

Abstract
AIM
The rhizome of turmeric, Curcuma longa (CL), is a herbal medicine used in many traditional prescriptions. It has previously been shown that CL treatment showed greater than 47% recovery from cisplatin-induced cell damage in human kidney HEK 293 cells. This study was conducted to evaluate the recovery mechanisms of CL that occur during cisplatin induced nephrotoxicity by examining the genome wide mRNA expression profiles of HEK 293 -cells.
METHOD
Recovery mechanisms of CL that occur during cisplatin-induced nephrotoxicity were determined by microarray, real-time PCR, immunofluorescent confocal microscopy and Western blot analysis.
RESULTS
The results of microarray analysis and real-time PCR revealed that NFκB pathway-related genes and apoptosis-related genes were down-regulated in CL-treated HEK 293 cells. In addition, immunofluorescent confocal microscopy and Western blot analysis revealed that NFκB p65 nuclear translocation was inhibited in CL-treated HEK 293 cells. Therefore, the mechanism responsible for the effects of CL on HEK 293 cells is closely associated with regulation of the NFκB pathway.
CONCLUSION
CL possesses novel therapeutic agents that can be used for the prevention or treatment of cisplatin-induced renal disorders.
Keywords: cisplatin, Curcuma longa, gene expression profile, HEK 293 cells
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
High doses of cisplatin more effectively suppress cancer, but high dose therapy is limited by nephrotoxic side effects, Nephrotoxicity is of critical concern during the early stages of drug development when selecting new drug candidates. Curcuma longa (CL) has been widely used for its anti-inflammatory, antioxidant and antitumour activities and has been taken orally to treat dyspepsia, flatulence and liver and urinary tract disease.
THIS STUDY ADDS
New information indicated that the mechanism responsible for the effects of CL on HEK 293 cells was closely associated with regulation of the NFκB pathway.
This study confirmed the association of CL with the NFκB pathway.
CL may be an effective therapeutic approach to the alleviation of the progression of renal disease through cell anti-apoptosis and proliferation that occurs via inhibition of the inflammatory cytokines and the NFκB signaling pathway.
Introduction
Cisplatin (cis-diammine II dichloroplatimum) is a DNA-damaging molecule that is widely used as a chemotherapeutic agent [1]. Cisplatin is frequently used to treat a variety of tumours, including those of the lung, bladder, head, neck, testis, ovary and breast [2]. Although high doses of cisplatin effectively suppress cancer [3–7], high dose therapy is limited by nephrotoxic side effects [8]. Specifically, cisplatin predominantly accumulates in the kidneys because they are the major route of excretion [9]. Nephrotoxicity is of critical concern during the early stages of drug development when selecting new drug candidates [10]. Due to its unique metabolism, the kidney is an important target of the toxic effects of xenobiotics, drugs and oxidative stress. However, supporters of herbal medicine claim that herbs can be used to both treat and prevent diseases [11]. Accordingly, products produced by medicinal plants were screened for use as new therapeutic agents for the treatment of renal diseases by evaluating the recovery activities of 239 spray-dried extracts of herbal medicines on cisplatin-induced cytotoxicity in HEK 293 cells [12]. Among these extracts, Curcuma longa (CL) showed the highest recovery activity. Therefore, this compound was evaluated in the present study. The rhizome of turmeric, CL, has been widely used for its anti-inflammatory, antioxidant and antitumour activities and has been taken orally to treat dyspepsia, flatulence and liver and urinary tract disease [13–16].
The goal of this study was to determine the mechanisms by which CL extract leads to recovery from cisplatin-induced cytotoxicity in HEK 293 cells. The results of these tests and the possible mechanisms by which they occurred are discussed herein.
Methods
Preparation of CL and cisplatin
CL was purchased from Sun Ten Pharmaceutical (Taipei, Taiwan), powdered to 0.1 g and then extracted by stirring in 10 ml of distilled water (DW) overnight at room temperature using a stirrer. Next, the sample was centrifuged for 10 min at 3000 rev min−1, after which the supernatant was removed and sterilized by passing it through a 0.22 µm syringe filter. The filtered supernatant was used for the subsequent experiments. A voucher specimen was deposited in the Herbarium of the College of Oriental Medicine, KyungHee University, Korea. Dr Minkyu Shin, the director of the herbarium, identified the plants and assigned the herbarium sheet number (No. PMP0081). Cisplatin was dissolved in 0.1% DMSO as a control.
Quantitative chromatographic analysis
HPLC analysis was conducted using a Waters system (Waters Co., Milford, MA, USA) with a 717+ autosampler, 2996 photodiode array detector (PDA) 2487 dual λ absorbance detector, and 1525 binary HPLC pump. In addition, a Waters Millennium 32 System (Waters Co., Milford, MA, USA) was used for data acquisition and integration. HPLC grade and other reagents (J.T. Baker Co., Ltd, Phillipsburg, NJ, USA) were used for HPLC analysis. All solvents were filtered and degassed before use. The CL was accurately weighed to 1.0 g and then dissolved in 20 ml of methanol, after which the sample was subjected to ultrasonic treatment for 60 min and subsequent centrifugation at 3000 rev min−1 for 10 min. The supernatant of the sample was then filtered through a 0.45 µm syringe filter (PVDF, Whatman). Quantitation of CL was conducted by comparison with curcumin purchased from Sigma as the standard material. The separation was conducted on a reverse phase system (Xterra RP C18 4.6 × 250 mm, 5 µm, ODS, Waters, USA). A gradient mobile phase consisting of A (0.1% formic acid in acetonitrile) and B (water) was used to run the separation. The elution programme was set as follows: from 100% B to 100% A in 20 min in a gradient system at a flow rate of 1.0 ml min−1 using an injection volume of 10 µl. The detector was a Photodiode Array with detection wavelengths at 420 nm. The calibration curve of curcumin was linear in the range of 2 µg–0.125 µg.
Cell culture
HEK 293 cells, which represent a primary human embryonal kidney cell line transfected with adenovirus 5 DNA, were obtained from the Korean Cell Line Bank (KCLB™). The HEK 293 cells were grown and maintained at 100% humidity under 5% CO2 at 37°C in Dulbecco's modified Eagles' medium (DMEM) supplemented with 10% fetal bovine serum (FBS), streptomycin and penicillin (Invitrogen Life Technologies, Rockville, USA). The HEK 293 cells were then seeded into tissue culture flasks (T-75 cm2) that contained hormonally defined DMEM media at a density of 1 × 107 ml−1 as described previously [12]. The medium was changed every 3 days until the cells became 80–90% confluent, at which point they were used for the experiments.
RNA preparation
HEK 293 cells were initially cultured in a 100 mm dish (2 × 106 ml−1) for 24 h at 37°C, after which they were treated with cisplatin for 1 h. Next, 0 or 1 µg ml−1 CL was added and the cells were incubated at 37°C for 23 h. The total RNA was then isolated from the HEK 293 cells using an Rneasy® mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions and subsequently quantified using a 1000 NanoDrop spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE USA).
Oligonucleotide chip microarray
Microarray analysis was conducted using single round RNA amplification protocols according to the Affymetrix specifications (Affymetrix GeneChip Expression Analysis Technical Manual). Briefly, 3 µg of total RNA was used to synthesize first-strand cDNA using oligonucleotide probes with oligo-dT plus T7 promoter as a primer (Proligo LLC, Boulder, CO) and the Superscript Choice System (Invitrogen Life Technologies, Rockville, USA). Double-stranded cDNA was then synthesized and purified by phenol-chloroform extraction, after which biotinylated antisense complimentary RNA (cRNA) was generated through in vitro transcription using a BioArray RNA High-Yield Transcript Labeling kit (ENZO Life Sciences Inc., Farmingdale, NY, USA). The biotinylated cRNA was then fragmented and 10 µg of total fragmented cRNA was hybridized to the Affymetrix Human Genome U133 Plus 2.0 GeneChip array (P/N900470, Affymetrix Inc., Santa Clara, CA, USA). The array was then washed, stained and scanned according to the manufacturer's instructions. The fluorescence was detected using the Genechip System Confocal Scanner (Hewlett-Packard, Santa Clara, CA, USA), and analysis of each GeneChip was conducted using the GeneChip 3.1 software produced by Affymetrix with the standard default settings. Global scaling was used to compare chips, with all probe sets being scaled to a user-defined target intensity of 150.
Data analysis
The MAS5 algorithm was used to evaluate the expression signals generated by the GeneChip. Global scaling normalization was then performed and the normalized data were log-transformed using base2. Next, fold change was applied to select the differentially expressed genes (DEGs) using a fold change threshold of 2.0 and P < 0.05 to indicate significance. Each probe set used in the GeneChip produces a detection call, with P (present call) indicating good quality, M (marginal call) indicating intermediate quality and A (absent call) indicating relatively low reliability. Therefore, probe sets that resulted in A calls in the compared groups were removed to filter false positives. The two-fold DEGs were clustered using the GenPlex™ v3.0 software (ISTECH Inc., Korea) using hierarchical clustering with Pearson's correlation as a similarity measure and complete linkage as the linkage method. Gene ontology significance analysis was conducted to investigate the functional relationships among the two-fold DEGs using high-throughput GoMiner [17]. The candidate gene list was selected by fold changes larger than two-fold (log2).
Real-time PCR (RTP) analysis
Microarray verification was performed by RTP analysis of selected genes using SYBR Green I Master Mix (Applied Biosystems, Foster City, CA, USA) and primers (Genotech Inc., Korea). cDNA was synthesized using 2 µg of RNA in a reverse transcription reaction. RTP quantitative mRNA analyses were performed with an Applied Biosystems 7300 Real time PCR System using the SYBR Green fluorescence quantification system (Applied Biosystems, Foster City, CA, USA) to quantify the amplicons. The PCR conditions were as follows: 40 cycles of 95°C for 15 s and 60°C for 1 min, after which a standard denaturation curve was produced. The sequences of the human primers were as follows: BCL2L11 (FW 5′-att tag cct ggt gcc tgt gt-3′; RW 5′-ggg agc ttg agg atg ctt ct-3′), CFLAR (FW 5′-gag ctg gga ttc aaa agc tg-3′; RW 5′-tcc agg tac tgg aga ccc ata-3′), CYP24A1 (FW 5′-tcc cat gcc ata att ttt ctg-3′; RW 5′- tgc atg cat ttc tgt gca tt-3′), IL1A (FW 5′-gcc tca aga tga agg caa ag-3′; RW 5′-gat gcc tgg tca cac tca ga-3′), PDCD4 (FW 5′-tgc ctt tca gat atg ccg ta-3′; RW 5′-tgg ata gtt ccc tac caa tga a-3′), RHOC (FW 5′-atc cct tct tcc agc cac tt-3′; RW 5′-tcg ggg aga cag ctt atg tt-3′), hCG (FW 5′-gac tag gtt tcc tgc ctc c.-3′; RW 5′-ttc tgg gat atc tgg gga aa-3′), TRADD (FW 5′-gct ttg gag atc agc ctc ac-3′; RW 5′-gta tct gca gca ccc agg at-3′), TRIM13 (FW 5′-gct tga tac act gat gct cca-3′; RW 5′-tca act cct gtg tga tgt gtg a-3′) and GAPDH (FW 5′-ttc acc acc atg gag aag gc-3′; RW 5′-ggc atg gac tgt ggt cat ga-3′). The PCR conditions for each target were optimized according to the primer concentration, the absence of primer dimer formation and the efficiency of amplification of both the target genes and the housekeeping gene control. PCR reactions were conducted in a PCR master mix with a total volume of 20 µl that contained 10 µl of 2× SYBR Green, 5 µm each of sense and antisense primer and 2 µl of 1:2 diluted cDNA adjusted to 20 µl with DEPC-treated H2O. To normalize the cDNA content of the samples, we used the comparative threshold (CT) cycle method, which consists of normalization of the number of target gene copies vs. the endogenous reference gene, GAPDH. The CT is defined as the fractional cycle number at which the fluorescence generated by cleavage of the probe passes a fixed threshold baseline when amplification of the PCR product is first detected.
Immunofluorescence confocal microscopy
HEK 293 cells were seeded on four-well Lab-Tek™ II Chambered Coverglasses (Nalge Nunc International, Naperville, Illinois) in culture medium. Twenty-four hours after seeding, 0 or 1 µg ml−1 CL was added and the cells were then incubated at 37°C for 24 h. Next, 14 µm cisplatin was added for 10 min or 30 min. Subsequently, the cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min, followed by washing in PBS with 0.1% Tween-20 (PBST). After fixation, the cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min and then blocked with 1% bovine serum albumin in PBS for 1 h at room temperature, after which they were incubated with NFκB p65 antibodies (Santa Cruz Biotechnology, CA, USA) at 4°C overnight. After washing with PBST, the cells were incubated with FITC-conjugated secondary antibodies (Sigma) diluted 5000-fold in blocking buffer for 1 h at room temperature. The cells were then washed with PBST, mounted in Vectashield mounting medium with DAPI (Vector Laboratories Inc., Burlingame, CA, USA) and analyzed by confocal microscopy (Zeiss LSM Pascal 5, Heidelberg, Germany). Settings of the Start Zeiss Software were kept constant within each experiment to allow comparison of the overall fluorescence intensities of the samples.
NFκB p65 Western blot analysis
HEK 293 cells were seeded onto a 100 mm dish in culture medium. Twenty-four hours after seeding, 0 or 1 µg ml−1 CL was added and the cells were incubated at 37°C for 24 h. Next, 14 µm cisplatin was added for 10 or 30 min. Subsequently, the cells were washed with PBS and lyzed in 400 µl of lysis buffer (Hepes (10 mm, pH 7.5), MgCl2 (2 mm), EDTA (1 mm), EGTA (1 mm), KCl (10 mm), DTT (1 mm), NaF (10 mm), Na3VO4 (0.1 mm) and 1× pharminogen–protease-cocktail), after which they were allowed to stand on ice for 15 min. Next, 25 µl of 10% NP40 was added and the samples were incubated on ice for an additional 5 min, followed by vigorous vortexing for 10 s and centrifugation at 13 000 rev min−1 for 30 s. The pelleted nuclei were then resuspended in 50 µl of nuclear extraction buffer (Hepes (25 mm, pH 7.5), NaCl (500 mm), DTT (1 mm), NaF (10 mm), glycerol (10%), NP40 (0.2%) and MgCl2 (5 mm)). After three rounds of sonication (30 s), the lysates were centrifuged at 13 000 rev min−1 for 5 min. The supernatants, which contained the nuclear proteins, were then collected and stored at −80°C until use. Next, 30 µg of protein were separated on tris-glycine by 4–12% (KOMA Biotech, Korea) gel electrophoresis and then electroblotted onto a nitrocellulose membrane. The membranes were then blocked with 5% (w/v) skim milk powder in 0.1% (v/v) Tween-20 PBS for 1 h at room temperature, after which they were incubated with anti-NFκB p65 (1:1000) or β-actin (1:1000) antibody overnight at 4°C. After washing three times in PBST, the blot was incubated with secondary antibodies (1:5000) for 1 h at room temperature, followed by visualization using the ECL chemiluminescence detection system (Amersham Biosciences, Luminogen™ TMA-6, Buckinghamshire, UK).
Statistical analysis
Statistical analysis of the data was conducted using the Prism 4.02 software (GraphicPad Software Inc., CA, USA). Data were analyzed by one-way analysis of variance (anova) and Tukey's multiple comparison test. Results with a P value <0.05 were considered to be statistically significant.
Results
Quantitative analysis of Curcuma longa
To evaluate the quality of the Curcuma longa, curcumin was used as a standard material. The relationship between the concentration and the peak area was measured by the minimum square method (r2 value). The standard calibration curve of curcumin was described by y = 75481x− 2576.8 (r2 = 0.9994). The average content of curcumin in the CL was determined to be 2.174 ± 0.116 µg g−1 (n = 3) using the above formula (Figure 1).
Figure 1.
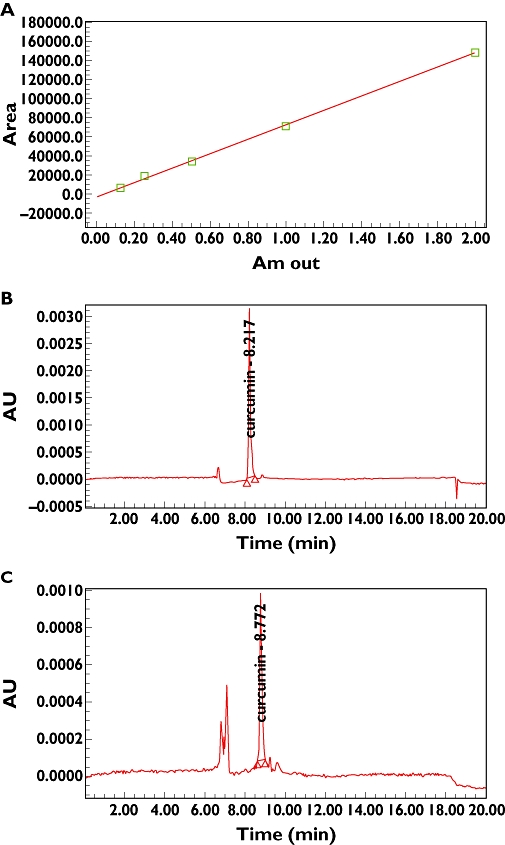
Quantitative analysis of herbal medicines. (A) Calibration curve of curcumin (r2 = 0.9994), (B) HPLC chromatogram of standard curcumin at 420 nm, (C) HPLC chromatogram of CL used to quantify curcumin (RT = about 8 min)
Gene expression profiles in HEK 293 cells
To determine the proper concentration of CL, a dose–response curve was generated. The results revealed that 1 µg ml−1 CL produced the maximal recovery effect in cisplatin-stimulated HEK293 cells (Figure 2). Although there is some debate regarding the origin of the HEK293 cells, they were selected for this study because they are derived from immortalized primary kidney cells and not from tumours.
Figure 2.
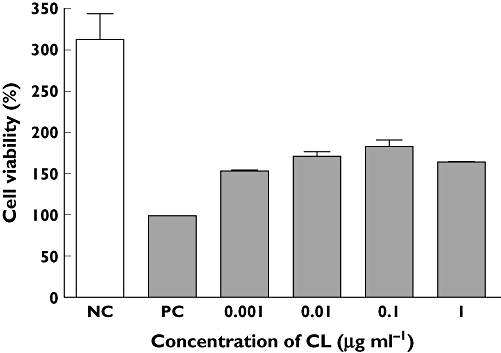
Effective dose of CL on cell damaged HEK293 cells. HEK293 cells were cultured with 14 µm cisplatin for 1 h. Next, HEK 293 cells were treated with various concentrations of CL for 24 h. The values of cell viability were normalized in proportion to the value of the control. Each point represents the mean ± SEM
Genes that showed more than a two-fold difference in expression in the experimental group (CL plus cisplatin treated) when compared with the control groups (NC non-treated control or PC cisplatin treated control) were initially selected for analysis. Overall, 384 DEGs were detected in the experimental group using approximately 45 100 oligonucleotide probes. These 384 DEGs were grouped into eight different expression clusters using a hierarchical clustering algorithm (Figure 3). Among these clusters, the genes in clusters 2, 7 and 8 were further analyzed because the expression of genes within these clusters was opposite to the expression pattern induced by cisplatin treatment. In addition to these genes, DNA repair, cell proliferation, apoptosis and immune and inflammatory response-related genes were selected based on differential expression (Table 1). Specifically, we detected the down-regulation of apoptosis-related genes including BCL2L11, CFLAR, IL1A, PDCD4, PDCD5, PHLDA1, TRADD, IQWD1 and CCR2, as well as down-regulation of NFκB signalling pathway-related genes such as CFLAR, RHOC, hCG, TRADD and TRIM13, and the up-regulation of cell proliferation-related genes such as EVI5 and TBC1D8 and the DNA repair-related gene RAD23B in CL-treated HEK 293 cells (Table 1, Figure 5). To assess the reliability of our microarray technique, we calculated the microarray reproducibility between triplicate RNA samples from three independent cell cultures. The correlation matrix plot from the control (NC or PC) cisplatin plus CL-treated HEK 293 cells provided an r2 value (Figure 4).
Figure 3.
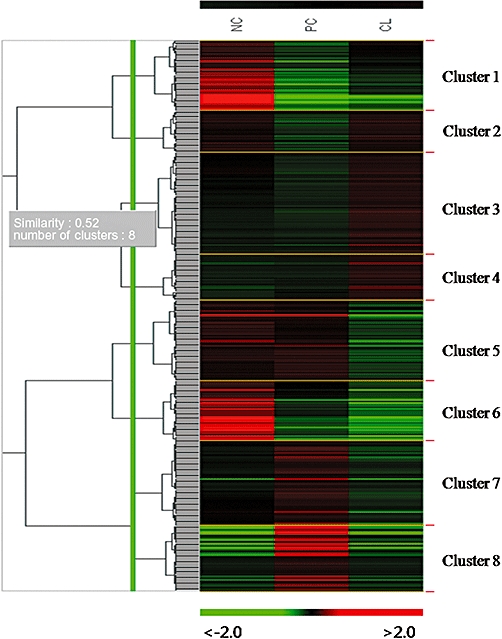
Overview of all gene expression profiles. Microarray data from control (non-treated HEK 293 cells) and experimental (cisplatin or cisplatin plus CL-treated HEK 293 cells) groups were combined and clustered. Each gene is represented by a single row of clustered boxes and each column represents the average expression levels of all genes in three independent hybridization samples. Experimental conditions (NC non-treated control, PC cisplatin-treated control, and CL cisplatin plus CL-treated) are shown at the top of the data matrix. A colour bar indicates higher levels of gene expression in the sample in red and lower levels in green when compared with those in the common reference RNA
Table 1.
Up- and down-regulation of genes based on comparison of gene expression between experimental (cisplatin plus Curcuma longa (CL)-treated) and control (non-treated (NC) or cisplatin-treated (PC)) HEK 293 cells
| Regulation profile and fold change | |||||
|---|---|---|---|---|---|
| Genes | Probe ID | Symbol | NC | PC | CL |
| Up-regulated genes | |||||
| Cell proliferation-related genes | |||||
| Ecotropic viral integration site 5 | 208297_s_at | EVI5 | 1.7 | 1 | 4.5 |
| TBC1 domain family, member 8 (with GRAM domain) | 204526_s_at | TBC1D8 | 7.2 | 1 | 2.7 |
| DNA repair-related genes | |||||
| RAD23 homolog B (S. cerevisiae) | 214422_at | RAD23B | 1.5 | 1 | 2.7 |
| Down-regulated genes | |||||
| Immune and inflammatory response-related genes | |||||
| Chemokine (C-C motif) receptor 2 | 206978_at | CCR2 | 0.3 | 1 | 0.3 |
| C-type lectin domain family 4, member C | 1552552_s_at | CLEC4C | 1.1 | 1 | 0.2 |
| Dual oxidase 1 | 1565795_at | DUOX1 | 0.6 | 1 | 0.3 |
| Glutamate receptor, ionotropic, N-methyl D-aspartate 2B | 210412_at | GRIN2B | 0.9 | 1 | 0.5 |
| Interferon induced with helicase C domain 1 | 1555464_at | IFIH1 | 0.9 | 1 | 0.1 |
| Ras homolog gene family, member C | 235742_at | RHOC | 0.6 | 1 | 0.3 |
| Ribosomal protein L17 | 214291_at | hCG_2004593 | 0.9 | 1 | 0.5 |
| Tripartite motif-containing 13 | 1569142_at | TRIM13 | 0.3 | 1 | 0.5 |
| Apoptosis-related genes | |||||
| BCL2-like 11 (apoptosis facilitator) | 225606_at | BCL2L11 | 0.8 | 1 | 0.5 |
| CASP8 and FADD-like apoptosis regulator | 214618_at | CFLAR | 0.7 | 1 | 0.3 |
| Interleukin 1, alpha | 210118_s_at | IL1A | 0.3 | 1 | 0.4 |
| Programmed cell death 4 (neoplastic transformation inhibitor) | 1557166_at | PDCD4 | 1.5 | 1 | 0.3 |
| Programmed cell death 5 | 227751_at | PDCD5 | 1.2 | 1 | 0.2 |
| Pleckstrin homology-like domain, family A, member 1 | 217999_s_at | PHLDA1 | 0.9 | 1 | 0.3 |
| TNFRSF1A-associated via death domain | 1729_at | TRADD | 2.3 | 1 | 0.5 |
| IQ motif and WD repeats 1 | 224372_at | IQWD1 | 0.5 | 1 | 0.4 |
| Other | |||||
| Cytochrome P450, family 24, subfamily A, polypeptide 1 | 206504_at | CYP24A1 | 1.7 | 1 | 0.2 |
Figure 5.
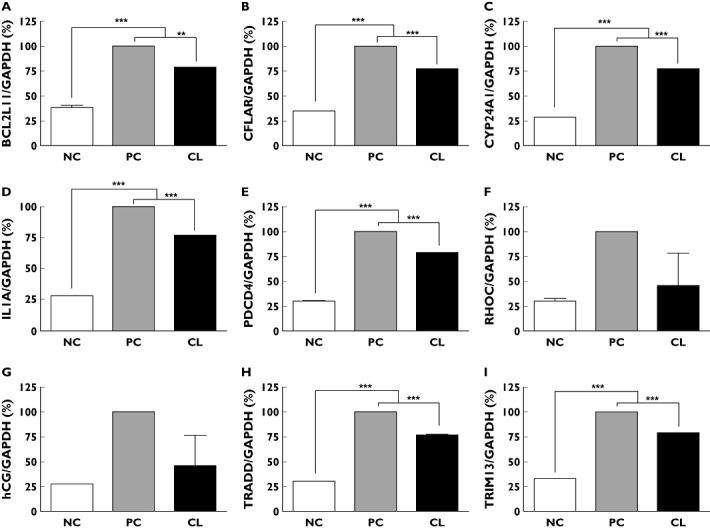
Effects of CL extract on mRNA concentrations as determined by real-time RT-PCR. (A) BCL2L11, (B) CFLAR, (C) CYP24A1, (D) IL1A, (E) PDCD4, (F) RHOC, (G) hCG, (H) TRADD, and (I) TRIM13. NC non-treated control, PC cisplatin-treated control, CL cisplatin plus CL-treated. Data are presented as the mean ± SEM (n = 3). **P < 0.01 and ***P < 0.001 when compared with control
Figure 4.
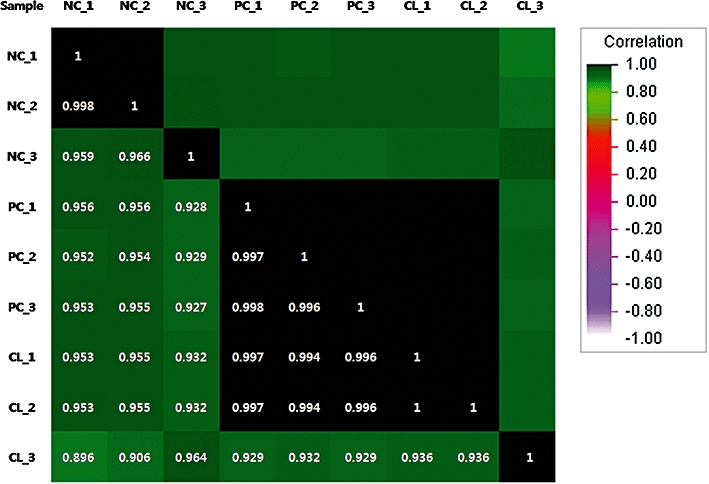
Correlation matrix plot analysis for calculation of the reproducibility between triplicates. The r2 value is based on the intensity of signals from all probe sets on the Affymetrix Human Genome U133 Plus 2.0 GeneChip array. Based on the r2 value, the microarray hybridization patterns were highly consistent among samples. NC non-treated control, PC cisplatin-treated control, CL cisplatin plus CL-treated
Validation of selected genes via real-time PCR
We normalized the relative expression levels of each gene by dividing the expression levels of GAPDH obtained by real-time PCR analysis because GAPDH was the most stably expressed housekeeping gene among the housekeeping genes evaluated in the microarray sets. The mean intensities of GAPDH in NC, PC, and CL were 20 035 ± 1208, 21 602 ± 629 and 24 398 ± 408, respectively. The SYBR Green assay was used to confirm changes in the expression of nine selected genes identified by microarray analysis in the CL-treated HEK 293 cells, BCL2L11, CFLAR, CYP24A1, IL1A, PDCD4, RHOC, hCG, TRADD and TRIM13. The real-time PCR assay yielded results that were in agreement with the microarray results (Figure 5).
The effects of CL on cisplatin-induced NFκB p65 nuclear translocation and NFκB p65 protein expression
We analyzed the nuclear translocation of NFκB p65. Immunocytochemical analysis revealed that cisplatin treatment with CL resulted in decreased nuclear accumulation of NFκb p65 when compared with cisplatin treatment alone in HEK293 cells (Figure 6A–C). Similarly, Western blot analysis revealed that the nuclear content of NFκb p65 was decreased in the CL treated HEK293 cells (Figure 6D). Taken together, these results revealed that CL extract could prevent NFκb activation, possibly by blocking the nuclear translocation of NFκb p65.
Figure 6.
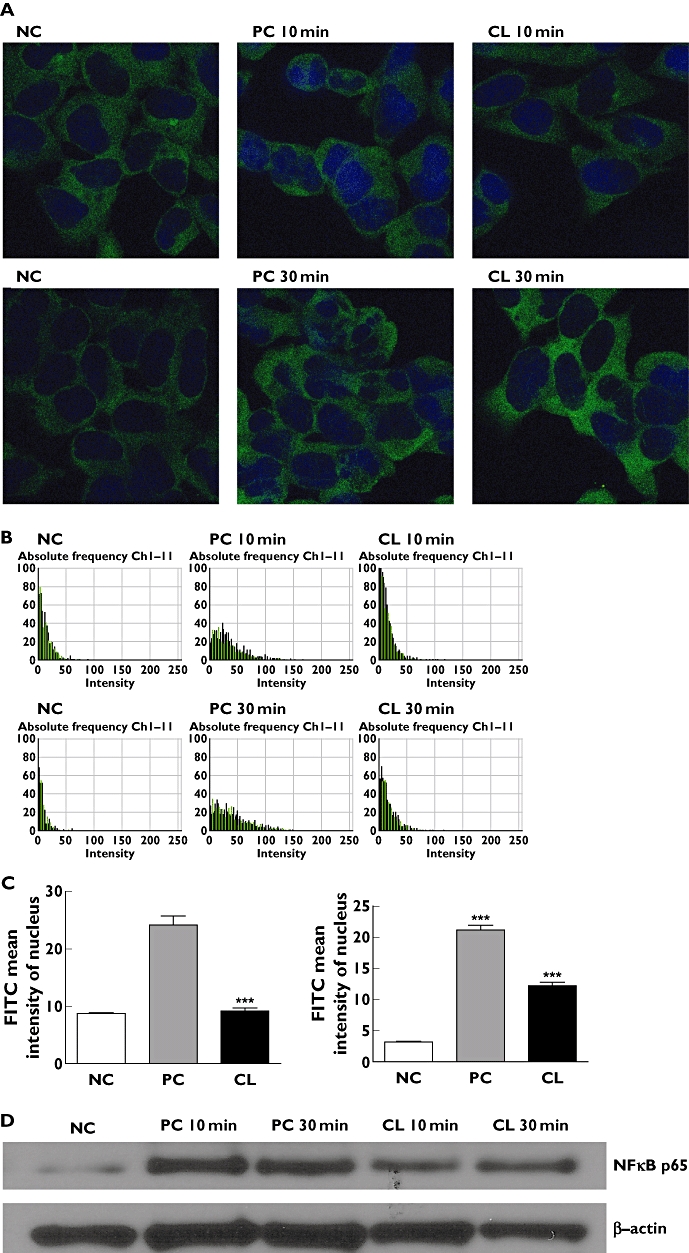
Effects of CL extract on NFκB p65 nuclear translocation and protein expression. (A) confocal microscopy images of fluorescence immunocytochemical analysis of NFκB p65 in HEK 293 cells (×1000). (B) Intensity images of fluorescence immunocytochemical analysis of NFκB p65 in the nucleus. (C) FITC mean intensity of NFκB p65 in the nucleus. (D) Western blot analysis of NFκB p65 and β-actin in nuclear extracts of HEK 293 cells. NC non-treated control, PC cisplatin-treated control, CL cisplatin plus CL-treated. ***P < 0.001 compared with control
Discussion
Previously, we demonstrated that cisplatin induced cytotoxicity in HEK 293 cells [12]. In addition, the recovery activities of 239 herbal medicines on cisplatin-induced cytotoxicity in HEK 293 cells were evaluated in an effort to identify new therapeutic agents for the treatment of renal disease [12]. CL was found to exert strong recovery effects (47%) on cisplatin-induced cytotoxicity. To investigate the mechanism behind the action of CL, the gene expression profiles of HEK 293 cells treated with CL were evaluated in this study. The results revealed specific and significant alterations in the expression profiles of CL-treated HEK 293 cells (Table 1).
Cisplatin acts as a DNA alkylator and cross-linker, forming cross-links between guanine bases. It is presumed that these cisplatin-DNA adducts initiate apoptosis in treated cells [18]. Recently, several studies have shown that cisplatin causes nephrotoxicity through multiple mechanisms, including hypoxia, the generation of free radicals, inflammation and apoptosis [19]. Specifically, apoptosis is an important mode of cell death in cisplatin nephrotoxicity, and many studies have demonstrated renal tubular cell apoptosis following cisplatin treatment [20]. A comet assay was used to detect genotoxic-related apoptosis of cisplatin in the cell culture [21], and the results revealed that cisplatin induced an increase in the median tail length. Previous studies revealed that 1 µg ml−1 CL induced a significant decrease in the median tail length in a comet assay [12]. In this experiment, we detected the down-regulation of apoptosis-related genes and the up-regulation of cell proliferation-related genes in CL-treated HEK 293 cells (Table 1, Figure 5A,B,D,E,H). Cisplatin has also been found to induce the expression of inflammatory cytokines and chemokines in cisplatin-induced renal injury [22–25]. These cytokines and chemokines contribute to the development and progression of renal injury with renal leukocyte infiltration. Specifically, CCR2 acts as a potent chemoattractant for monocytes and macrophages to promote migration from the peripheral circulation to sites of inflammation [26–28]. This gene encodes two isoforms of a receptor for monocyte chemoattractant protein-1 (MCP-1). The MCP-1 gene is mediated by transcription factor NFκB in the kidney [29, 30]. Further activation of the NFκB signalling pathway leads to renal dysfunction [31]. NFκB, which normally exists in an inactive cytoplasmic complex, is a heterodimer that consists of the p50 and p65 subunits complexed with the inhibitory subunit IκB. NFκB is activated in response to primary (bacteria, viruses, UV) or secondary (inflammatory cytokines) pathogenic stimuli [32, 33] via a mechanism that involves the degradation of IκB, which frees NFκB to translocate to the nucleus. Once in the nucleus, NFκB binds to the DNA at a specific κB site, where it regulates the expression of a large number of genes associated with immune and inflammatory responses [34–36]. In this experiment, we detected down-regulation of CCR2, as well as down-regulation of NFκB signaling pathway-related genes in CL-treated HEK 293 cells (Table 1, Figure 5B,F–I). In addition, NFκB p65 nuclear translocation and NFκB p65 protein expression were inhibited in CL-treated HEK 293 cells (Figure 6). It should be noted that there have been several reports showing that the major component of CL, curcumin, down-regulates NFκB and has anti-cancer effects [37, 38]. Furthermore, dual oxidase 1 (Duox1) mRNA expression is induced by the Th2-specific cytokines, interleukin-4 and interleukin-13 [39], and in this experiment Duox1 was found to be down-regulated in CL-treated HEK 293 cells (Table 1). Curcumin is a major constituent of CL that exerts a variety of biological effects including anti-cancer and anti-inflammatory effects [34–38]. Considering that the achievable concentration of curcumin in humans was around 1 µm, which was far higher than the maximum effective dosage of curcumin observed in this study, the protective effects of CL on cisplatin-induced nephrotoxicity could be achieved at the clinical level via proper product formulation consisting of CL [40].
In summary, the results of this study suggest that cisplatin induced the expression of inflammatory cytokines and apoptosis-related genes in HEK 293 cells, and that these genes were significantly inhibited by treatment with CL. In addition, CL down-regulates the genes related to the NFκB signalling pathway in cisplatin-stimulated HEK 293 cells. Taken together, these findings indicate that the use of CL might be an effective therapeutic approach to alleviating renal dysfunction through cell anti-apoptosis and proliferation that occurs via inhibition of inflammatory cytokines and the NFκB signalling pathway.
Competing interests
There are no competing interests to declare.
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant, funded by the Korea government (MEST) (No. R13-2007-019-00000-0).
REFERENCES
- 1.Brabec V, Kasparkova J. Molecular aspects of resistance to antitumor platinum drugs. Drug Resist Updat. 2002;5:147–61. doi: 10.1016/s1368-7646(02)00047-x. [DOI] [PubMed] [Google Scholar]
- 2.Lebwohl D, Canetta R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur J Cancer. 1998;34:1522–34. doi: 10.1016/s0959-8049(98)00224-x. [DOI] [PubMed] [Google Scholar]
- 3.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 4.Pratibha R, Sameer R, Rataboli PV, Bhiwgade DA, Dhume CY. Enzymatic studies of cisplatin induced oxidative stress in hepatic tissue of rats. Eur J Pharmacol. 2006;532:290–3. doi: 10.1016/j.ejphar.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Iraz M, Ozerol E, Gulec M, Tasdemir S, Idiz N, Fadillioglu E, Naziroglu M, Akyol O. Protective effect of caffeic acid phenethyl ester (CAPE) administration on cisplatin-induced oxidative damage to liver in rat. Cell Biochem Funct. 2006;24:357–61. doi: 10.1002/cbf.1232. [DOI] [PubMed] [Google Scholar]
- 6.Martins NM, Santos NA, Curti C, Bianchi ML, Santos AC. Cisplatin induces mitochondrial oxidative stress with resultant energetic metabolism impairment, membrane rigidification and apoptosis in rat liver. J Appl Toxicol. 2007;28:337–44. doi: 10.1002/jat.1284. [DOI] [PubMed] [Google Scholar]
- 7.Hsu DZ, Chen KT, Lin TH, Li YH, Liu MY. Sesame oil attenuates cisplatin-induced hepatic and renal injuries by inhibiting nitric oxide-associated lipid peroxidation in mice. Shock. 2007;27:199–204. doi: 10.1097/01.shk.0000238063.54332.06. [DOI] [PubMed] [Google Scholar]
- 8.Wolfgang GH, Dominick MA, Walsh KM, Hoeschele JD, Pegg DG. Comparative nephrotoxicity of a novel platinum compound, cisplatin, and carboplatin in male Wistar rats. Fundam Appl Toxicol. 1994;22:73–9. doi: 10.1006/faat.1994.1010. [DOI] [PubMed] [Google Scholar]
- 9.Yao X, Panichpisal K, Kurtzman N, Nugent K. Cisplatin nephrotoxicity: a review. Am J Med Sci. 2007;334:115–24. doi: 10.1097/MAJ.0b013e31812dfe1e. [DOI] [PubMed] [Google Scholar]
- 10.Uehara T, Miyoshi T, Tsuchiya N, Masuno K, Okada M, Inoue S, Torii M, Yamate J, Maruyama T. Comparative analysis of gene expression between renal cortex and papilla in nedaplatin-induced nephrotoxicity in rats. Hum Exp Toxicol. 2007;26:767–80. doi: 10.1177/0960327107084069. [DOI] [PubMed] [Google Scholar]
- 11.Stickel F, Schuppan D. Herbal medicine in the treatment of liver diseases. Dig Liver Dis. 2007;39:293–304. doi: 10.1016/j.dld.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Sohn SH, Lee H, Nam JY, Kim SH, Jung HJ, Kim Y, Shin M, Hong M, Bae H. Screening of herbal medicines for the recovery of cisplatin-induced nephrotoxicity. Environ Toxicol Pharmacol. 2009;28:206–12. doi: 10.1016/j.etap.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Ringman JM, Frautschy SA, Cole GM, Masterman DL, Cummings JL. A potential role of the curry spice curcumin in Alzheimer's disease. Curr Alzheimer Res. 2005;2:131–6. doi: 10.2174/1567205053585882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber WM, Hunsaker LA, Abcouwer SF, Deck LM, Vander Jagt DL. Anti-oxidant activities of curcumin and related enones. Bioorg Med Chem. 2005;13:3811–20. doi: 10.1016/j.bmc.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 15.Cao H, Sasaki Y, Fushimi H, Komatsu K. Molecular analysis of medicinally-used Chinese and Japanese curcuma based on 18S rRNA gene and trnK gene sequences. Biol Pharm Bull. 2001;24:1389–94. doi: 10.1248/bpb.24.1389. [DOI] [PubMed] [Google Scholar]
- 16.Maheshwari RK, Singh AK, Gaddipati J, Srimal RC. Multiple biological activities of curcumin: a short review. Life Sci. 2006;78:2081–7. doi: 10.1016/j.lfs.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiss RB, Christian MC. New cisplatin analogues in development. A review. Drugs. 1993;46:360–77. doi: 10.2165/00003495-199346030-00003. [DOI] [PubMed] [Google Scholar]
- 19.Kang DG, Lee AS, Mun YJ, Woo WH, Kim YC, Sohn EJ, Moon MK, Lee HS. Butein ameliorates renal concentrating ability in cisplatin-induced acute renal failure in rats. Biol Pharm Bull. 2004;27:366–70. doi: 10.1248/bpb.27.366. [DOI] [PubMed] [Google Scholar]
- 20.Tsuruya K, Ninomiya T, Tokumoto M, Hirakawa M, Masutani K, Taniguchi M, Fukuda K, Kanai H, Kishihara K, Hirakata H, Iida M. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int. 2003;63:72–82. doi: 10.1046/j.1523-1755.2003.00709.x. [DOI] [PubMed] [Google Scholar]
- 21.Follmann W, Birkner S. The use of cultured primary bovine colon epithelial cells as a screening model to detect genotoxic effects of heterocyclic aromatic amines in the comet assay. J Toxicol Environ Health A. 2008;71:947–53. doi: 10.1080/15287390801988962. [DOI] [PubMed] [Google Scholar]
- 22.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835–42. doi: 10.1172/JCI15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramesh G, Reeves WB. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol. 2003;285:F610–8. doi: 10.1152/ajprenal.00101.2003. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki H, Inoue T, Matsushita T, Kobayashi K, Horii I, Hirabayashi Y, Inoue T. In vitro gene expression analysis of hepatotoxic drugs in rat primary hepatocytes. J Appl Toxicol. 2008;28:227–36. doi: 10.1002/jat.1328. [DOI] [PubMed] [Google Scholar]
- 25.Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, Oh DJ, Lu L, Klein CL, Dinarello CA, Edelstein CL. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther. 2007;322:8–15. doi: 10.1124/jpet.107.119792. [DOI] [PubMed] [Google Scholar]
- 26.Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci USA. 1994;91:2752–6. doi: 10.1073/pnas.91.7.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J Exp Med. 1989;169:1485–90. doi: 10.1084/jem.169.4.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshimura T, Robinson EA, Tanaka S, Appella E, Kuratsu J, Leonard EJ. Purification and amino acid analysis of two human glioma-derived monocyte chemoattractants. J Exp Med. 1989;169:1449–59. doi: 10.1084/jem.169.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rovin BH, Dickerson JA, Tan LC, Hebert CA. Activation of nuclear factor-kappa B correlates with MCP-1 expression by human mesangial cells. Kidney Int. 1995;48:1263–71. doi: 10.1038/ki.1995.410. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz-Ortega M, Bustos C, Hernandez-Presa MA, Lorenzo O, Plaza JJ, Egido J. Angiotensin II participates in mononuclear cell recruitment in experimental immune complex nephritis through nuclear factor-kappa B activation and monocyte chemoattractant protein-1 synthesis. J Immunol. 1998;161:430–9. [PubMed] [Google Scholar]
- 31.Kuhad A, Chopra K. Attenuation of diabetic nephropathy by tocotrienol: involvement of NFkB signaling pathway. Life Sci. 2009;84:296–301. doi: 10.1016/j.lfs.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 32.Thanos D, Maniatis T. NF-kappa B: a lesson in family values. Cell. 1995;80:529–32. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 33.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–79. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 34.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 35.Ray A, Siegel MD, Prefontaine KE, Ray P. Anti-inflammation: direct physical association and functional antagonism between transcription factor NF-KB and the glucocorticoid receptor. Chest. 1995;107(3 Suppl):139S. doi: 10.1378/chest.107.3_supplement.139s. [DOI] [PubMed] [Google Scholar]
- 36.Baeuerle PA, Baichwal VR. NF-kappa B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv Immunol. 1997;65:111–37. [PubMed] [Google Scholar]
- 37.Everett PC, Meyers JA, Makkinje A, Rabbi M, Lerner A. Preclinical assessment of curcumin as a potential therapy for B-CLL. Am J Hematol. 2007;82:23–30. doi: 10.1002/ajh.20757. [DOI] [PubMed] [Google Scholar]
- 38.Jobin C, Bradham CA, Russo MP, Juma B, Narula AS, Brenner DA, Sartor RB. Curcumin blocks cytokine-mediated NF-B activation and proinflammatory gene expression by inhibiting inhibitory Factor I-B Kinase activity. J Immunol. 1999;163:3474–83. [PubMed] [Google Scholar]
- 39.Harper RW, Xu C, Eiserich JP, Chen Y, Kao CY, Thai P, Setiadi H, Wu R. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005;579:4911–7. doi: 10.1016/j.febslet.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, Ko JY, Lin JT, Lin BR, Ming-Shiang W, Yu HS, Jee SH, Chen GS, Chen TM, Chen CA, Lai MK, Pu YS, Pan MH, Wang YJ, Tsai CC, Hsieh CY. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21:2895–900. [PubMed] [Google Scholar]