortical neurons grown on a coverslip were placed inside a 6-well microplate insert, which has a 0.02 μm Anopore® membrane in the bottom (161395; Nunc) and allow diffusion of soluble factors into the media. The inserts were then placed inside individual wells in 6-well plate, which had Cxcr1+/+, Cx3cr1−/− or no primary microglia, and incubated for 24 h. The neurons were lysed in 1x Lithium dodecyl sulfate (LDS) buffer, and the proteins were resolved in 4–12% Bis-Tris NuPage gels (Invitrogen) and Western immunoblotting was performed as described above. All the experiments were done in triplicates with independent cultures.
Microglial conditioned media treatment
One fourth of the media (25%) present in the primary neurons at 21 DIV was replaced with equal volume of conditioned media (CM) obtained from primary microglial cultures. CM from no microglial cells served as controls. For the analysis of heat-inactivation, the CM was microwaved until boiling and cooled and applied to neurons. For the analysis of specific p38 MAPK inhibitors, 21 DIV cortical neurons were pre-incubated for 30 min or 15 min with 20 μM SB203580 (S8307; Sigma-Aldrich) or IL1RA (Kineret, 50 ng/ml; Amgen) respectively prior to CM treatment. After 24 h incubation, the neuronal lysates were prepared and Western blotted as mentioned above. All the experiments were done in triplicates with independent cultures.
Gene expression analysis
RNA from the hemi-brain was extracted using the Tri Reagent as described by the manufacturer (Invitrogen). Total RNA (50ng/uL) was converted to cDNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems Inc., ABI) and amplified using specific TaqMan probes (See supplemental information) and GAPDH as a house keeping gene for normalization, on the ABI 7300 Real-Time PCR System.
Sarkosyl extraction
Sarkosyl-insoluble fraction of MAPT was isolated from hippocampal tissues as described before (Greenberg and Davies, 1990) with minor modifications. The experimental details are provided in the Supplemental Information.
Gallyas silver staining
The Gallyas silver staining on 30 μm free-floating sections were performed as described (Braak and Braak, 1991).
Behavioral analysis
A symmetrical Y maze was used to evaluate spatial working memory (Hughes, 2004). Experimental details and calculation of percent spontaneous alternations are mentioned in the Supplemental Information.
Statistical analysis
Data are presented as mean ± s.e.m. Unless otherwise noted, the Student’s t-test (two-tailed; unpaired) at 95% confidence interval (for two group comparison) or one-way ANOVA followed by Tukey or Dunnett’s posthoc test (for multiple comparisons) was utilized for statistical analysis. Statistical significance was determined at p < 0.05.
Supplementary Material
Acknowledgments
We thank Guixiang Xu, Nicole Maphis and Angeliki Maria Nikolakopoulou for their assistance in cell culture experiments; Dr. Sanjay Pimplikar for providing antibodies, critical discussions and valuable suggestions in manuscript preparation. We thank Drs. Robert Fairchild and Bruce Trapp for providing Il1r−/− and Tlr4−/− mice, respectively and Dr. Xiaoxia Li for providing IL1RA (Kineret). This work is supported by CurePSP (Society for Progressive Supranuclear Palsy) to K.B.; an Alzheimer’s Association, American Health Assistance Foundation and NIH (AG023012) grant to B.T.L.; Dana Foundation to R.M.R; National Multiple Sclerosis Society Career Transition Award to A.C.
Footnotes
Supplemental information includes three supplemental figures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. doi: 10.1186/1750-1326-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, Rawlins JN, Perry VH. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65:304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A, Ferenczi S, Halasz J, Kornyei Z, Kovacs KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects. Microsc Res Tech. 2001;54:47–58. doi: 10.1002/jemt.1120. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006;21:89–93. doi: 10.1002/mds.20668. [DOI] [PubMed] [Google Scholar]
- Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62. doi: 10.1016/s0014-5793(97)00483-3. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzler AW, Zhu X, Siedlak SL, Castellani RJ, Avila J, Perry G, Smith MA. The p38 pathway is activated in Pick disease and progressive supranuclear palsy: a mechanistic link between mitogenic pathways, oxidative stress, and tau. Neurobiol Aging. 2002;23:855–859. doi: 10.1016/s0197-4580(02)00029-5. [DOI] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Lukovic L, Bigaud M, Stoeckel ME. Brain inflammatory response induced by intracerebroventricular infusion of lipopolysaccharide: an immunohistochemical study. Brain Res. 1998;794:211–224. doi: 10.1016/s0006-8993(98)00227-3. [DOI] [PubMed] [Google Scholar]
- Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesbery WR, et al. p38 kinase is activated in the Alzheimer’s disease brain. J Neurochem. 1999;72:2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol. 2001;60:647–657. doi: 10.1093/jnen/60.6.647. [DOI] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Peters CM, Mejia NA, Ghilardi JR, Kuskowski MA, Mantyh PW. Sensory neurons and their supporting cells located in the trigeminal, thoracic and lumbar ganglia differentially express markers of injury following intravenous administration of paclitaxel in the rat. Neurosci Lett. 2006;405:62–67. doi: 10.1016/j.neulet.2006.06.043. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher I, Garwood C, Hanger DP, Anderton BH, Noble W. Kinase activities increase during the development of tauopathy in htau mice. J Neurochem. 2007;103:2256–2267. doi: 10.1111/j.1471-4159.2007.04930.x. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s Disease mouse models. Am J Pathol. 2010 doi: 10.2353/ajpath.2010.100265. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003;23:1605–1611. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masocha W. Systemic lipopolysaccharide (LPS)-induced microglial activation results in different temporal reduction of CD200 and CD200 receptor gene expression in the brain. J Neuroimmunol. 2009;214:78–82. doi: 10.1016/j.jneuroim.2009.06.022. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schonig K, Bujard H, Mandelkow E, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W, Garwood C, Stephenson J, Kinsey AM, Hanger DP, Anderton BH. Minocycline reduces the development of abnormal tau species in models of Alzheimer’s disease. FASEB J. 2009;23:739–750. doi: 10.1096/fj.08-113795. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Orellana DI, Gonzalez-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. 2004;295:245–257. doi: 10.1016/j.yexcr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Saez TE, Pehar M, Vargas M, Barbeito L, Maccioni RB. Astrocytic nitric oxide triggers tau hyperphosphorylation in hippocampal neurons. In Vivo. 2004;18:275–280. [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WS. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltys Z, Ziaja M, Pawlinski R, Setkowicz Z, Janeczko K. Morphology of reactive microglia in the injured cerebral cortex. Fractal analysis and complementary quantitative methods. J Neurosci Res. 2001;63:90–97. doi: 10.1002/1097-4547(20010101)63:1<90::AID-JNR11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Sun A, Liu M, Nguyen XV, Bing G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp Neurol. 2003;183:394–405. doi: 10.1016/s0014-4886(03)00180-8. [DOI] [PubMed] [Google Scholar]
- Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296(Pt 1):15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Varvel NH, Bhaskar K, Kounnas MZ, Wagner SL, Yang Y, Lamb BT, Herrup K. NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J Clin Invest. 2009;119:3692–3702. doi: 10.1172/JCI39716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.