

Introduction
Irrespective of whether a drug is in development or already available on the market, the current paradigm for paediatric dose selection does not guarantee safe and effective dosing recommendation for children. The normalization of the adult dose according to age, body weight or any other demographic covariate without prior evidence of how these factors contribute to differences in drug exposure may lead to poor and unsafe estimates of the paediatric dose. Nevertheless, the implications of such common practice remain unquestioned. No matter how easy and simple a dosing regimen may be for clinical investigators or prescribers, the continuous use of empirical approaches for dose selection cannot be justified by the current understanding of how developmental growth affects pharmacokinetics and exposure–response relationships [1]. The recent advances in quantitative methodology for the analysis of clinical pharmacology data offer researchers and prescribers the appropriate tools for establishing what dose is right for children.
Current paediatric prescription practice
In contrast to the use of off-label prescription entrenched in clinical practice, the introduction of the paediatric regulation by the European Union, together with the renewal of the Pediatric Rule by the Food and Drug Administration on the requirements for paediatric labelling, imposes special attention to dose selection in paediatric clinical trials. The main objective of these guidelines is to ensure that effective and safe doses are evaluated in children [2, 3]. However, the design and implementation of paediatric trials remain challenging and are often difficult to accomplish. Ethical, practical and even economic considerations have caused the evaluation of efficacy and safety of drugs in children to be based on empirical extrapolations from clinical trials in adults. Despite the many flaws of this approach and mounting evidence [4–7] from quantitative pharmacological methods, very few examples exist where exposure–response relationships obtained in children are used to define dosing regimens in the paediatric population. Thus far, empirical scaling from adults to children continues to be the mainstream method for dose selection in children, with adjustment for body weight as the most commonly used approach.
The rationale for dose adjustment in paediatric indications may be determined by differences in pharmacokinetics, pharmacodynamics, disease or a combination of these factors. Pharmacokinetics of drugs in children may differ from adults for several reasons: variability due to age, gender, body composition, functionality of liver and kidneys and maturation of enzymatic systems throughout the life span from neonates to adults are all potential sources of pharmacokinetic differences [5]. Assuming similar exposure–response relationships between adult and children, efficacy in children is warranted if the same exposure can be achieved in either population. To meet this requirement, physiological differences between adults and children must be taken into account when selecting the paediatric dose [6].
In spite of the aforementioned considerations, dose scaling in paediatric trials remains an open issue, from both a clinical perspective and a drug development standpoint. Given that children may not be subject to dose-finding studies similar to those carried out in the adult population, some initial estimation of the paediatric dose must be obtained via extrapolation approaches [7]. As a consequence, the dose selected for a considerable number of drugs disseminates into clinical practice, irrespective of consensus about the appropriate dosing recommendation. This phenomenon is illustrated by scientific publications showing different dosing requirements (e.g. pain management, paediatric oncology) and by differences in prescription practice in many hospitals (e.g. heart failure, pulmonary hypertension), which have their own protocols based on the empirical experience of its staff [8, 9]. Some exceptions exist, such as the British National Formulary for Children (http://bnfc.org) and the Dutch Children's Formulary (http://www.kinderformularium.nl), but these guidelines rely on dosing recommendations primarily from clinical experience and off-label use rather than on prospective studies or randomized clinical trials.
A similar scenario is observed in drug development, where empiricism also prevails. Usually, the adult dose is divided by a fixed (scaling) factor, under the assumption that the appropriate efficacy/safety profile can be assured. These empirical procedures are often referred to as ‘bridging’. It is evident that such an approach has some serious disadvantages, amongst which are the risk of toxicity due to lack of understanding of the ontogeny of metabolic pathways, as for example in neonates and toddlers, or poor efficacy due to suboptimal dosing.
It is clear that a shift in paradigm is required that focuses on the differences in (physiological) function between populations, rather than differences in size between adults and children.
The correlation between dose and demographic covariates is not linear
Probably, the most common method for dose adjustment in children in paediatric clinical practice is to normalize the adult dose by body weight (i.e. mg kg−1), assuming a linear relationship between weight and dose. This means that the dose doubles with a twofold increase in the weight of a child.
Another method for dose adjustment is based on age: the paediatric population is divided into subcategories (preterm newborns, term newborns, infants, toddlers, children and adolescents) and the dose is selected according to a child's age. This method does not take into account the changes due to developmental growth that occur within each age group. Even though the hepatic metabolic capacity of a 5-year-old child is completely different from that of a neonate, this approach fails in describing the maturation of the metabolism between 1 and 6 months of age. On the other hand, no differences in drug metabolism may exist between adolescents and adults. Furthermore, categorizing dosing regimens by age ranges creates an artificial discontinuity in the dose–exposure relationship across each age group, hardly substantiated by scientific evidence [10].
Scaling the dose from adults can also be performed by normalization based on body surface area (BSA), under the assumption that metabolic processes in humans are constant when expressed as a function of BSA. However, a few disadvantages limit the application of this method: the difficulty in calculating BSA (due to the complexity and inaccuracy of the formulae that can be used) and the tendency to overdose neonates and infants [11]. There is also little justification for BSA from a pharmacokinetic perspective: the change in pharmacokinetic parameters across the paediatric population does not change proportionally with BSA, because BSA is not a descriptor of metabolic function (e.g. scaling with BSA cannot predict the lack of enzymes at birth, leading to overdosing neonates [12]).
From the above, it becomes comprehensible that the assumption of a linear relationship between body size and drug exposure or response is not always justifiable and that size itself may not be a surrogate for developmental growth. Implicitly and most importantly, one must realize that the use of demographic variables also implies unidirectional increase of the dose with body size, which constrains the paediatric dose to be always smaller than in adults, irrespective of the relevance of physiological and disease factors.
Scaling for function, not for size
Currently, evidence suggests that a more reliable way to establish how dose relates to body weight is through the use of nonlinear relationships, such as, for example, allometric scaling (see Equation 1) [13].
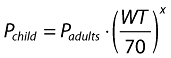 |
(1) |
where P is the parameter of interest, WT the bodyweight of the individual child and x the allometric exponent.
Different examples show that this approach yields the most accurate results in terms of exposure in children [5, 14, 15]. Nevertheless, the qualitative description of this relationship with a nonlinear function is only the tip of the iceberg: how such a relationship should be described mathematically is still subject to intense debate. Some authors defend the use of ‘pure’ allometry, fixing the exponent in the equation (e.g. 0.75 for clearance). They are de facto still scaling for size, since the relationship between parameter and weight is decided a priori [16–18]. Other authors prefer to estimate the exponent based on the available clinical data. In this case, body weight can be considered a surrogate for the physiological function (which is not always directly measurable) [19–21].
Besides allometric scaling, a more mechanistic approach is lacking for paediatric dosing recommendation that can counter the empiricism in current clinical practice. Such an approach must identify which physiological factors alter pharmacokinetics and how these (might) differ across the paediatric population(s), without relying on a priori assumptions about the correlation between pharmacokinetic parameters and demographic covariates. For these reasons, we strongly suggest using a physiologically based scaling approach, which we describe as scaling for function.
The concept of scaling for function relies on inferences from pharmacokinetic parameter distributions in children. This is equivalent to the rationale for assessing drug exposure in other special populations such as obese and hepatically impaired patients. In this way, dosing requirements are derived primarily from a model-based analysis of pharmacokinetic or pharmacokinetic–pharmacodynamic data [22, 23]. In fact, it is known that under steady-state conditions, total clearance (CL) determines systemic exposure. CL itself may be dependent on liver blood flow (LBF) and/or glomerular filtration rate (GFR). However, these processes may vary not only with developmental growth but also with different (patho)physiological conditions [24].
In addition to GFR and LBF, ontogeny (the development and maturation of metabolic pathways) is proven to have considerable effects on drug elimination. However, enzymatic maturation (i.e. metabolic capacity) is completely unrelated to body weight, and as such does not follow developmental growth. Each enzyme system has its own phenotype, showing different times of onset and maturation rates: some enzymes are present at birth (CYP3A7, UGT), whereas others are not (CYP2E1, CYP2D6, CYP3A4, CYP2C9), some mature to their maximum adult activity in just a few days (UGT2B7), whereas some require months to years to reach complete maturation (UGT1A6). Finally, some enzymes may be critical to the metabolism of certain drugs during early age, but their role may become less relevant with developmental growth in favour of other metabolic pathways (CYP3A7, CYP2C9) [25, 26]. Consequently, ontogeny cannot be described simply by body weight differences. Even if a nonlinear relationship is used to correlate exposure to weight, weight alone does not capture age-dependent nonlinearities in metabolic capacity.
Although a scientific rationale for dose recommendation in children is desirable and necessary, awareness is lacking with regard to the implications it may have on prescription practice. As stated previously, most of paediatric labels report doses normalized by body weight. Many renowned researchers and regulatory agencies still defend such normalization, as it eliminates the apparent need for dosing algorithms [27–29]. It makes prescription easy and simple, allegedly reducing the risk of prescription errors [30].
These views ignore, however, the nonlinearity in the relationship between exposure and body weight. This was the case, for example, in the accumulation of chloramphenicol, which causes the grey baby syndrome. Chloramphenicol is mainly metabolized by UDP-glucuronyl-transferase enzyme, but this system is immature in newborns and renal excretion of the unconjugated drug is limited. When chloramphenicol is linearly scaled according to body weight, the resulting exposure in newborns is fivefold higher than that reached in adults, causing the well-known adverse reactions. Dose adjustment according to a nonlinear correlation between dose and body weight (Figure 1) was enough to avoid the adverse events (i.e. neonates and infants up to 1 month are given half of the dose recommended to the other groups) [31–34].
Figure 1.
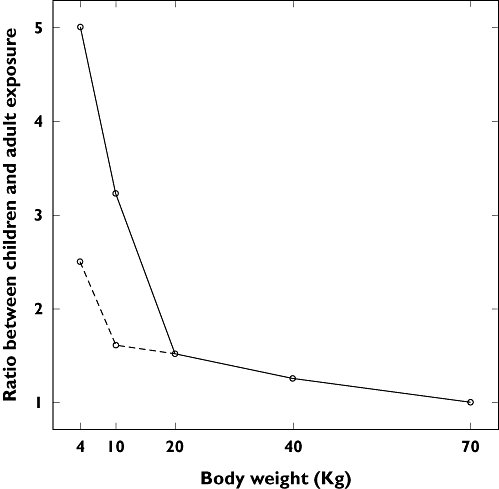
Ratio of exposures to chloramphenicol between children and adults. Solid line: original dose (50 mg kg−1 day−1). Dashed lined: dose adjustment to avoid grey baby syndrome (25 mg kg−1 day−1 for babies up to 1 month). Empirical scaling of the paediatric dose based on body weight has led to overexposure to chloramphenicol
On the other hand, linear scaling based on body weight may lead to subtherapeutic exposures in children. This is illustrated by the use of carbamazepine in the treatment of epileptic seizures. Carbamazepine clearance is largely dependent on CYP3A4, which is known to show increased activity in children compared with adults. These differences result in a higher weight-adjusted dose of carbamazepine to achieve comparable therapeutic plasma levels [35, 36]. Other examples of common medications for which dose scaling by body weight may be inappropriate are phenytoin [26], propofol [37] and aminoglycosides [38]. One must also consider the presence of comorbidities, which can affect pharmacokinetics and pharmacodynamics. These interactions are usually uncorrelated with and independent of demographic covariates. The use of higher doses of tobramycin in the presence of cystic fibrosis is one of the best examples of the influence of comorbidities on pharmacokinetics [39]. The implications of nonlinearity are further exemplified by busulfan, enfuvirtide, oseltamivir and nelfinavir, for which dosing algorithms have been introduced. Dosing requirements for these drugs are presented in their label as tables, categorized by weight and/or other characteristics [40–43].
It is also important to highlight that some drugs may not require scaling at all and children should receive the dose recommended for adults (e.g. vaccines, antidotes) and that there are cases in which the recommended dose is similar (e.g. telithromycin, desloratadine, olopatadine) or even higher (e.g. digoxin) than the dosing regimen in adults [44–48]. A summary of the paediatric dosing recommendation for these drugs is presented in Table 1.
Table 1.
Examples of drugs commonly used in paediatric medicine for which the paediatric dose is not linearly correlated with body weight
| Drug | Therapeutic indication | Adult dose | Paediatric dose |
|---|---|---|---|
| Chloramphenicol | Bacterial infection | 50 mg kg−1 day−1 | 50 mg kg−1 day−1 |
| neonates: 25 mg kg−1 day−1 | |||
| Carbamazepine | Epilepsy | 5–8 mg kg−1 every 12 h | >12 years: 5–8 mg kg−1 every 12 h |
| Children: 3–10 mg kg−1 every 8 h | |||
| Infants: 3–10 mg kg−1 every 8 h | |||
| Phenytoin | Epilepsy | 2 mg kg−1 every 12 h | Children: 2.3–2.6 mg−1 kg every 8 h |
| Infants: 2.3 mg kg−1 every 8 h | |||
| Neonates: 2.5–4.0 mg kg−1 every 12 h | |||
| Propofol | Anaesthesia | <55 years: 6–12 mg kg−1 h−1 | 2 months–16 years: 7.5–18 mg kg−1 h−1 |
| >55 years: 3–6 mg kg−1 h−1 | |||
| Busulfan | Cancer | 0.8 mg kg−1 every 6 h | ≤12 kg: 1.1 mg kg−1 every 6 h |
| >12 kg: 0.8 mg kg−1 every 6 h | |||
| Tobramycin | Bacterial infection | 3 mg kg−1 day−1 | Children: 6–7.5 mg kg−1 day−1 |
| <2 weeks: 4 mg kg−1 day−1 | |||
| With cystic fibrosis: 10 mg kg−1 day−1 | |||
| Enfuvirtide | HIV | 180 mg day−1 | 11–15.5 kg: 54 mg day−1 |
| 15.6–20 kg: 72 mg day−1 | |||
| 20.1–24.5 kg: 90 mg day−1 | |||
| 24.6–29 kg: 108 mg day−1 | |||
| 29.1–33.5 kg: 126 mg day−1 | |||
| 33.6–38 kg: 144 mg day−1 | |||
| 38.1–42.5 kg: 162 mg day−1 | |||
| Oseltamivir | Influenza | 150 mg day−1 | <15 kg: 60 mg day−1 |
| 15–23 kg: 90 mg day−1 | |||
| 23–40 kg: 120 mg day−1 | |||
| Nelfinavir | HIV | 2.5 g day−1 | 7.5–8.5 kg: 0.8 g day−1 |
| 8.5–10.5 kg: 1 g day−1 | |||
| 10.5–12 kg: 1.2 g day−1 | |||
| 12–14 kg: 1.4 g day−1 | |||
| 14–16 kg: 1.6 g day−1 | |||
| 16–18 kg: 1.8 g day−1 | |||
| 18–22 kg: 2.1 g day−1 | |||
| Digoxin | Heart failure | 1.4–4.0 µg kg−1 day−1 | Children: 3–8 µg kg−1 day−1 |
| Infants: 7.5–12 µg kg−1 day−1 | |||
| Neonates: 4–8 µg kg−1 day−1 |
References for each drug are provided in the text together with further details about the clinical implication of nonlinearity between drug exposure and descriptors of body size.
Based on the evidence provided above, it is understandable that the rationale for dose adjustment entails more than the assumption of linearity between body size and drug exposure. In fact, one should not generalize the requirements for paediatric dose recommendation without further understanding of the physiological phenomena associated with developmental growth.
It is evident that empiricism cannot continue as the mainstream method for clinical research in children. Dosing recommendation in children must be derived from an integrated (model-based) analysis of pharmacokinetic and pharmacodynamic data, accounting for the role of disease factors as well as developmental growth. Moreover, optimal dosing in children ought to include an assessment of the impact of potential differences in mode of administration, pharmaceutical formulation and delivery devices.
The future
Concerted efforts into two distinct areas of paediatric pharmacology research are required to ensure accurate selection of doses for children. The first involves revisiting dosing recommendations for those drugs that are already on the market but are used off-label in children. In this context, one should take advantage of the available pharmacokinetic, safety and efficacy data in adults and across the various age ranges in children. Used in conjunction with appropriate research tools, these data can confirm current clinical practice or provide the appropriate scaling factor to account for the differences associated with developmental growth. Critical to this evaluation are methodologies such as pharmacokinetic–pharmacodynamic modelling [49] and physiologically based pharmacokinetic approaches [50–52]. It is unfortunate that a communication gap still exists between paediatricians and clinical pharmacologists, who can apply the aforementioned methodologies to validate current prescription practice, in many cases without the need for additional prospective trials. Institutional and cultural differences about the individualization of dosing regimens, and more often ignorance of modelling and simulation concepts, prevent these efforts from becoming a paradigm in paediatric medicine.
The second area of attention refers to early drug development, for which there are no previous data in children. Not by chance, this subject has also become the focus of regulatory guidelines and policies [1–3]. Under the assumption of comparable exposure–response relationships between adults and children, a model-based bridging approach can be used that relies on the assessment of primary pharmacokinetic parameter distributions (i.e. clearance, volume of distribution). Simulation scenarios can then be derived to explore the implications of nonlinearity between exposure and demographic covariates [53]. Such scenarios can also incorporate differences in pharmacodynamics and in the exposure–response relationship, if applicable. Among other advantages, the use of modelling and simulation tools provides an algorithm for dose selection and prospective evaluation of safety and efficacy in a paediatric clinical trial. In conjunction with sparse blood sampling and adaptive trial designs, this approach ensures that accurate dosing recommendations are provided in the label at the time of launch.
Paediatric prescribers, including primary and secondary care physicians, are living in the 21st century, but for many diseases paediatric drug prescription still dwells in the empiricism of foregone times. It is time to change it. Despite the opportunities offered by the existing regulations and better quantitative methods in clinical pharmacology, the silent assent of current practices and beliefs in paediatric research seems to undermine the achievement of an unmet medical need, i.e. allowing children to be given the right dose.
Competing interests
There are no competing interests to declare.
REFERENCES
- 1.Manolis E, Pons G. Proposals for model based paediatric medicinal development within the current EU regulatory framework. Br J Clin Pharmacol. 2009;68:493–501. doi: 10.1111/j.1365-2125.2009.03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. European Parliament and Council of the European Union. Regulation (EC) No. 1901/2006 on medicinal products for paediatric use. 12 December 2006.
- 3.FDAAA. Title IV: Pediatric Research Equity Act of 2007 (PREA) and Title V: Best Pharmaceuticals for Children Act of 2007 (BPCA). September 27, 2007 (Public Law No. 110-85)
- 4.Anderson GD, Lynn AM. Optimizing pediatric dosing: a developmental pharmacologic approach. Pharmacotherapy. 2009;29:680–90. doi: 10.1592/phco.29.6.680. [DOI] [PubMed] [Google Scholar]
- 5.Mahmood I. Prediction of drug clearance in children: impact of allometric exponents, body weight, and age. Ther Drug Monit. 2007;29:271–8. doi: 10.1097/FTD.0b013e318042d3c4. [DOI] [PubMed] [Google Scholar]
- 6.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology – drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–67. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 7.Johnson TN. Modelling approaches to dose estimation in children. Br J Clin Pharmacol. 2005;59:663–9. doi: 10.1111/j.1365-2125.2005.02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cimpello LB, Khine H, Avner JR. Practice patterns of pediatric versus general emergency physicians for pain management of fractures in pediatric patients. Pediatr Emerg Care. 2004;20:228–32. doi: 10.1097/01.pec.0000121242.99242.e0. [DOI] [PubMed] [Google Scholar]
- 9.Conroy S, Choonara I, Impicciatore P, Mohn A, Arnell H, Rane A, Knoeppel C, Seyberth H, Pandolfini C, Raffaelli MP, Rocchi F, Bonati M, Jong G, de Hoog M, van den Anker J, on behalf of the European Network for Drug Investigation in Children Survey of unlicensed and off label drug use in paediatric wards in European countries. BMJ. 2000;320:79–82. doi: 10.1136/bmj.320.7227.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45:931–56. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 11.Meine Jansen CF. Treatment of symptomatic congenital cytomegalovirus infection with valganciclovir. J Perinat Med. 2005;33:364–6. doi: 10.1515/JPM.2005.065. [DOI] [PubMed] [Google Scholar]
- 12.Lack JA, Stuart-Taylor ME. Calculation of drug dosage and body surface area of children. Br J Anaesth. 1997;78:601–5. doi: 10.1093/bja/78.5.601. [DOI] [PubMed] [Google Scholar]
- 13.Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30:329–32. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 14.Mahmood I. Prediction of drug clearance in children from adults: a comparison of several allometric methods. Br J Clin Pharmacol. 2006;61:545–57. doi: 10.1111/j.1365-2125.2006.02622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–32. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 16.Habtemariam B, Sallas W, Sunkara G, Kern S, Jarugula V, Pillai G. Population pharmacokinetics of valsartan in pediatrics. Drug Metab Pharmacokinet. 2009;24:145–52. doi: 10.2133/dmpk.24.145. [DOI] [PubMed] [Google Scholar]
- 17.Allegaert K, de Hoon J, Verbesselt R, Naulaers G, Murat I. Maturational pharmacokinetics of single intravenous bolus of propofol. Paediatr Anaesth. 2007;17:1028–34. doi: 10.1111/j.1460-9592.2007.02285.x. [DOI] [PubMed] [Google Scholar]
- 18.Standing JF, Howard RF, Johnson A, Savage I, Wong IC. Population pharmacokinetics of oral diclofenac for acute pain in children. Br J Clin Pharmacol. 2008;66:846–53. doi: 10.1111/j.1365-2125.2008.03289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knibbe CA, Krekels EH, van den Anker JN, DeJongh J, Santen GW, van Dijk M, Simons SH, van Lingen RA, Jacqz-Aigrain EM, Danhof M, Tibboel D. Morphine glucuronidation in preterm neonates, infants and children younger than 3 years. Clin Pharmacokinet. 2009;48:371–85. doi: 10.2165/00003088-200948060-00003. [DOI] [PubMed] [Google Scholar]
- 20.El-Tahtawy A, Kokki H, Reidenberg BE. Population pharmacokinetics of oxycodone in children 6 months to 7 years old. J Clin Pharmacol. 2006;46:433–42. doi: 10.1177/0091270006286433. [DOI] [PubMed] [Google Scholar]
- 21.Chhun S, Jullien V, Rey E, Dulac O, Chiron C, Pons G. Population pharmacokinetics of levetiracetam and dosing recommendation in children with epilepsy. Epilepsia. 2009;50:1150–7. doi: 10.1111/j.1528-1167.2008.01974.x. [DOI] [PubMed] [Google Scholar]
- 22.Green B, Duffull SB. What is the best size descriptor to use for pharmacokinetic studies in the obese? Br J Clin Pharmacol. 2004;58:119–33. doi: 10.1111/j.1365-2125.2004.02157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Areberg J, Christophersen JS, Poulsen MN, Larsen F, Molz KH. The pharmacokinetics of escitalopram in patients with hepatic impairment. AAPS J. 2006;8:E14–9. doi: 10.1208/aapsj080102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peeters MY, Bras LJ, DeJongh J, Wesselink RM, Aarts LP, Danhof M, Knibbe CA. Disease severity is a major determinant for the pharmacodynamics of propofol in critically ill patients. Clin Pharmacol Ther. 2008;83:443–51. doi: 10.1038/sj.clpt.6100309. [DOI] [PubMed] [Google Scholar]
- 25.De Wildt SN, Ito S, Koren G. Challenges for drug studies in children: CYP3A phenotyping as example. Drug Discov Today. 2009;14:6–15. doi: 10.1016/j.drudis.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology – drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–67. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 27.Sharma V, McNeill JH. To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol. 2009;157:907–21. doi: 10.1111/j.1476-5381.2009.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson BJ, Meakin GH. Scaling for size: some implications for paediatric anaesthesia dosing. Paediatr Anaesth. 2002;12:205–19. doi: 10.1046/j.1460-9592.2002.00616.x. [DOI] [PubMed] [Google Scholar]
- 29.Johnson TN. The problems in scaling adult drug doses to children. Arch Dis Child. 2008;93:207–11. doi: 10.1136/adc.2006.114835. [DOI] [PubMed] [Google Scholar]
- 30.Conroy S, Sweis D, Planner C, Yeung V, Collier J, Haines L, Wong IC. Interventions to reduce dosing errors in children: a systematic review of the literature. Drug Saf. 2007;30:1111–25. doi: 10.2165/00002018-200730120-00004. [DOI] [PubMed] [Google Scholar]
- 31.Raichgot P, Prober C, Soldin S, Golas C, Good F, Harding L, MacLeod S. Chloramphenicol pharmacokinetics in the newborn. Dev Pharmacol Ther. 1983;6:305–14. doi: 10.1159/000457331. [DOI] [PubMed] [Google Scholar]
- 32.Lugo Goytia G, Lares-Asseff I, Pérez Guillé MG, Pérez AG, Mejía CL. Relationship between clinical and biologic variables and chloramphenicol pharmacokinetic parameters in pediatric patients with sepsis. Ann Pharmacother. 2000;34:393–7. doi: 10.1345/aph.19050. [DOI] [PubMed] [Google Scholar]
- 33.Kokwaro GO, Muchohi SN, Ogutu BR, Newton CR. Chloramphenicol pharmacokinetics in African children with severe malaria. J Trop Pediatr. 2006;52:239–43. doi: 10.1093/tropej/fmi082. [DOI] [PubMed] [Google Scholar]
- 34.Acharya GP, Davis TM, Ho M, Harris S, Chataut C, Acharya S, Tuhladar N, Kafle KE, Pokhrel B, Nosten F, Dance DA, Smith A, Weber A, White NJ. Factors affecting the pharmacokinetics of parenteral chloramphenicol in enteric fever. J Antimicrob Chemother. 1997;40:91–8. doi: 10.1093/jac/40.1.91. [DOI] [PubMed] [Google Scholar]
- 35.Pynnönen S, Sillanpäa M, Frey H, Iisalo E. Carbamazepine and its 10,11-epoxide in children and adults with epilepsy. Eur J Clin Pharmacol. 1977;11:129–33. doi: 10.1007/BF00562904. [DOI] [PubMed] [Google Scholar]
- 36.Riva R, Contin M, Albani F, Perucca E, Procaccianti G, Baruzzi A. Free concentration of carbamazepine and carbamazepine-10,11-epoxide in children and adults: influence of age and phenobarbitone co-medication. Clin Pharmacokinet. 1985;10:524–31. doi: 10.2165/00003088-198510060-00005. [DOI] [PubMed] [Google Scholar]
- 37.Mazoit JX. Pharmacokinetic/pharmacodynamic modeling of anesthetics in children: therapeutic implications. Paediatr Drugs. 2006;8:139–50. doi: 10.2165/00148581-200608030-00001. [DOI] [PubMed] [Google Scholar]
- 38.MCracken GH., Jr Aminoglycoside toxicity in infants and children. Am J Med. 1986;80:172–8. doi: 10.1016/0002-9343(86)90497-3. [DOI] [PubMed] [Google Scholar]
- 39.Kelly HB, Menendez R, Fan L, Murphy S. Pharmacokinetics of tobramycin in cystic fibrosis. J Pediatr. 1982;100:318–21. doi: 10.1016/s0022-3476(82)80664-1. [DOI] [PubMed] [Google Scholar]
- 40. Product information of Busilvex®. Available at http://www.emea.europa.eu/humandocs/Humans/EPAR/busilvex/busilvex.htm (last accessed 5 March 2009)
- 41. Product information of Fuzeon®. Available at http://www.emea.europa.eu/humandocs/Humans/EPAR/fuzeon/fuzeon.htm (last accessed 5 March 2009)
- 42. Product information of Tamiflu®. Available at http://www.emea.europa.eu/humandocs/Humans/EPAR/tamiflu/tamiflu.htm (last accessed 5 March 2009)
- 43. Product information of Viracept®. Available at http://www.emea.europa.eu/humandocs/Humans/EPAR/viracept/viracept.htm (last accessed 5 March 2009)
- 44. Product information of Ketek®. Available at http://www.emea.europa.eu/humandocs/Humans/EPAR/ketek/ketek.htm (last accessed 5 March 2009)
- 45. Product information of Neoclarityn®. Available at http://www.emea.europa.eu/humandocs/Humans/EPAR/neoclarityn/neoclarityn.htm (last accessed 5 March 2009)
- 46. Product information of Opatanol®. Available at http://www.emea.europa.eu/humandocs/Humans/EPAR/opatanol/opatanol.htm (last accessed 5 March 2009)
- 47.Baker MD. Antidotes for nerve agent poisoning: should we differentiate children from adults? Curr Opin Pediatr. 2007;19:211–5. doi: 10.1097/MOP.0b013e328012cba2. [DOI] [PubMed] [Google Scholar]
- 48. Product information of Lanoxin®. Available at http://www.gsk.com/products/prescription_medicines/uk/lanoxin.htm (last accessed 30 June 2009)
- 49.Ince I, de Wildt SN, Tibboel D, Danhof M, Knibbe CA. Tailor-made drug treatment for children: creation of an infrastructure for data-sharing and population PK–PD modeling. Drug Discov Today. 2009;14:316–20. doi: 10.1016/j.drudis.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Ginsberg G, Hattis D, Russ A, Sonawane B. Physiologically based pharmacokinetic (PBPK) modeling of caffeine and theophylline in neonates and adults: implications for assessing children's risks from environmental agents. J Toxicol Environ Health. 2004;67:297–329. doi: 10.1080/15287390490273550. [DOI] [PubMed] [Google Scholar]
- 51.Hsu DT. Biological and psychological differences in the child and adolescent transplant recipient. Pediatr Transplant. 2005;9:416–21. doi: 10.1111/j.1399-3046.2005.00352.x. [DOI] [PubMed] [Google Scholar]
- 52.Jamei M, Dickinson GL, Rostami-Hodjegan A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘Bottom-Up’ Vs ‘Top-Down’ recognition of covariates. Drug Metab Pharmacokinet. 2009;24:53–75. doi: 10.2133/dmpk.24.53. [DOI] [PubMed] [Google Scholar]
- 53.Cella M, Gorter de Vries F, Burger D, Danhof M, Della Pasqua O. A model-based approach to dose selection in early paediatric development. Clin Pharmacol Ther. 2009 doi: 10.1038/clpt.2009.234. doi: 10.1038/clpt.2009.234. [DOI] [PubMed] [Google Scholar]