

Abstract
Background
Alcohol abuse increases the risk for acute respiratory distress syndrome (ARDS). Efferocytosis, the clearance of apoptotic cells, is important in the resolution of inflammation and is regulated by RhoA and rho kinase (ROCK) activation. The effects of alcohol on pulmonary Rho pathway activation and efferocytosis have not been determined. We hypothesize that acute and chronic alcohol exposure impair pulmonary efferocytosis, leading to heightened inflammation during ARDS.
Methods
For in vivo experiments, C57BL/6 mice received either a single, intraperitoneal injection of alcohol or chronic ethanol-in-water for 8 weeks prior to intratracheal instillation of apoptotic cells or lipopolysaccharide (LPS). Bronchoalveolar lavage (BAL) was performed for cells counts, calculation of the phagocytic index (PI), and Rho activity measurements. For in vitro studies, primary alveolar macrophages were cultured in alcohol (25-100mM) and then co-cultured with apoptotic cells. RhoA activity was determined following alcohol exposure, and the PI was determined before and after treatment with the ROCK inhibitor, Y27632.
Results
Acute alcohol exposure was associated with impaired efferocytosis. Following LPS exposure, acute alcohol exposure was also associated with increased BAL neutrophils. Chronic alcohol exposure alone did not alter efferocytosis. However, following exposure to LPS, chronic alcohol exposure was associated with both impaired efferocytosis and increased BAL neutrophils. In vitro alcohol exposure caused a dose-dependent decrease in efferocytosis. Despite the fact that RhoA activity was decreased by alcohol exposure and RhoA inhibition did not alter the effects of alcohol on efferocytosis, treatment with the Rho kinase inhibitor, Y27632, reversed the effects of alcohol on efferocytosis.
Conclusions
Acute alcohol exposure impairs pulmonary efferocytosis, while exposure to chronic alcohol is only associated with impaired efferocytosis following LPS-induced lung injury. Both forms of alcohol exposure are associated with increased alveolar neutrophil numbers in response to LPS. The acute effects of alcohol on efferocytosis appear to be mediated, at least in part, by RhoA-independent activation of ROCK. Further studies are needed to dissect the differences between the effects of acute and chronic alcohol exposure on efferocytosis and to determine the effects of alcohol on alternative activators of ROCK.
Keywords: Alcohol, ARDS, Efferocytosis, RhoA, Rho kinase
Introduction
The acute respiratory distress syndrome (ARDS) is characterized by increased permeability of the alveolar capillary membrane, diffuse alveolar damage, the accumulation of proteinaceous interstitial and intra-alveolar edema, and an intense inflammatory response. These pathological changes are accompanied by physiological alterations, including severe hypoxemia, an increase in the mean pulmonary dead space fraction, and a decrease in pulmonary compliance. ARDS is a common diagnosis in patients who require mechanical ventilation for greater than 24 hours, occurring in approximately 200,000 individuals in the United States each year (Rubenfeld et al. 2005b). Despite advances in the field, the mortality from ARDS remains high (Phua et al. 2008).
Two large epidemiological studies involving nearly 600 critically ill patients have shown that a history of alcohol abuse is associated with an increased susceptibility to the development of ARDS (Moss et al. 1996;Moss et al. 2003a). Importantly, the association between alcohol abuse and ARDS is common, occurring in nearly 50% of patients with ARDS. In those patients who survive ARDS, alcohol abuse is associated with an increased duration of mechanical ventilation and prolonged ICU length of stay (Moss et al. 2003b), suggesting that alcohol abuse may alter the resolution phase of this disease. Follow-up studies have validated the deleterious effects of alcohol on the development of ARDS, showing an increased incidence and severity of ARDS in patients following surgical lung resection and blood transfusion (Gajic et al. 2007;Licker et al. 2003). Many critically ill patients with a history of alcohol abuse are acutely intoxicated at the time of hospital admission. However, the relative contribution of chronic alcohol abuse and superimposed acute intoxication to the development of ARDS has not been determined.
ARDS is characterized by an influx of inflammatory cells into the alveolar space (Ware & Matthay 2000), and resolution requires the removal of these inflammatory cells from the lung. During normal resolution of ARDS, the clearance of apoptotic inflammatory cells is a highly efficient process, as the proportion of apoptotic neutrophils in BAL remain low throughout the course of resolution (Matute-Bello et al. 1997b). Alveolar macrophages and other phagocytes clear apoptotic cells from the lung and thereby prevent their lysis and the consequent release of toxic or immunogenic intracellular components (Vandivier, Henson, & Douglas 2006). In the case of neutrophils, in vitro studies have shown that lysed neutrophils exhibit pro-inflammatory effects in an elastase-dependent manner (Fadok et al. 2001). In contrast, the interaction between apoptotic cells and phagocytes is anti-inflammatory, decreasing the innate immune response and promoting resolution of inflammation by suppressing the production of inflammatory mediators (Henson, Bratton, & Fadok 2001;Huynh, Fadok, & Henson 2002).
The intracellular balance of Rho GTPases has been shown to be important in the regulation of efferocytosis (Ravichandran & Lorenz 2007a). These small GTPases cycle between the inactive GDP-bound state and the active GTP-bound state, and their activation and inactivation occur during efferocytosis. Importantly, RhoA has a negative effect on engulfment, such that the loss or suppression of RhoA activity promotes engulfment (Ravichandran & Lorenz 2007b). Activation of RhoA also results in activation of its downstream effector, Rho kinase (ROCK), a serine/threonine kinase that plays an important role in a variety of cellular functions, including contraction, adhesion, migration, proliferation, and apoptosis (Shi & Wei 2007a). ROCK has also been shown to be important in the regulation of pulmonary efferocytosis, as treatment with the ROCK inhibitor, Y27632, improves efferocytosis in the setting of oxidative stress, cigarette smoke exposure, and deficient cystic fibrosis transmembrane conductance regulator (McPhillips et al. 2007;Richens et al. 2009;Vandivier et al. 2009). While RhoA is the most common activator of ROCK, there are many Rho-independent mechanisms of ROCK activation, including lipid mediators (arachidonic acid and sphingosylphosphorylcholine), inositol phospholipids, Granzyme B, and caspases (Caspase-2 and Caspase-3) (Shi & Wei 2007c).
Alcohol impairs phagocytosis of bacteria by neutrophils (Zhang et al. 1997) and alveolar macrophages (Brown et al. 2007b;Rimland & Hand 1980), two important cell types involved in the pathogenesis of ARDS. In the liver, chronic alcohol exposure impairs the ability of hepatocytes to phagocytose apoptotic cells (McVicker et al. 2002) and alters the anti-inflammatory effects of apoptotic cells (McVicker et al. 2007). However, the effects of acute and chronic alcohol exposure on the phagocytosis of apoptotic cells within the alveolar compartment are unknown. The effects of alcohol on Rho pathway signaling are varied. Both in vivo and in vitro alcohol exposure have been shown to activate RhoA in cerebellar granule neurons (Joshi et al. 2006) and astrocytes (Minambres et al. 2006). In contrast, chronic ethanol treatment of rat astrocytes is associated with reduced levels of activated RhoA (Martinez et al. 2007). The effects of acute alcohol exposure on RhoA and ROCK activity in the lung are yet to be described.
Therefore, we hypothesized that, similar to its effects on the phagocytosis of bacteria, both acute and chronic alcohol exposure would impair the process by which dying cells are phagocytosed (efferocytosis) during ARDS, leading to impaired resolution of inflammation. As more apoptotic cells are allowed to undergo necrosis, a pro-inflammatory environment would be promoted, resulting in worsening lung injury. The effects of alcohol on pulmonary efferocytosis have not been previously described. Because alcohol abuse patterns are complex, often including both acute intoxication and long-term, chronic abuse, it was important that we describe the effects of each type of exposure on this system. The focus of this manuscript was to (1) define the effects of acute and chronic alcohol exposure on efferocytosis, (2) determine whether these effects were associated with increased numbers of inflammatory cells during LPS-induced lung injury, and (3) determine whether the effects of alcohol on efferocytosis may be mediated in part by activation of RhoA and ROCK.
Materials and Methods
Reagents
Cell culture media (including Dulbecco's Modified Eagle Medium (DMEM), Hanks' Balanced Salt Solution (HBSS), and RPMI 1640), Penicillin/Streptomycin/L-Glutamine, EDTA, Diff-Quik stain, Permount, and modified finding needles were obtained from Fisher Scientific (Pittsburgh, PA). Thioglycolate and ethanol were obtained from Sigma Aldrich (St. Louis, MO). Fetal bovine serum (FBS) was obtained from Atlantic Biologicals (Lawrenceville, GA). The human Jurkat leukemia T cell line was obtained from the American Type Culture Collection (Manassas, VA). Isofluorane was obtained through the animal facility of the University of Colorado Denver. C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). Lipopolysaccharide (LPS) from Escherichia coli O111:B4 was obtained from List Biological Laboratories (Campbell, CA). The Rho kinase inhibitor, Y27632, was obtained from Calbiochem (Gibbstown, NJ), while the cell permeable Rho inhibitor, C3 transferase, was obtained from Cytoskeleton (Denver, CO). The RhoA activity assay was obtained from Cytoskeleton (Denver, CO).
Experimental Animals
All experiments were performed with female, C57BL/6 mice, which were maintained on a standard laboratory diet and housed in a controlled environment with a 12h light/dark cycle. All studies were conducted under the Institutional Animal Care and Use Committee-approved protocols in the animal facility of the University of Colorado Denver.
In vivo Acute Alcohol Model
In vivo animal experiments were performed with 7-8 wk-old mice. Acute ethanol intoxication was induced by intraperitoneal (i.p.) injection of 20% ethanol (3 g/kg) in phosphate-buffered saline (PBS). Control rats received an equal volume of PBS intraperitoneally. Thirty minutes after intraperitoneal injection of ethanol or PBS, intratracheal (i.t.) instillation of apoptotic thymocytes or LPS was performed as previously described (Morimoto et al. 2006;Teder et al. 2002;Vandivier et al. 2002). Briefly, mice were anesthetized with isofluorane, followed by intratracheal instillation of either apoptotic thymocytes (107 cells per mouse in 50uL PBS) or LPS (20ug/mouse) using a modified finding needle (Fisher Scientific). Mice were subsequently sacrificed with pentobarbital, and BAL samples were obtained (three separate lavages of 1ml each with PBS containing 5mM EDTA) for pulmonary cell counts, phagocytosis assay, and RhoA activity assay (see below).
In vivo Chronic Alcohol Model
Chronic alcohol exposure was employed using the chronic ethanol-in-water model, a method that has been extensively validated in C57Bl/6 mice (Cook et al. 2007). For this model, mice in the ethanol group are given, as the only water source, 10% w/v ethanol ad libitum for 2 days, 15% w/v ethanol for 5 days, and 20% w/v ethanol thereafter for 8 weeks. All mice continued to have access to standard rodent chow for nutrition. This model results in significant elevations in blood alcohol levels without high serum corticosteroid concentrations or weight gain abnormalities for >20 weeks. In our hands, we have treated mice with this diet for 8 wks, with no differences in weight or significant morbidity or mortality. Following exposure to alcohol for 8 weeks, i.t. instillation of either apoptotic thymocytes or LPS was performed as described above.
In vitro Alcohol Model
Female, 7-8 week-old mice were sacrificed with pentobarbital, and BAL samples were obtained (ten separate lavages of 1ml each with PBS containing 5mM EDTA). BAL cells were pooled, counted, and plated in 96-well culture plates (100,000 cells/well) in DMEM media containing 10% FBS supplemented with 2 mM L-glutamine, 100 U/ml penicillin, and 100 g/ml streptomycin. Media was changed 60 minutes later, and cells were cultured overnight before starting experimental treatments. Next, cells were treated with physiologically relevant concentrations of alcohol (25-100mM, or 115-460 mg/dL) for a period of 24h. Cells were then washed with warm media prior to the addition of apoptotic Jurkat T cells. To determine the importance of Rho pathway activation in this system, PM's were treated with the Rho kinase inhibitor, Y27632 (1-10μM), or Rho inhibitor, C3 transferase (1-10μg/ml) 2h prior to the addition of apoptotic cells and calculation of the PI. The reported half-maximal inhibitory concentration (IC50) for Y27632 is 0.7-1.7 μM (Sward et al. 2000;Uehata et al. 1997), while the typical effective range for C3 transferase when used in the presence of 10% serum is 2.5-10μg/ml.
Cell isolation and Induction of Apoptosis
For in vivo experiments, murine thymocytes were isolated from the thymi of 3-4 wk-old C57BL/6 mice by first passing thymi through a 40 um strainer to separate individual cells. Thymocytes were washed with PBS, resuspended in RPMI media containing 10% FCS at 3×106 cells/ml, exposed to UV irradiation at 254 nm for 12.5 min, and cultured for 3h in 5% CO2 at 37°C before use. For in vitro studies, Jurkat T cells were cultured in RPMI media (RPMI 1640 with 10% heat-inactivated FBS and supplemented with 2 mM L-glutamine, 100 U/ml penicillin, and 100 g/ml streptomycin) and incubated at 37°C in 5% CO2. Cells were then exposed to UV irradiation (Fotodyne Inc.) at 254 nm for 10 min and cultured for 3h in 5% CO2 at 37°C before use. By these methods, cells are typically 70-90% apoptotic (Hoffmann et al. 2001b).
Phagocytosis Assay
Phagocytosis assays were performed as previously described (Hoffmann et al. 2001a). For in vitro experiments, apoptotic Jurkat T cells (2.5×106 cells in 0.5ml DMEM media) were added to each well and co-cultured with alveolar macrophages at 37°C in 10% CO2 for four hours. Following incubation, cells were washed three times with PBS and stained with a modified Wright-Giemsa stain. For in vivo experiments, BAL was performed sixty minutes following instillation of apoptotic cells. Cell monolayers were prepared by cytocentrifugation and stained with a modified Wright-Giemsa stain. To determine the effects of alcohol on the phagocytosis of apoptotic cells, the phagocytic index (PI) was calculated using light microscopy. Figure 1 shows a representative photomicrograph alveolar macrophages following exposure to apoptotic cells. Ingested cells can be easily visualized and quantified. Of note, apoptotic cells that have not been ingested are often destroyed by the cytospin process that is required for in vivo experiments. In figure 1, the arrowheads demonstrate remnants of apoptotic cells that have been destroyed during the cytospin process. The presence of these cell remnants not only confirms successful intratracheal instillation of apoptotic cells, but also further reinforces the fragility of these cells and their propensity to undergo cell lysis. A minimum of 400 AM's were counted per condition in duplicate. In all cases, during analysis, the reader was blinded to the sample identification. The PI was calculated using the following formula: ((number of apoptotic bodies)/(total macrophages)) × 100. Each condition was tested in duplicate and repeated at least three times.
Figure 1.
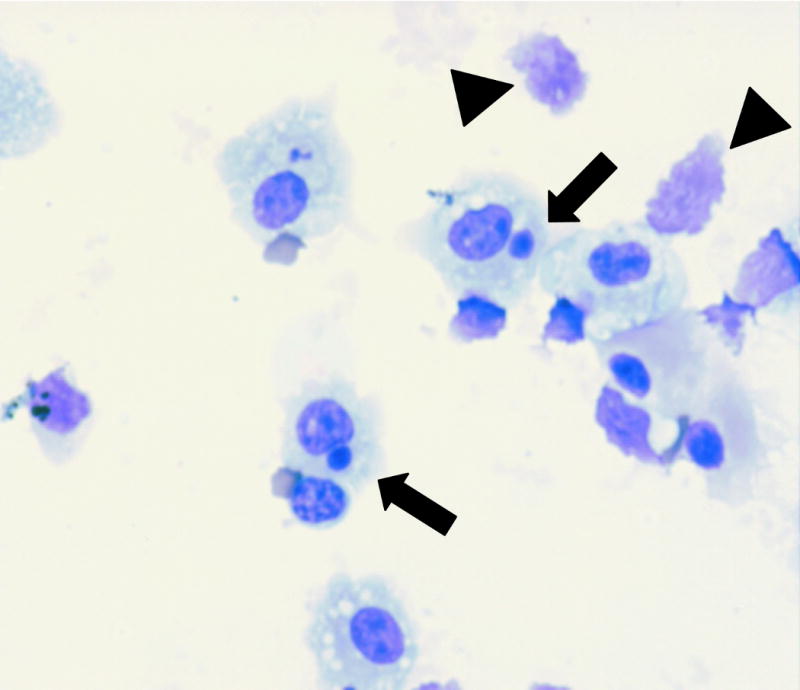
Representative photomicrograph (60×) demonstrating phagocytosis of apoptotic thymocytes by alveolar macrophages. Ingestions are indicated by arrows, which are manually counted to determine the phagocytic index. The arrowheads demonstrate remnants of apoptotic thymocytes that have been destroyed during the cytospin process.
RhoA Activity Assay
Activation of RhoA was determined in both in vitro and in vivo models. For the in vitro model, alveolar macrophages were harvested as described above and plated in 6-well plates (250,000 cells/well). Cells were cultured in the presence of either control media or media containing 100mM alcohol for 0-30 minutes prior to assay. For in vivo experiments, BAL was performed 60 minutes following i.p. injection of either PBS or PBS containing 20% alcohol (5 mice per experimental group; BAL with a total of 10ml per animal in 1ml increments). Cells were washed and plated on 6-well plates, followed by incubation for 30min at 10% CO2 at 37°C. Non-adherent cells were then removed prior to assay. For both in vitro and in vivo experiments, cell lysates were collected using a cell scraper. RhoA activity was determined using the G-LISA assay kit according to the manufacturer's instructions. At least three independent experiments were performed according to the manufacturer's specifications.
Statistical Analysis
The means were analyzed using ANOVA for multiple comparisons. When ANOVA indicated significance, a Dunnett's method was used to compare groups with an internal control. For all other experiments in which two conditions were being compared, a Student's t-test assuming equal variance was used. All data were analyzed using JMP (version 3) Statistical Software for the Macintosh (SAS Institute Inc., Cary, North Carolina, USA) and are presented as the mean ± SEM.
Results
In vitro alcohol exposure impairs efferocytosis
Alveolar macrophages were cultured in the presence and absence of alcohol (25-100mM) for 24h. Macrophages in all treatment groups were >97% viable, and culture in the presence of alcohol did not alter cell viability, as determined by trypan blue exclusion (data not shown). Alcohol exposure impaired efferocytosis at all doses of alcohol used, and the effect of alcohol on efferocytosis was dose-dependent, such that increasing doses of alcohol resulted in decreasing levels of phagocytosis (figure 2). Of note, the concentrations of alcohol used for these experiments (25-100 mM, or 115-460 mg/dL) are comparable to blood alcohol levels commonly seen in individuals at the time of presentation to the hospital(Winek et al. 2004).
Figure 2.

In vitro exposure of alveolar macrophages to alcohol for 24h impairs phagocytosis of apoptotic cells in a dose dependent fashion. Data shown represent combined data from three separate experiments, each performed in duplicate. *p<0.05 when compared to control.
In vivo acute alcohol intoxication impairs efferocytosis
Many critically ill patients with a history of alcohol abuse are acutely intoxicated at the time of hospital admission with diagnoses that place them at increased risk for ARDS, such as aspiration or bacterial pneumonia. Previous studies have shown that human neutrophils undergo apoptosis very quickly, with approximately 40% becoming apoptotic within 6 hours of culture(Matute-Bello et al. 1997c). Because neutrophils become apoptotic in such a short period of time, the effects of acute alcohol intoxication on their clearance may be of critical importance during the early stages of ARDS. To determine whether in vivo, acute alcohol exposure impairs efferocytosis, apoptotic cells were instilled i.t. 30 minutes following a single i.p. injection of either PBS or 20% ethanol (3g/kg total dose). In vivo treatment with alcohol was not associated with altered numbers of total cells, macrophages, or neutrophils (data not shown). Sixty minutes following instillation of apoptotic cells, the PI was calculated from cytospins of the BAL fluid. A single dose of alcohol was associated with a significant decrease in efferocytosis (figure 3).
Figure 3.
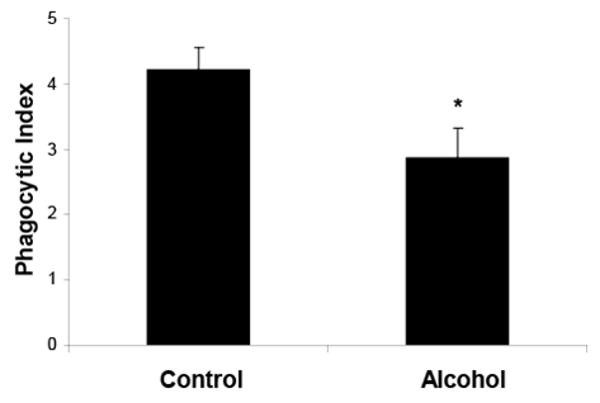
In vivo acute alcohol exposure is associated with impaired phagocytosis of apoptotic cells by alveolar macrophages. Data shown represent combined data from three separate experiments. *p<0.05 when compared to control.
Acute alcohol intoxication is associated with an increased number of neutrophils in the alveolar space following LPS-induced acute lung injury
Alcohol intoxication is associated with decreased alveolar neutrophil numbers at 4 hours following intratracheal challenge with lipopolysaccharide (LPS) (Nelson, Bagby, & Summer 1989). However, little is known about the effects of such intoxication on the accumulation of inflammatory cells at later time points following an inflammatory stimulus. Based on our hypothesis that alcohol impairs efferocytosis, we expected that alveolar neutrophil numbers would be paradoxically increased during the later stages of lung injury due to ineffective removal. To test this hypothesis, i.t instillation of LPS (20ug/mouse) was performed 30 min following a single i.p. injection of saline or 20% ethanol (3g/kg), and BAL inflammatory cells were measured. BALF neutrophils were significantly increased at 24h in the alcohol group, while alveolar macrophage numbers were similar in both groups at all time points tested (figure 4). This effect could be due to either (1) increased late chemotaxis of neutrophils into the alveolar compartment during acute lung injury, (2) impaired phagocytosis and clearance of neutrophils from the alveolar compartment, or (3) both.
Figure 4.
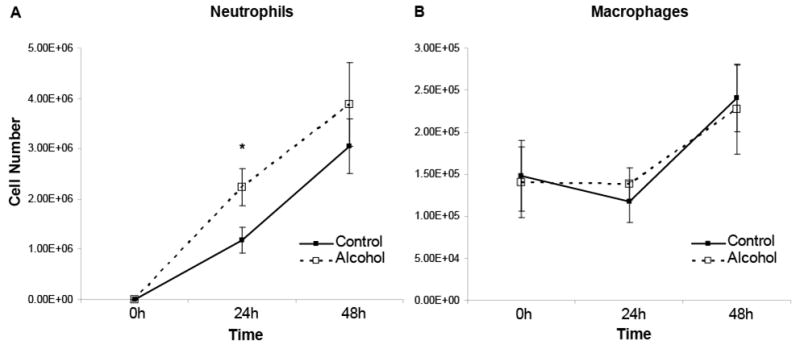
In vivo acute alcohol exposure is associated with increased numbers of alveolar neutrophils (A) but does not alter alveolar macrophage (B) numbers following LPS-induced lung injury. n=6-8 for each experimental group. *p<0.05 when compared to time-matched control.
In vivo chronic alcohol exposure impairs efferocytosis
Data from our lab and others have consistently shown that chronic alcohol abuse decreases pulmonary anti-oxidant capacity by decreasing pulmonary concentrations of the antioxidant glutathione (Bechara et al. 2004;Brown et al. 2001a;Brown et al. 2001b;Brown et al. 2007a;Burnham et al. 2003;Guidot et al. 1999;Guidot et al. 2000;Holguin et al. 1998;Moss et al. 2000a;Moss et al. 2000b;Moss & Burnham 2006). LPS instillation, a well-described model of lung injury, produces a pulmonary environment that is rich in oxidative stress; therefore, we hypothesized that the effects of chronic alcohol exposure on efferocytosis may be more profound during LPS-induced lung injury. To test this hypothesis, we measured the effects of alcohol on pulmonary efferocytosis in the presence and absence of LPS instillation. In addition, we measured efferocytosis during two general times following LPS instillation, one during the early stages of inflammation, and the other at a time when inflammation is resolving. Following exposure to alcohol for eight weeks, i.t. instillation of apoptotic thymocytes was performed, and the PI was calculated 60min later from cytospins of the BAL fluid. Surprisingly, chronic alcohol exposure alone had no affect on efferocytosis at 8 weeks (figure 5). To determine the effects of chronic alcohol exposure in the setting of lung injury, experiments were repeated 24 hours and 7 days following i.t. instillation of LPS. Similar to baseline data in the absence of lung injury, chronic alcohol exposure did not significantly alter efferocytosis 24h following instillation of LPS. In contrast, efferocytosis was significantly decreased in the alcohol group 7 days following instillation of LPS (figure 5). These data suggest that the effects of alcohol on recruited macrophages are different that those on resident phagocytes.
Figure 5.
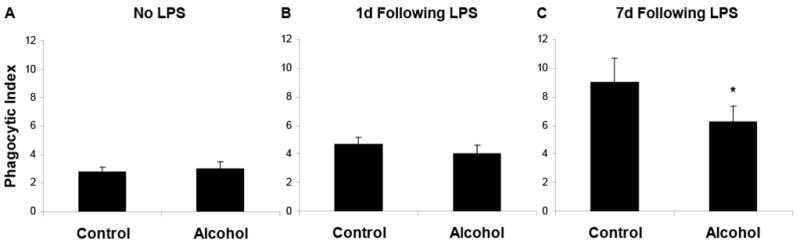
In vivo chronic alcohol exposure did not alter phagocytosis of apoptotic cells by alveolar macrophages at baseline (A) or 24h following i.t. LPS instillation (B). However, at 7 days following LPS, chronic alcohol exposure was associated with impaired efferocytosis (C). Data shown represent combined data from three separate experiments. *p<0.05 when compared to time-matched control.
Chronic alcohol exposure is associated with an increased number of neutrophils in the alveolar space following LPS-induced acute lung injury
To determine whether chronic alcohol exposure also alters the inflammatory response to lung injury, mice received chronic ethanol in water for 8 weeks. Following alcohol exposure, there was no difference in animal weight or appearance (animal weight: 21.2 g +/- 0.28 for control, 20.9 g +/- 0.25 for alcohol; p>0.05). BALF neutrophil numbers were similar 24h following LPS. However, chronic alcohol exposure was associated with increased numbers of BALF neutrophils at 48 and 72 hours following LPS (figure 6). Of interest, alveolar macrophage numbers were reduced in the alcohol group prior to instillation of LPS but increased similarly in both groups at later time points (figure 6). Therefore, in the case of chronic alcohol exposure, alveolar macrophage numbers are decreased at baseline, although their ability to phagocytose apoptotic cells is not affected. In contrast, in the setting of LPS-induced lung injury, alveolar macrophage numbers are similar, although their ability to phagocytose apoptotic cells is impaired. These data suggest that the pulmonary phagocytic capacity is decreased at baseline through a decrease in the total number of available phagocytes. Following LPS exposure, alcohol impairs the ability of alveolar macrophages to phagocytose apoptotic cells. Similar to data from our acute model, chronic exposure to alcohol exposure is associated with increased neutrophil numbers during lung injury.
Figure 6.
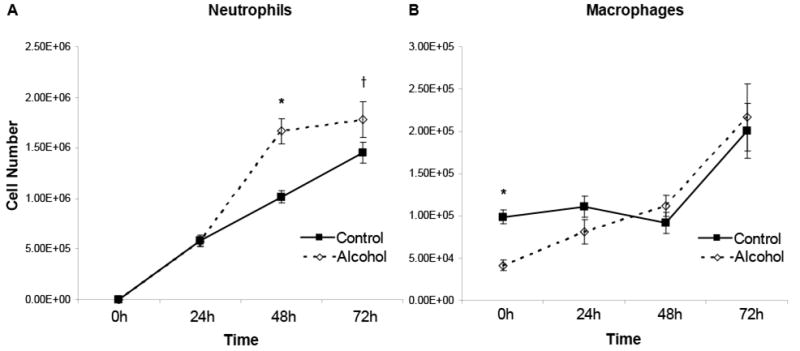
In vivo chronic alcohol exposure is associated with increased numbers of alveolar neutrophils (A) at 48h and 72h following i.t. instillation of LPS. Chronic alcohol exposure was also associated with decreased numbers of alveolar macrophages (B) at baseline, although alveolar macrophage numbers were similar at all times measured following LPS instillation. n=6-8 for each experimental group. *p<0.05 when compared to time-matched control. † p=0.07.
The effects of alcohol on efferocytosis are mediated, in part, by Rho kinase activation
Based on data from our acute model of alcohol exposure, it is clear that short-term exposure to alcohol is associated with impaired efferocytosis, even in the absence of lung injury. To investigate some of the mechanisms involved in this alcohol-associated defect, our in vitro model was employed. Prior to addition of apoptotic cells, AMs were treated with the ROCK inhibitor, Y27632 (0.1-10μM). Treatment with this agent reversed the effects of alcohol on efferocytosis at all concentrations used, suggesting ROCK activation may be an important mechanism by which alcohol impairs efferocytosis (figure 7).
Figure 7.

In vitro exposure of alveolar macrophages to alcohol for 24h impairs phagocytosis of apoptotic cells. This effect is reversed by pre-treatment with the Rho kinase inhibitor, Y27632 (1-10μM). Data shown represent combined data from three separate experiments, each performed in duplicate. *p<0.05 when compared to control.
The effects of alcohol on efferocytosis occur independently of RhoA activation
A well-described activator of ROCK is the Rho GTPase, RhoA. We hypothesized that alcohol-induced activation of ROCK occurs through activation of RhoA. To determine the role of Rho activation on alcohol-mediated suppression of efferocytosis, AMs were treated in vitro with the Rho inhibitor, C3 transferase (1-10μg/ml), prior to the addition of apoptotic cells. In contrast to inhibiton of ROCK activation, inhibition of Rho activation did not reverse the effects of alcohol on efferocytosis (figure 8a), suggesting that the effects of alcohol on efferocytosis occur independent of Rho activation. To determine the effects of both in vitro and in vivo alcohol exposure on RhoA activation, RhoA activity was determined using the G-LISA assay kit in cell lysates from AMs following in vitro and in vivo exposure to alcohol. Contrary to our hypothesis, RhoA activity was not increased following in vitro (figure 8b) or in vivo (figure 8c) exposure to alcohol. In fact, RhoA activity was significantly decreased by alcohol in our in vivo model, suggesting that alcohol-induced activation of ROCK is not secondary to activation of RhoA.
Figure 8.

In vitro exposure of alveolar macrophages to alcohol for 24h impairs phagocytosis of apoptotic cells. This effect is not reversed by pre-treatment with the cell permeable Rho inhibitor, C3 transferase (1-10μg/ml) (A). RhoA activity is not increased by either in vitro (B) or in vivo (C) exposure to alcohol, suggesting that the ROCK-dependent effects of alcohol on efferocytosis are independent of RhoA. In fact, in vivo alcohol exposure is associated with significantly decreased RhoA activity. *p<0.05 when compared to control.
Discussion
In the present study, we demonstrate that in vivo acute alcohol exposure impaired the process by which dying cells are phagocytosed (efferocytosis). Interestingly, while chronic alcohol exposure was associated with decreased numbers of alveolar macrophages at baseline, efferocytosis was only impaired in the setting of lung injury, suggesting important differences between the acute and chronic effects of alcohol exposure. Both acute and chronic alcohol exposure were associated with increased alveolar neutrophil numbers during LPS-induced lung injury. Our in vitro studies confirmed that alcohol exposure, at clinically relevant concentrations, caused a dose-dependent decrease in efferocytosis. In this model, treatment with the ROCK inhibitor, Y27632, reversed the adverse effects of alcohol on efferocytosis. However, these effects were not mediated through activation of RhoA, as RhoA activity was decreased following both in vivo and in vitro exposure to alcohol, Rho inhibition had no effect on alcohol-mediated suppression of efferocytosis. Taken together, our data demonstrate a novel mechanism by which both acute and chronic alcohol exposure may promote a pro-inflammatory environment, resulting in an increased susceptibility and severity of ARDS. Specifically, alcohol exposure impairs the clearance of inflammatory cells from the lungs, which may result in higher numbers of inflammatory neutrophils within the alveolar space during the later stages of lung injury. Epidemiological studies linking alcohol abuse to ARDS have shown a clear relationship between a history of alcohol abuse and increased susceptibility to ARDS. However, it remains unclear whether it is the short-term or long-term effects of alcohol that play the most critical role in increasing one's susceptibility to ARDS. Because patients with a history of chronic alcohol abuse are also more likely to be acutely intoxicated on presentation to the hospital, further work is required to delineate these conditions and determine the specific role that each type of exposure plays in the development of ARDS. Because both forms of alcohol abuse are known to cause alterations in the pulmonary inflammatory response (Szabo & Mandrekar 2009), it is likely that both forms of abuse are important in the pathophysiology of ARDS. The current study highlights important differences between the effects of acute and chronic alcohol exposure on efferocytosis and alveolar cell numbers following LPS instillation. Further studies will be needed to explore the mechanisms underlying these differences, as well as to determine their role in the pathophysiology of ARDS.
The process by which inflammation is resolved during lung injury is complex. One key step in this process is the timely phagocytosis and clearance of dying cells from the site (efferocytosis), before the pro-inflammatory components within these cells are released. Efferocytosis is a highly efficient process (Matute-Bello et al. 1997a) that is regulated, in part, by RhoA and ROCK activation. Factors that activate RhoA and ROCK impair efferocytosis and are associated with an increased pro-inflammatory environment. However, the effects of Rho pathway activation and impaired efferocytosis on the pathophysiology of ARDS have yet to be determined. In our model, the effects of alcohol on efferocytosis appear to be due, at least in part, to ROCK activation. However, ROCK activation occurred in the absence of RhoA activation, suggesting that alternative activators of ROCK may be important in the alcohol-induced effects on efferocytosis (such as lipid mediators, inositol phospholipids, Granzyme B, and caspases)(Shi & Wei 2007b).
The current study not only highlights some of the important effects of alcohol on efferocytosis, but it also demonstrates important differences between the pulmonary effects of acute and chronic alcohol exposure. ARDS affects approximately 200,000 individuals in the United States each year (Rubenfeld et al. 2005a), and nearly 50% of those individuals have a history of alcohol abuse (Moss et al. 2003c). Those patients who abuse alcohol have an increased duration of mechanical ventilation, prolonged ICU length of stay, and increased mortality (Moss et al. 2003d). As we gain more insight into the mechanisms by which alcohol predisposes patients to the development of ARDS, additional novel therapies will be identified that will improve the incidence and severity of ARDS in this important patient population While our findings are intriguing, there are several important limitations of this study that are worth noting. First, while both acute and chronic alcohol exposure were associated with impaired efferocytosis and increased numbers of alveolar neutrophils, a direct causal relationship has yet to be established. Further investigations will be required to fully establish this relationship. Second, while activation of the ROCK is an important mechanism by which alcohol alters efferocytosis in our model, this occurs through a mechanism that does not involve increased activation of RhoA. This is a novel finding that raises additional questions regarding the mechanism by which alcohol increases ROCK activation. As there are many Rho-independent activators of ROCK, further studies of the effects of alcohol on these various agents within the lung will be required. Finally, several studies have highlighted important gender differences between the effects of alcohol on inflammatory and immune responses(Grossman et al. 1993;Kovacs & Messingham 2002;Lee & Rivier 1996;Li et al. 1998;Spitzer 1999;Spitzer et al. 2002;Spitzer & Spitzer 2000;Spitzer & Zhang 1996b). For example, in the absence of alcohol exposure, inflammatory and immune responses are stronger in females than males(Grossman, Nienaber, Mendenhall, Hurtubise, Roselle, Rouster, Weber, Schmitt, & Gartside 1993;Spitzer 1999;Spitzer & Zhang 1996b;Spitzer & Zhang 1996a). However, this effect is nullified by alcohol exposure(Grossman, Nienaber, Mendenhall, Hurtubise, Roselle, Rouster, Weber, Schmitt, & Gartside 1993;Li, Grossman, Mendenhall, Hurtubise, Rouster, Roselle, & Gartside 1998;Spitzer & Zhang 1996b). Because the current studies were performed in female mice only, it is unknown whether the effects described also apply to male mice, or if there may be important gender differences between the effects of alcohol on efferocytosis.
In conclusion, our study demonstrates that both acute and chronic alcohol exposure impair efferocytosis, an important process in the normal resolution of inflammation, and that this effect is mediated, in part, by RhoA-independent activation of ROCK. While this may be an important mechanism by which alcohol increases patient susceptibility to ARDS, further studies will be needed to characterize the nature of this alcohol-induced effect and to determine the mechanisms by which this occurs. Also, there are differences between the effects of acute and chronic exposure on efferocytosis that warrant further investigation. Ultimately, by better understanding the mechanisms by which alcohol delays the resolution of inflammation, novel therapies may be developed to reverse this process. Given the widespread use of alcohol in our critically ill patients, the clear role of alcohol abuse in increasing the risk for ARDS, and the current lack of specific treatments for individuals with alcohol abuse and ARDS, further investigations into the mechanisms by which alcohol predisposes to ARDS and the effects of specific therapies are necessary.
Acknowledgments
Support: This work was supported by grant K24 HL 089223 from the National Institute of Health as well as funding from the American Thoracic Society Fellow Career Development Award.
References
- Bechara RI, Brown LA, Roman J, Joshi PC, Guidot DM. Transforming growth factor beta1 expression and activation is increased in the alcoholic rat lung. Am J Respir Crit Care Med. 2004;170(2):188–194. doi: 10.1164/rccm.200304-478OC. [DOI] [PubMed] [Google Scholar]
- Brown LA, Harris FL, Bechara R, Guidot DM. Effect of chronic ethanol ingestion on alveolar type II cell: glutathione and inflammatory mediator-induced apoptosis. Alcohol Clin Exp Res. 2001b;25(7):1078–1085. [PubMed] [Google Scholar]
- Brown LA, Harris FL, Bechara R, Guidot DM. Effect of chronic ethanol ingestion on alveolar type II cell: glutathione and inflammatory mediator-induced apoptosis. Alcohol Clin Exp Res. 2001a;25(7):1078–1085. [PubMed] [Google Scholar]
- Brown LA, Ping XD, Harris FL, Gauthier TW. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am J Physiol Lung Cell Mol Physiol. 2007a;292(4):L824–L832. doi: 10.1152/ajplung.00346.2006. [DOI] [PubMed] [Google Scholar]
- Brown LA, Ping XD, Harris FL, Gauthier TW. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am J Physiol Lung Cell Mol Physiol. 2007b;292(4):L824–L832. doi: 10.1152/ajplung.00346.2006. [DOI] [PubMed] [Google Scholar]
- Burnham EL, Brown LA, Halls L, Moss M. Effects of chronic alcohol abuse on alveolar epithelial barrier function and glutathione homeostasis. Alcohol Clin Exp Res. 2003;27(7):1167–1172. doi: 10.1097/01.ALC.0000075821.34270.98. [DOI] [PubMed] [Google Scholar]
- Cook RT, Schlueter AJ, Coleman RA, Tygrett L, Ballas ZK, Jerrells TR, Nashelsky MB, Ray NB, Haugen TH, Waldschmidt TJ. Thymocytes, pre-B cells, and organ changes in a mouse model of chronic ethanol ingestion--absence of subset-specific glucocorticoid-induced immune cell loss. Alcohol Clin Exp Res. 2007;31(10):1746–1758. doi: 10.1111/j.1530-0277.2007.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Guthrie L, Henson PM. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J Immunol. 2001;166(11):6847–6854. doi: 10.4049/jimmunol.166.11.6847. [DOI] [PubMed] [Google Scholar]
- Gajic O, Rana R, Winters JL, Yilmaz M, Mendez JL, Rickman OB, O'Byrne MM, Evenson LK, Malinchoc M, DeGoey SR, Afessa B, Hubmayr RD, Moore SB. Transfusion-related acute lung injury in the critically ill: prospective nested case-control study. Am J Respir Crit Care Med. 2007;176(9):886–891. doi: 10.1164/rccm.200702-271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman CJ, Nienaber M, Mendenhall CL, Hurtubise P, Roselle GA, Rouster S, Weber N, Schmitt G, Gartside PS. Sex differences and the effects of alcohol on immune response in male and female rats. Alcohol Clin Exp Res. 1993;17(4):832–840. doi: 10.1111/j.1530-0277.1993.tb00850.x. [DOI] [PubMed] [Google Scholar]
- Guidot D, Moss M, Holguin F, Lois M, Brown L. Ethanol ingestion impairs alveolar epithelial glutathione homeostasis and function, and predisposes to endotoxin-mediated acute lung injury. Chest. 1999;116(1 Suppl):82S. doi: 10.1378/chest.116.suppl_1.82s. [DOI] [PubMed] [Google Scholar]
- Guidot DM, Modelska K, Lois M, Jain L, Moss IM, Pittet JF, Brown LA. Ethanol ingestion via glutathione depletion impairs alveolar epithelial barrier function in rats. Am J Physiol Lung Cell Mol Physiol. 2000;279(1):L127–L135. doi: 10.1152/ajplung.2000.279.1.L127. [DOI] [PubMed] [Google Scholar]
- Henson PM, Bratton DL, Fadok VA. Apoptotic cell removal. Curr Biol. 2001;11(19):R795–R805. doi: 10.1016/s0960-9822(01)00474-2. [DOI] [PubMed] [Google Scholar]
- Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol. 2001a;155(4):649–659. doi: 10.1083/jcb.200108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol. 2001b;155(4):649–659. doi: 10.1083/jcb.200108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holguin F, Moss I, Brown LA, Guidot DM. Chronic ethanol ingestion impairs alveolar type II cell glutathione homeostasis and function and predisposes to endotoxin-mediated acute edematous lung injury in rats. J Clin Invest. 1998;101(4):761–768. doi: 10.1172/JCI1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109(1):41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Guleria RS, Pan J, Bayless KJ, Davis GE, Dipette D, Singh US. Ethanol impairs Rho GTPase signaling and differentiation of cerebellar granule neurons in a rodent model of fetal alcohol syndrome. Cell Mol Life Sci. 2006;63(23):2859–2870. doi: 10.1007/s00018-006-6333-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs EJ, Messingham KA. Influence of alcohol and gender on immune response. Alcohol Res Health. 2002;26(4):257–263. [PMC free article] [PubMed] [Google Scholar]
- Lee S, Rivier C. Gender differences in the effect of prenatal alcohol exposure on the hypothalamic-pituitary-adrenal axis response to immune signals. Psychoneuroendocrinology. 1996;21(2):145–155. doi: 10.1016/0306-4530(95)00038-0. [DOI] [PubMed] [Google Scholar]
- Li X, Grossman CJ, Mendenhall CL, Hurtubise P, Rouster SD, Roselle GA, Gartside P. Host response to mycobacterial infection in the alcoholic rat: male and female dimorphism. Alcohol. 1998;16(3):207–212. doi: 10.1016/s0741-8329(98)00004-4. [DOI] [PubMed] [Google Scholar]
- Licker M, de P M, Spiliopoulos A, Robert J, Diaper J, Chevalley C, Tschopp JM. Risk factors for acute lung injury after thoracic surgery for lung cancer. Anesth Analg. 2003;97(6):1558–1565. doi: 10.1213/01.ANE.0000087799.85495.8A. [DOI] [PubMed] [Google Scholar]
- Martinez SE, Lazaro-Dieguez F, Selva J, Calvo F, Piqueras JR, Crespo P, Claro E, Egea G. Lysophosphatidic acid rescues RhoA activation and phosphoinositides levels in astrocytes exposed to ethanol. J Neurochem. 2007;102(4):1044–1052. doi: 10.1111/j.1471-4159.2007.04581.x. [DOI] [PubMed] [Google Scholar]
- Matute-Bello G, Liles WC, Radella F, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997a;156(6):1969–1977. doi: 10.1164/ajrccm.156.6.96-12081. [DOI] [PubMed] [Google Scholar]
- Matute-Bello G, Liles WC, Radella F, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997b;156(6):1969–1977. doi: 10.1164/ajrccm.156.6.96-12081. [DOI] [PubMed] [Google Scholar]
- Matute-Bello G, Liles WC, Radella F, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997c;156(6):1969–1977. doi: 10.1164/ajrccm.156.6.96-12081. [DOI] [PubMed] [Google Scholar]
- McPhillips K, Janssen WJ, Ghosh M, Byrne A, Gardai S, Remigio L, Bratton DL, Kang JL, Henson P. TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J Immunol. 2007;178(12):8117–8126. doi: 10.4049/jimmunol.178.12.8117. [DOI] [PubMed] [Google Scholar]
- McVicker BL, Tuma DJ, Kharbanda KK, Kubik JL, Casey CA. Effect of chronic ethanol administration on the in vitro production of proinflammatory cytokines by rat Kupffer cells in the presence of apoptotic cells. Alcohol Clin Exp Res. 2007;31(1):122–129. doi: 10.1111/j.1530-0277.2006.00270.x. [DOI] [PubMed] [Google Scholar]
- McVicker BL, Tuma DJ, Kubik JA, Hindemith AM, Baldwin CR, Casey CA. The effect of ethanol on asialoglycoprotein receptor-mediated phagocytosis of apoptotic cells by rat hepatocytes. Hepatology. 2002;36(6):1478–1487. doi: 10.1053/jhep.2002.37137. [DOI] [PubMed] [Google Scholar]
- Minambres R, Guasch RM, Perez-Arago A, Guerri C. The RhoA/ROCK-I/MLC pathway is involved in the ethanol-induced apoptosis by anoikis in astrocytes. J Cell Sci. 2006;119(Pt 2):271–282. doi: 10.1242/jcs.02723. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Janssen WJ, Fessler MB, McPhillips KA, Borges VM, Bowler RP, Xiao YQ, Kench JA, Henson PM, Vandivier RW. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J Immunol. 2006;176(12):7657–7665. doi: 10.4049/jimmunol.176.12.7657. [DOI] [PubMed] [Google Scholar]
- Moss M, Bucher B, Moore FA, Moore EE, Parsons PE. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA. 1996;275(1):50–54. [PubMed] [Google Scholar]
- Moss M, Burnham EL. Alcohol abuse in the critically ill patient. Lancet. 2006;368(9554):2231–2242. doi: 10.1016/S0140-6736(06)69490-7. [DOI] [PubMed] [Google Scholar]
- Moss M, Guidot DM, Wong-Lambertina M, Ten HT, Perez RL, Brown LA. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am J Respir Crit Care Med. 2000a;161(2 Pt 1):414–419. doi: 10.1164/ajrccm.161.2.9905002. [DOI] [PubMed] [Google Scholar]
- Moss M, Guidot DM, Wong-Lambertina M, Ten HT, Perez RL, Brown LA. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am J Respir Crit Care Med. 2000b;161(2 Pt 1):414–419. doi: 10.1164/ajrccm.161.2.9905002. [DOI] [PubMed] [Google Scholar]
- Moss M, Parsons PE, Steinberg KP, Hudson LD, Guidot DM, Burnham EL, Eaton S, Cotsonis GA. Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med. 2003c;31(3):869–877. doi: 10.1097/01.CCM.0000055389.64497.11. [DOI] [PubMed] [Google Scholar]
- Moss M, Parsons PE, Steinberg KP, Hudson LD, Guidot DM, Burnham EL, Eaton S, Cotsonis GA. Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med. 2003d;31(3):869–877. doi: 10.1097/01.CCM.0000055389.64497.11. [DOI] [PubMed] [Google Scholar]
- Moss M, Parsons PE, Steinberg KP, Hudson LD, Guidot DM, Burnham EL, Eaton S, Cotsonis GA. Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med. 2003a;31(3):869–877. doi: 10.1097/01.CCM.0000055389.64497.11. [DOI] [PubMed] [Google Scholar]
- Moss M, Parsons PE, Steinberg KP, Hudson LD, Guidot DM, Burnham EL, Eaton S, Cotsonis GA. Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med. 2003b;31(3):869–877. doi: 10.1097/01.CCM.0000055389.64497.11. [DOI] [PubMed] [Google Scholar]
- Nelson S, Bagby G, Summer WR. Alcohol suppresses lipopolysaccharide-induced tumor necrosis factor activity in serum and lung. Life Sci. 1989;44(10):673–676. doi: 10.1016/0024-3205(89)90472-4. [DOI] [PubMed] [Google Scholar]
- Phua J, Badia JR, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, Scales DC, Stather DR, Li A, Jones A, Gattas DJ, Hallett D, Tomlinson G, Stewart TE, Ferguson ND. Has Mortality from Acute Respiratory Distress Syndrome Decreased Over Time? A Systematic Review. Am J Respir Crit Care Med. 2008 doi: 10.1164/rccm.200805-722OC. [DOI] [PubMed] [Google Scholar]
- Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007a;7(12):964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007b;7(12):964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- Richens TR, Linderman DJ, Horstmann SA, Lambert C, Xiao YQ, Keith RL, Boe DM, Morimoto K, Bowler RP, Day BJ, Janssen WJ, Henson PM, Vandivier RW. Cigarette Smoke Impairs Clearance of Apoptotic Cells Through Oxidant-dependent Activation of RhoA. Am J Respir Crit Care Med. 2009 doi: 10.1164/rccm.200807-1148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimland D, Hand WL. The effect of ethanol on adherence and phagocytosis by rabbit alveolar macrophages. J Lab Clin Med. 1980;95(6):918–926. [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005b;353(16):1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005a;353(16):1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- Shi J, Wei L. Rho kinase in the regulation of cell death and survival. Arch Immunol Ther Exp (Warsz) 2007b;55(2):61–75. doi: 10.1007/s00005-007-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wei L. Rho kinase in the regulation of cell death and survival. Arch Immunol Ther Exp (Warsz) 2007c;55(2):61–75. doi: 10.1007/s00005-007-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wei L. Rho kinase in the regulation of cell death and survival. Arch Immunol Ther Exp (Warsz) 2007a;55(2):61–75. doi: 10.1007/s00005-007-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer JA. Gender differences in some host defense mechanisms. Lupus. 1999;8(5):380–383. doi: 10.1177/096120339900800510. [DOI] [PubMed] [Google Scholar]
- Spitzer JA, Spitzer JJ. Lipopolysaccharide tolerance and ethanol modulate hepatic nitric oxide production in a gender-dependent manner. Alcohol. 2000;21(1):27–35. doi: 10.1016/s0741-8329(99)00098-1. [DOI] [PubMed] [Google Scholar]
- Spitzer JA, Zhang P. Gender differences in phagocytic responses in the blood and liver, and the generation of cytokine-induced neutrophil chemoattractant in the liver of acutely ethanol-intoxicated rats. Alcohol Clin Exp Res. 1996b;20(5):914–920. doi: 10.1111/j.1530-0277.1996.tb05271.x. [DOI] [PubMed] [Google Scholar]
- Spitzer JA, Zhang P. Gender differences in neutrophil function and cytokine-induced neutrophil chemoattractant generation in endotoxic rats. Inflammation. 1996a;20(5):485–498. doi: 10.1007/BF01487041. [DOI] [PubMed] [Google Scholar]
- Spitzer JA, Zhang P, Bagby GJ, Stouwe CV, Nelson S. Sex differences in the modulation by ethanol of lung chemotaxis. Alcohol. 2002;28(2):95–102. doi: 10.1016/s0741-8329(02)00271-9. [DOI] [PubMed] [Google Scholar]
- Sward K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. Inhibition of Rho-associated kinase blocks agonist-induced Ca2+ sensitization of myosin phosphorylation and force in guinea-pig ileum. J Physiol. 2000;522(Pt 1):33–49. doi: 10.1111/j.1469-7793.2000.0033m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33(2):220–232. doi: 10.1111/j.1530-0277.2008.00842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science. 2002;296(5565):155–158. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389(6654):990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129(6):1673–1682. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, Walport MJ, Fisher JH, Henson PM, Greene KE. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol. 2002;169(7):3978–3986. doi: 10.4049/jimmunol.169.7.3978. [DOI] [PubMed] [Google Scholar]
- Vandivier RW, Richens TR, Horstmann SA, deCathelineau AM, Ghosh M, Reynolds SD, Xiao YQ, Riches DW, Plumb JD, Vachon E, Downey GP, Henson PM. Dysfunctional Cystic Fibrosis Transmembrane Conductance Regulator Inhibits Phagocytosis of Apoptotic Cells with Pro-inflammatory Consequences. Am J Physiol Lung Cell Mol Physiol. 2009 doi: 10.1152/ajplung.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Winek CL, Wahba WW, Windisch RM, Winek CL., Jr Serum alcohol concentrations in trauma patients determined by immunoassay versus gas chromatography. Forensic Sci Int. 2004;139(1):1–3. doi: 10.1016/s0379-0738(03)00262-7. [DOI] [PubMed] [Google Scholar]
- Zhang P, Nelson S, Summer WR, Spitzer JA. Acute ethanol intoxication suppresses the pulmonary inflammatory response in rats challenged with intrapulmonary endotoxin. Alcohol Clin Exp Res. 1997;21(5):773–778. [PubMed] [Google Scholar]