

Abstract
Salvinorin A is a naturally-occurring potent and selective kappa opioid receptor agonist, and smoking salvinorin A produces the most intense hallucinogenic effects in human. Eight neoclerodane diterpene derivatives were isolated from the smoke of salvinorin A, and their structures were identified by spectroscopic methods. The major structural changes include epimerizations, eliminations, and rearrangements.
Keywords: Salvinorin A, Neoclerodane diterpenoid, Smoke, Opioid receptor
Salvia divinorum is a rare member of the Lamiaceae (mint) family and has been traditionally used by the Mazatec Indians of northeastern Oaxaca in spiritual practices.1 S. divinorum leaf and leaf preparations are widely available in Western Europe and the USA, notably on Internet sites.2 Because of its hallucinogenic effects, S. divinorum (also known as magic mint) has been increasingly used as a marijuana substitute in non-ethnopharmacological settings.2-4 Salvinorin A (1), a non-nitrogenous neoclerodane diterpenoid isolated from S. divinorum,5,6 was characterized as a potent and selective kappa (κ) opioid receptor (KOR) agonist and responsible for the observed psychoactive effects.1,7-10 Inhalation of the vaporized smoke of 1 was proven to be the most efficient method for its hallucinogenic effects in human.1 The effective dose as low as 200 to 500 μg produces profound hallucinations with similar potency as hallucinogen LSD (lysergic acid dirthylamide).1 However, the active principles in the smoke of 1 are still unclear. In order to better understand the biological and pharmacological activities of 1, we report the isolation and structural elucidation of eight neoclerodane diterpene derivatives resulted from the smoke extract of 1.
Salvinorin A (1, 1.0 g) was heated in a glass flask immersed in an oil bath. When the temperature reached at 245 °C, the visible white vapor rose to the cold upper inner glass wall and condensed into solid. The excess vapor was collected by passing through a short tube into a chloroform solution. The heating process lasted for 10 min at 248±3 °C. The condensed solid was washed out with chloroform, and the combined chloroform solution was concentrated under reduced pressure and the temperature of the water bath was kept below 40 °C. The residue (150 mg) was chromatographed over silica gel and eluted with hexane-ethyl acetate to give eight compounds 1-8 (Figure 1). 1 and 8-epi-1 (2) are the major components, while 3-8 are minor compounds. The known compounds 1-4 were identified by comparison with the their published spectroscopic data.11,12
Figure 1.

Compounds 1-8 isolated from the smoke of salvinorin A
Compound 5 is a colorless semi-solid. Its molecular formula was determined to be C23H28O8 on the basis of HR ESI-MS at m/z 455.1679 [M+Na]+ (cald 455.1682). The 1H and 13C data of 5 were closely related to those of 8-episalvinorin A (2),11 indicating that 5 is a derivative of 2. The difference between 5 and 2 was observed in the 1H and 13C NMR spectra, in which the 1H and 13C chemical shifts of the ring A of 5 were different from those of 2. Especially, the coupling pattern of H-2 (δ 4.88, t, J = 3.0 Hz) in 5 is indicative of equatorial orientation and the acetyl group at C-2 must have β-configuration. This was further confirmed by comparison of NMR data with those of 2-epi-salvinorin A.13 The 1H and 13C signals of 5 were assigned by extensive 2D-NMR methods (Table 1). This conclusion was furthermore supported by the synthesis of 5 from 2 via intermediate 8-episalvinorin B (9)14 following the literature procedures (Scheme 1).13,15 Consequently, the structure of 5 was established as 2-epi-8-epi-salvinorin A.
Table 1.
1H (300 MHz) and 13C NMR Data (75 MHz) for 5 and 6 in CDCl3
| No. C | 5 |
6 |
||
|---|---|---|---|---|
| δ H | δ C | δ H | δ C | |
| 1 | 204.4 | 198.5 | ||
| 2 | 4.88 (t, 3.0) | 76.1 | 5.94 (dd, 3.3,10.2) | 130.0 |
| 3 | 2.35 (m) | 30.6 | 6.78 (ddd, 1.8, 5.7, 9.9) | 145.9 |
| 2.10-2.25 (m) | ||||
| 4 | 2.90 (dd, 3.6, 13.2) | 50.0 | 2.37 (dt, 3.0,18.9) | 45.4 |
| 2.10-2.20 (m) | ||||
| 5 | 42.9 | 38.5 | ||
| 6 | 2.10-2.18 (m) | 34.1 | 1.80-2.02 (m) | 35.6 |
| 1.55 (dt, 3.3, 10.5) | 1.51-1.61 (m) | |||
| 7 | 2.15-2.25 (m) | 17.5 | 2.10-2.20 (m) | 17.3 |
| 1.87 (dt, 4.5, 14.1) | 1.80-2.02 (m) | |||
| 8 | 2.46 (d, 3.6) | 45.4 | 2.51 (br s) | 45.4 |
| 9 | 34.2 | 34.2 | ||
| 10 | 2.59 (s) | 62.1 | 2.36 (s) | 63.3 |
| 11 | 2.35 (m) | 47.8 | 2.66 (dd, 1.8, 15.3) | 48.9 |
| 1.40 (dd, 12.3, 14.7) | 1.72 (dd, 12.3, 15.3) | |||
| 12 | 5.26 (d, 12.0) | 69.9 | 5.27 (dd, 0.9, 12.3) | 69.9 |
| 13 | 123.4 | 123.6 | ||
| 14 | 6.36 (dd, 0.9, 1.8) | 108.4 | 6.40 (t, 0.9) | 108.4 |
| 15 | 7.40 (t, 1.8) | 143.6 | 7.39 (t, 1.2) | 143.2 |
| 16 | 7.44 (d, 0.9) | 139.7 | 7.46 (d, 0.6) | 139.4 |
| 17 | 173.7 | 174.2 | ||
| 18 | 172.5 | |||
| 19 | 1.07 (s) | 14.9 | 1.10 (s) | 19.8 |
| 20 | 1.84 (s) | 24.5 | 1.49 (s) | 23.7 |
| CO2CH3 | 3.69 (s) | 51.7 | ||
| -COCH3 | 169.6 | |||
| -COCH3 | 2.14 (s) | 21.1 | ||
Scheme 1.

Conversion of 2 to 5
Compound 6, a colorless semi-solid, gave a pseudomolecular ion peak at m/z 315.1599 [(M + H)]+ in HR ESI-MS suggesting a molecular formula of C19H22O4. The characteristic 1H NMR spectrum revealed the presence of two tertiary methyl groups (δ 1.10 and 1.49), an oxygenated methine proton [δ 5.27 (dd, J = 0.9 Hz and 12.3 Hz)], and five olefinic protons [δ 5.94 (dd, J = 3.3 Hz and 10.2 Hz), 6.40 (t, J = 0.9 Hz), 6.78 (ddd, J = 1.8 Hz, 5.7 Hz and 9.9 Hz), 7.39 (t, J = 1.2 Hz) and 7.46 (d, J = 0.6 Hz)]. The 13C NMR spectrum of 6 revealed 19 carbon signals, which were classified by DEPT as two methyls (δ 19.8 and 23.7), four methylenes (δ 17.3, 35.6, 45.4 and 48.9), eight methines (δ 45.4, 63.3, 69.9, 108.4, 130.0, 139.4, 143.2 and 145.9), three quaternary carbons (δ 34.2, 38.5 and 123.6), and two carbonyl carbons (δ 174.2 and 198.5). Comparison of the 1H and 13C NMR spectra of 6 with those of 5 suggested that the B-, C-, and furan-rings of these compounds were structurally similar. The most striking features of 6 were the absence of acetoxy and methyl ester groups and the presence of two additional olefinic signals. HMQC analysis indicated that the olefinic carbons at δ 130.0 and 145.9 were attached directly to the protons at δ 5.94 and 6.78, respectively. Interpretation of HMBC data (Figure 2) showed the following significant correlations: H-3 (δ 6.78) with C-5 (δ 38.5); H-4 (δ 2.10-2.20) with C-2 (δ 130.0), C-3 (δ 145.9) and C-10 (δ 63.3); and CH3-19 (δ 1.10) with C-4 (δ 45.4), C-5 (δ 38.5), C-6 (δ 35.6) and C-10 (δ 63.3). This, together with the upfield shift of C-1 (δ 198.5), led to the establishment of the partial structure – a conjugated carbonyl group in A-ring. Table 1 showed the full assignments of the 1H and 13C signals of 6 based on the COSY, HMQC and HMBC analysis. Furthermore, in the NOESY spectrum, H-20 showed cross peaks to H-8 and H-19, which fully supported that the relative stereochemistry of H-8 is α-orientation. On the basis of these data, the structure of 6 was proposed.
Figure 2.
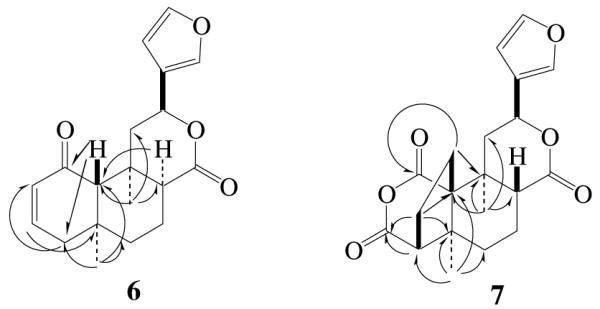
Selected HMBC correlations of 6 and 7
Compound 7 was obtained as colorless needles, and its molecular formula was deduced as C20H22O6 by a pseudomolecular ion peak at m/z 359.1496 [(M + H)]+ in its HR ESI-MS spectrum. The 1H NMR spectrum displayed signals of two tertiary methyls (δ 1.19 and 1.25), one oxygenated methines (δ 5.57) and three olefinic protons (δ 6.63, 7.66 and 7.75). The remaining protons were observed in aliphatic region from δ 1.2 to δ 3.0. The 13C NMR spectrum of 7 showed 20 carbon signals ascribed to two methyls, five methylenes, six methines, four quaternary carbons, and three carbonyl carbons. Assignments of the 1H and 13C signals were performed by extended 2D NMR methods including 1H-1H COSY, HMQC and HMBC spectra (Table 2 and Figure 2), which indicated that the A-ring is a five member ring, and an additional anhydride is formed between C-1 and C-18. In the HMBC spectrum, the methylene group resonating at δ 2.01 and 2.71 (H-2) was correlated with C-1 (δ 170.0), C-5 (δ 43.1), C-9 (δ 36.3) and C-10 (δ 58.8), indicating that the methylene group was located at C-10 position (Figures 1 and 2). The coupling constants of H-8 (dd, J = 3.0 Hz and 10.8 Hz) and H-12 (dd, J = 4.8 Hz and 11.7 Hz) is in agreement with those of natural salvinorins, suggesting that H-8 was β-orientation.11,16 In the NOESY spectrum, H-8 (δ 2.94) showed significant cross peaks to H-2 (δ 2.71), while H-12 (δ 5.57) related to H-20 (δ 1.25). The stereochemistry of 7 was further confirmed by X-ray analysis (Figure 3).17 From these data, the structure of 7 was determined.
Table 2.
1H (300 MHz) and 13C NMR Data (75 MHz) for 7 and 8
| No. C | 7a |
8b |
||
|---|---|---|---|---|
| δ H | δ C | δ H | δ C | |
| 1 | 170.0 | 170.0 | ||
| 2 | 2.71 (dt, 4.5,11.4) | 29.3 | 2.10-2.40 (m) | 33.2 |
| 2.01 (m) | ||||
| 3 | 2.24 (m) | 21.9 | 2.10-2.40 (m) | 22.0 |
| 1.79 (dt, 4.2, 9.6) | 1.80-1.90 (m) | |||
| 4 | 2.84 (d, 7.2) | 55.2 | 2.80 (d, 6.0) | 55.9 |
| 5 | 43.1 | 43.1 | ||
| 6 | 1.44-1.54 (m) | 28.8 | 1.90-2.00 (m) | 25.9 |
| 1.37 (m) | 1.22-1.36 (m) | |||
| 7 | 1.88-1.95 (m) | 17.4 | 2.20-2.40 (m) | 16.6 |
| 1.55-1.65 (m) | 1.90-2.00 (m) | |||
| 8 | 2.94 (dd, 3.0, 10.8) | 44.6 | 2.54 (br s) | 43.4 |
| 9 | 36.3 | 36.8 | ||
| 10 | 58.8 | 60.5 | ||
| 11 | 2.55 (dd, 4.8, 13.2) | 38.5 | 2.52 (d, 15.6) | 43.9 |
| 1.90-2.00 (m) | 2.10-2.30 (m) | |||
| 12 | 5.57 (dd, 4.8, 11.7) | 70.8 | 5.30 (d, 12.0) | 69.8 |
| 13 | 125.5 | 123.3 | ||
| 14 | 6.63 (t, 1.8) | 109.2 | 6.43 (br s) | 108.4 |
| 15 | 7.66 (t, 1.5) | 144.0 | 7.44 (br s) | 143.7 |
| 16 | 7.75 (t, 0.9) | 140.4 | 7.50 (br s) | 139.7 |
| 17 | 171.7 | 174.9 | ||
| 18 | 169.7 | 168.8 | ||
| 19 | 1.19 (s) | 20.8 | 1.32 (s) | 20.3 |
| 20 | 1.25 (s) | 15.8 | 1.58 (s) | 26.2 |
7 was measured in DMSO-d6
8 were measured in CDCl3.
Figure 3.

Crystal structure of 7 showing 50% probability displacement ellipsoids
Compound 8 was confirmed to have the same molecular formula as 7 by measurement of its HR ESI-MS. The 1H and 13C NMR data of 8 were closely related to those of 7, suggesting 8 to be an isomer of 7. Using 2D NMR techniques, including COSY, HMQC and HMBC, permitted the assignments of all 1H and 13C NMR chemical shifts as shown in Table 2. In the 1H NMR spectrum of 8, the coupling constants of H-8 (br s) and H-12 (d, J = 12.0 Hz) suggested that H-8 was α-orientated. This was also evidenced by the correlations of H-20 with H-8 and H-19 in its NOESY spectrum. Accordingly, the structure of 8 was established as 8-epimer of 7.
In conclusion, eight neoclerodane diterpene derivatives were isolated from the smoke of salvinorin A. The major structural changes of salvinorin A include epimerizations at C-2 and C-8, eliminations of acetyoxy and methyl ester groups, and carbon-carbon rearrangements at C-1, C-2 and C-10. Compounds 7 and 8 are unique salvinorin derivatives with five member rings and anhydrides confirmed by x-ray analysis. Our findings provided information on the chemical components of smoke generated by heating salvinorin A. The biological data of these novel products will be reported in due course.
Acknowledgement
The authors wish to thank Dr. Yongxuan Su, University of California, San Diego for HR-MS measurements, and Dr. Shao-Liang Zheng, Center for Crystallographic Studies, Department of Chemistry and Chemical Biology, Harvard University for X-ray analysis. This work was supported by research grant (R01-DA019688) from NIDA, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Siebert DJ. J. Ethnopharmacol. 1994;43:53–56. doi: 10.1016/0378-8741(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 2.Butelman ER, Prisinzano TE, Deng H, Rus S, Kreek MJ. J. Pharmacol. Exp. Ther. 2009;328:588–597. doi: 10.1124/jpet.108.145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harding WW, Tidgewell K, Schmidt M, Shah K, Dersch CM, Snyder J, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Org. Lett. 2005;7:3017–3020. doi: 10.1021/ol0510522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.González D, Riba J, Bouso JC, Gómez-Jarabo G, Barbanoj MJ. Drug Alcohol Depend. 2006;85:157–162. doi: 10.1016/j.drugalcdep.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Ortega A, Blount JF, Manchand PS. J. Chem. Soc., Perkin Trans. 1. 1982:2505–2508. [Google Scholar]
- 6.Valdes LJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda M. J. Org. Chem. 1984;49:4716–4720. [Google Scholar]
- 7.Prisinzano TE. J. Nat. Prod. 2009;72:581–587. doi: 10.1021/np8005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prisinzano TE, Rothman RB. Chem. Rev. 2008;108:1732–1743. doi: 10.1021/cr0782269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roth BL, Baner K, Westkaemper R, Siebert DJ, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc. Natl. Acad. Sci. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheffler DJ, Roth BL. Trends Pharmacol. Sci. 2003;24:107–109. doi: 10.1016/S0165-6147(03)00027-0. [DOI] [PubMed] [Google Scholar]
- 11.Ma Z, Lee DYW. Tetrahedron Lett. 2007;48:5461–5464. doi: 10.1016/j.tetlet.2007.05.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Gilmour B, Navarro H, Rothman RB, Prisinzano TE. J. Nat. Prod. 2006;69:107–112. doi: 10.1021/np050398i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beguin C, Richards MR, Li J-G, Wang Y, Xu W, Liu-Chen L-Y, Carlezon WA, Cohen BM. Bioorg. Med. Chem. Lett. 2006;16:4679–4685. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- 14.Munro TA, Duncan KK, Staples RJ, Xu W, Liu-Chen L-Y, Beguin C, Carlezon WA, Cohen BM. Beilstein J. Org. Chem. 2007;3 doi: 10.1186/1860-5397-3-1. No PP. given. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tidgewell K, Harding WW, Schmidt M, Holden KG, Murry DJ, Prisinzano TE. Bioorg. Med. Chem. Lett. 2004;14:5099–5122. doi: 10.1016/j.bmcl.2004.07.081. [DOI] [PubMed] [Google Scholar]
- 16.Ma Z, Lee DYW. Tetrahedron Lett. 2008;49:1782–1785. doi: 10.1016/j.tetlet.2008.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.X-ray crystallography of 7: CCDC 782913 contains the supplementary crystallographic data. These can be obtained free of charge from the Cambridge Crystallographic Data Centre via; www.ccdc.cam.ac.uk/data_request/cif. [Google Scholar]