

Abstract
Phenotypic diversity is shaped by both genetic and epigenetic mechanisms that program tissue specific patterns of gene expression. Cells, including neurons, undergo massive epigenetic reprogramming during development through modifications to chromatin structure, and by covalent modifications of the DNA through methylation. There is evidence that these changes are sensitive to environmental influences such as maternal behavior and diet, leading to sustained differences in phenotype. For example, natural variations in maternal behavior in the rat that influence stress reactivity in offspring induce long-term changes in gene expression, including in the glucocorticoid receptor, that are associated with altered histone acetylation, DNA methylation, and NGFI-A transcription factor binding. These effects can be reversed by early postnatal cross-fostering, and by pharmacological manipulations in adulthood, including Trichostatin A (TSA) and L-methionine administration, that influence the epigenetic status of critical loci in the brain. Because levels of methionine are influenced by diet, these effects suggest that diet could contribute significantly to this behavioral plasticity. Recent data suggest that similar mechanisms could influence human behavior and mental health. Epidemiological data suggest indeed that dietary changes in methyl contents could affect DNA methylation and gene expression programming. Nutritional restriction during gestation could affect epigenetic programming in the brain. These findings provide evidence for a stable yet dynamic epigenome capable of regulating phenotypic plasticity through epigenetic programming.
Keywords: DNA methylation, Demethylation, Maternal care, Nutrition, Methionine, TSA, HDAC inhibitor, Mental health, Psychopathology, Human brain, Rodent, Gene environment interaction, Stress, Glucocorticoid receptor, Histone acetylation, NGFI-A
1. Genes, gene expression programs, diet and mental health
Different cell types execute distinct patterns of gene expression that are highly responsive to developmental, physiological, pathological and environmental cues. The combination of mechanisms that confers long-term programming to genes leading to a change in gene function without a change in gene sequence is termed here epigenetic. The epigenetic programming of gene expression is somewhat dynamic in response to environmental exposures — especially though perhaps not exclusively during fetal development and early in life. Thus, much of the phenotypic variation seen in human populations might be caused by differences in long-term programming of gene function rather than the genetic sequence per se. Any analysis of inter-individual phenotypic diversity should take into account epigenetic variations in addition to genetic sequence polymorphisms (Meaney and Szyf, 2005b).
Some critical environmental exposures such as variations in maternal behavior and diet could alter the progression of epigenetic programming during development postnatally as well as in utero. Thus, variation in environmental exposures during these critical periods could result in epigenetic and therefore phenotypic differences later in life. It stands to reason that exposure to nutritional deprivation would affect the epigenetic machinery during development. Recent data suggest that psychosocial exposures early in life also impact the epigenome resulting in differences in epigenetic program and as a consequence in behavioral differences later in life (Meaney and Szyf, 2005a). Thus, certain behavioral pathologies might be a consequence of early in life exposures that alter epigenetic programming.
It is important to understand the mechanisms driving variations in epigenetic programming in order to identify the behavioral pathologies that result from such mechanisms. Unlike genetic mechanisms, epigenetic mechanisms are dynamic and thus potentially reversible and amenable to therapeutic intervention (Szyf, 2001). Because various drugs used in the treatment of psychiatric disorders such as schizophrenia and mood disorders have known epigenetic effects, interventions targeting the epigenetic machinery could have important consequences for normal cognitive function. Thus, components of diet that influence the epigenetic machinery should be considered interventions that could affect mental as well as physical health. Once the rules governing the effects of environmental exposures on epigenetic processes are understood, it might be possible to design behavioral and nutritional strategies to prevent and reverse deleterious environmentally driven epigenetic alterations.
2. The epigenome
The epigenome consists of chromatin, a protein-based structure around which wrapped the DNA, and its modifications as well as a covalent modification of cytosines residing at the dinucleotide sequence CG in DNA itself by methylation (Razin, 1998). These modifications determine the accessibility of the transcriptional machinery to the genome. Recently, an additional level of epigenetic regulation by small non-coding RNAs termed microRNA has been discovered (Bergmann and Lane, 2003). microRNA expression is itself regulated by epigenetic factors such as DNA methylation and chromatin structure (Saito and Jones, 2006). Therefore microRNAs should be considered under the headings of chromatin and DNA methylation, as they also act by changing chromatin structure (Chuang and Jones, 2007).
2.1. Chromatin structure and the histone code
The basic building block of chromatin is the nucleosome, which is formed of an octamer of histone proteins. The octamer structure of the nucleosome is composed of a H3–H4 histone protein tetramer flanked on either side with a H2A–H2B histone protein dimer (Finch et al., 1977). The N-terminal tails of these histones are extensively modified by methylation (Jenuwein, 2001), phosphorylation, acetylation (Wade et al., 1997), sumoylation (Shiio and Eisenman, 2003) and ubiquitination (Shilatifard, 2006). The specific pattern of histone modifications was proposed to form a ‘histone code’, that delineates the parts of the genome to be expressed at a given point in time in a given cell type (Jenuwein and Allis, 2001). Similar to a genetic mutation, a change in the state of modification of histone tails around a regulatory region of a gene can silence an active gene, resulting in “loss of function”, or activate a silent gene, leading to “gain of function”. In addition, such modifications can also enhance or impair levels of gene expression in the absence of complete gene silencing or activation.
2.2. Chromatin remodeling and targeting
Chromatin remodeling complexes can alter the position of nucleosomes around the transcription initiation site and define its accessibility to the transcription machinery (Varga-Weisz and Becker, 2006). It is becoming clear that both gene activating complexes and gene repression complexes contain chromatin remodeling activities (Xue et al., 1998). Remodeling might be required for facilitating the interaction between histone tails and chromatin-modifying enzymes.
The state of modification at specific loci is defined through recruitment of chromatin-modifying enzymes by sequence-specific factors to specific loci. Histone modifications are catalyzed by histone-modifying enzymes such as histone acetyltransferases (HAT), which acetylate histone tails and histone deacetylases (HDAC) that deacetylate histone tails (Kuo and Allis, 1998). Another group of important enzymes are the histone methyltransferases (HMT) and the histone demethylases (Shi et al., 2004; Tsukada et al., 2006). The balance of these activities determines the state of histone modification and thus the level of expression of the associated genes.
An important point that is emerging from current studies is that the state of modification of chromatin is not dependent exclusively on the overall levels of the histone-modifying enzymes but also on the targeting of these enzymes to specific genes. Specific transcription factors and transcription repressors recruit histone-modifying enzymes to specific genes and thus define the gene-specific profile of histone modification (Jenuwein and Allis, 2001). Some environmentally regulated alterations of histone acetylation in specific promoter sequences following seizures (Huang et al., 2002; Tsankova et al., 2004) or learning (Guan et al., 2002) are likely to be caused by neurotransmitter activation of multiple signalling pathways (Crosio et al., 2003). However, such histone modifications are transient and cannot directly explain enduring early environmental programming effects. A more likely, highly stable candidate could involve modification of the genome itself.
2.3. DNA methylation and consequences for transcription
DNA methylation is part of the covalent structure of the DNA (Razin and Riggs, 1980). This differentiates it from chromatin, which is associated with DNA but is not part of the DNA molecule itself. DNA methyltransferases (DNMTs) catalyze the transfer of a methyl group from the methyl donor S-adenosylmethionine (SAM) onto the 5′ position of the cytosine ring residing in most cases at the dinucleotide sequence CG (Adams et al., 1975; Cheng et al., 1993; Ho et al., 1991; Wu and Santi, 1985). DNMT1, known as a “maintenance” methyltransferase, has a preference for a hemimethylated substrate and is involved in copying DNA methylation patterns during cellular replication (Razin and Riggs, 1980). What distinguishes DNA methylation in vertebrates is the fact that not all CGs are methylated, but there is a cell-specific pattern of distribution of methylation on CG dinucleotides (Razin and Szyf, 1984).
Several lines of new data point to a model whereby DNA methylation patterns are actively maintained by DNMTs, which are targeted to methylated sequences. First, maintenance methylation of repetitive elements was shown to require the cooperation of the so called “de novo” methyltransferases DNMT3A and DNMT3B (Liang et al., 2002). Second, DNMT1 and DNMT3B were found in same complexes (Kim et al., 2002) in somatic cells. It would be difficult to explain this co-occurrence if copying the DNA methylation pattern during replication was the only methylation activity required in somatic cells. Third, not only are DNMTs targeted to specific genes by sequence-specific factors but they are also required to reside on these sequences to maintain their methylation state (Brenner et al., 2005; Burgers et al., 2006; Di Croce et al., 2002; Fuks et al., 2001; Vire et al., 2006). The targeting of DNMTs suggests that maintenance methylation is not just automatic copying of a template pattern, but it requires the positive identification of a specific sequence.
DNA methylation in critical sites silences genes by two principal mechanisms. First, methylation in critical sites inhibits the binding of transcription factors to their recognition elements (Comb and Goodman, 1990; Inamdar et al., 1991). Second, methylation of a regulatory region of DNA recruits methylated DNA binding proteins such as MeCP2 to the gene (Fujita et al., 1999; Hendrich and Bird, 1998; Nan et al., 1997; Ng et al., 1999) and chromatin modification enzymes such as HDACs which in turn introduce histone modifications, resulting in the silencing of chromatin. Thus, any random or programmed event of DNA methylation in critical sites in response to an environment insult or trigger might result in a change in phenotype similar to a mutation in the same sequence. It is possible that the dynamic equilibrium is altered by either pathological or adaptive mechanisms in response to extra and intracellular signaling.
2.4. Reversibility of DNA methylation in somatic tissues
There is general agreement that during development both de novo methylation and demethylation events shape and sculpt the mature cell-specific DNA methylation pattern (Brandeis et al., 1993; Frank et al., 1990; Kafri et al., 1993; Razin and Shemer, 1995; Razin et al., 1984). There is also evidence that DNA methylation patterns are dynamic in neurons (Levenson et al., 2006; Miller and Sweatt, 2007; Weaver et al., 2004; 2005). However, the precise mechanisms by which this occurs continue to be a subject of debate. There has been reluctance to accept the idea that an enzymatic activity removes methyl groups directly from the cytosine ring (Wolffe et al., 1999). We previously proposed that the METHYLATED DOMAIN DNA BINDING PROTEIN 2 (MBD2) bears a demethylation activity (Bhattacharya et al., 1999; Detich et al., 2002; 2003a; 2003b). However, other groups disputed this finding (Ng et al., 1999). A number of indirect mechanisms for demethylation have also been proposed which do not require direct removal of the methyl bond (Barreto et al., 2007; Jost, 1993; Zhu et al., 2000). We propose that, as with DNMT action, targeting plays an important role in demethylase action — activity that is intimately linked to chromatin structure.
2.5. The relationship between chromatin structure and DNA methylation
There is a well established bidirectional relationship between DNA methylation and chromatin structure (Razin and Cedar, 1977). Since it has been known for some time that chromatin configuration is dynamic and responsive to cellular signaling pathways, this relationship provides a link between the extracellular environment and the state of DNA methylation. That is, signaling pathways that activate chromatin-modifying enzymes could potentially result in altering DNA methylation patterns. There are genetic and epigenetic data linking chromatin modeling and modifying enzymes to DNA methylation (Fuks et al., 2000; 2003; Rountree et al., 2000; Vire et al., 2006). We propose that if a sequence-specific factor which targets DNMT is inactivated, DNMT is removed from the gene and the DNA methylation equilibrium is tilted toward DNA demethylation by demethylases. For example, in cancer, the histone methyltransferase EZH2 targets DNMTs to specific sequences in DNA (Di Croce et al., 2002; Vire et al., 2006). EZH2 associates with DNMTs in silencing of tumor suppressor genes (Schlesinger et al., 2007). The targeting factors are responsive to cellular signaling pathways, thus creating a conduit between cellular and extracellular signals and the epigenetic state. Thus, maintenance of the DNA methylation pattern is at least partly an active and targeted process rather than an automatic process, as the pattern of methylation is maintained by the constitutive presence of these sequence selective factors on the target genes. Some of these factors might be responsive to intracellular signaling pathways (Fig. 1). The requirement for targeting molecules which could be responsive to different signaling pathways might explain how environmental signals and oncogenic signals might affect the DNA methylation pattern in a dynamic way throughout life. It also implies that DNA methylation patterns could be therapeutically manipulated in the absence of cell division which has significance for brain targeted DNA methylation therapeutics. At the present time, this is an idea that requires further study with respect to signaling pathways in the brain.
Fig. 1.
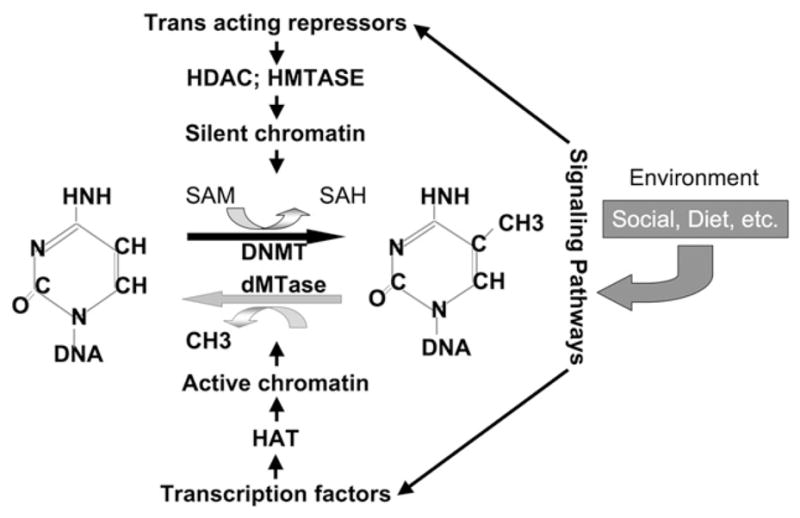
The dynamic and responsive DNA methylation pattern; a model. A balance of methylation and demethylation reactions determines the DNA methylation state. Active chromatin facilitates DNA demethylation while silent chromatin facilitates methylation. Different environmental signals trigger pathways in the cell that activate sequence-specific factors which recruit chromatin-modifying enzymes to specific loci, resulting in either activation or silencing of chromatin.
Similar to DNA methylation, demethylation is targeted by transacting factors to specific genes (Kirillov et al., 1996), and demethylation is facilitated by histone acetylation (Cervoni et al., 2002; Cervoni and Szyf, 2001). Pharmacological acetylation using HDAC inhibitors (HDACi) such as TSA (Cervoni and Szyf, 2001) or valproic acid (Detich et al., 2003a) triggers replication-independent active demethylation of transiently transfected in vitro methylated plasmids and causes genomic demethylation (Milutinovic et al., 2007; Ou et al., 2007).
The pharmacological data with HDACi might explain why certain transcription factors target DNA demethylation to specific genes. Several transcription factors recruit HATs to genes and their mode of action is similar to TSA. By increasing histone acetylation, these factors facilitate the access of demethylation activities to their target genes, an example of which is the ubiquitous transcription factor CREB binding protein (CBP) (Braganca et al., 2003; Ogryzko et al., 1996; Purucker et al., 1990; Uchida et al., 2002; Weaver et al., 2007). As we will discuss further, such a mechanism provides a conduit through which both the chemical and the social environment could affect our epigenome and thus gene expression and function, including in neurons (Fig. 1).
2.6. The dynamic pattern of DNA methylation in neurons
It stands to reason that certain chemicals would interfere with DNA methylation enzymes and thus result in an alteration in DNA methylation. It is also widely accepted that chemicals as well as altered dietary intake would affect DNA methylation during gestation (Simmons, 2007) especially during gametogenesis (Anway et al., 2005) at a point when methylation machineries are highly active and cells are undergoing rapid cell division. It has been more difficult to accept, however, that environmental agents could affect DNA methylation patterns throughout life well after tissues and organs are formed and their methylation pattern is established. The model proposed here offers a possible mechanism for alterations in methylation in adult tissue by proposing that the DNA methylation machinery remains active throughout life and thus sensitive to, for example, the social environment and the effects of diet. Two relatively recent lines of data provide some support for this hypothesis. One line of evidence comes from our study of epigenetic programming of stress responses by maternal care (Meaney and Szyf, 2005a). Another line of evidence comes from the role of nutritional constituents such as methionine that reverse such epigenetic alterations, and that influence behavior and mental health.
3. Epigenetic programming of the stress response: the role of maternal behavior and diet
3.1. Maternal care as an epigenetic regulator of the stress response
In the rat, the adult offspring of mothers that exhibit increased levels of pup licking/grooming (i.e., High LG mothers) over the first week of life show increased hippocampal GR expression, enhanced glucocorticoid feedback sensitivity, decreased hypothalamic corticotrophin releasing factor expression, and more modest HPA stress responses compared to animals reared by Low LG mothers (Francis et al., 1999; Liu et al., 1997). Cross-fostering studies suggest direct effects of maternal care on both gene expression and stress responses (Francis et al., 1999; Liu et al., 1997). These studies support an epigenetic mechanism, since the fostering mother and not the biological genetic mother define the stress response of its adult offspring. We have demonstrated that, for example, the GR exon 17 promoter is programmed differently in the hippocampus of offspring of the High and Low LG mothers and that differences which emerge between day 1 and 8 after birth remain stable thereafter. These differences include histone acetylation, DNA methylation, and the occupancy of the promoter with the transcription factor NERVE GROWTH FACTOR-INDUCIBLE PROTEIN A (NGFI-A) (Weaver et al., 2004). A comprehensive analysis of the hippocampal transcriptome of the adult offspring of High and Low LG mothers revealed differences in a few hundred genes (Weaver et al., 2005). This suggests a change in epigenetic programming in the brain of the offspring as a consequence of maternal care.
This programming by maternal behavior is stable and long lasting, but as will be discussed in the next section, is reversible by agents that interfere with either the methylation or histone deacetylation machinery (Weaver et al., 2004; 2005). Thus, the maternal care model typifies the first principles of epigenetic programming, which are stability and relative plasticity.
3.2. Epigenetic programming by maternal care is reversible in adulthood
The idea that epigenetic programming can be reversible in adulthood depends upon the assumption that the enzymatic machineries required to generate new methylation pattern are present in adult tissue. There is evidence from studies in cultured cells that this is indeed the case. TSA induces replication-independent demethylation in cell culture (Cervoni and Szyf, 2001). TSA induces histone acetylation by inhibiting HDACs (Yoshida et al., 1990) and thus tilting the histone acetylation equilibrium toward acetylation. We proposed that this open chromatin structure induced by hyperacetylation facilitated the interaction of demethylases with methylated DNA and thus tilted the DNA methylation equilibrium toward demethylation (Cervoni and Szyf, 2001). We therefore addressed the question of whether the epigenetic programming early in life could be modulated during adulthood.
We injected the HDACi TSA into the brain to test the hypothesis that the machineries required for the modulation of chromatin and DNA methylation were found in neurons and associated with the GR exon 17 promoter and that the epigenetic state was an equilibrium of modifying and demodifying enzymes. TSA injected into brains of adult offspring of Low LG maternal care increased acetylation, reduced methylation, activated GR exon 17 promoter to levels indistinguishable from adult offspring of High LG maternal care and reduced stress responsivity to the levels of offspring of High LG (Weaver et al., 2004).
We similarly reasoned that if the DNA methylation and chromatin state is in a dynamic equilibrium even in adult neurons, it should be possible to revert the epigenetic programming in the other direction toward increased methylation, leading to a reversal of the maternal programming of GR expression and HPA responses to stress. We therefore injected methionine, the precursor of SAM, into the brain of the adult offspring of different maternal care mothers. Dietary methionine is converted by methionine adenosyltransferase into SAM (Cantoni, 1975; Mudd and Cantoni, 1958), which serves as the donor of methyl groups for DNA methylation. SAM was shown to inhibit active demethylation (Detich et al., 2003b) and to stimulate methylation (Pascale et al., 1991). Importantly, the synthesis of SAM is dependent on the local availability of methionine (Cooney, 1993).
Methionine treatment has been previously shown to increase SAM and DNA methylation levels in the brain (Guidotti et al., 2007; Tremolizzo et al., 2002). Adult offspring of High and Low LG mothers were infused into the lateral ventricles with methionine (100μg/ml) or saline vehicle once a day for 7 consecutive days. Methionine treatment of the offspring of High LG mothers changed the DNA methylation state of GR exon 17 promoter and expression of GR in the hippocampus as well as increasing their stress responsiveness and reducing the time that these animals spent in the center of an open field, a measure of anxiety (Weaver et al., 2005; 2006).
An important question here is whether the effects of methionine are limited to a subset of genes such as GR or whether they disrupted the DNA methylation patterns across the entire genome. Surprisingly, results from gene expression microarray analysis performed on hippocampal tissue from a separate cohort of methionine-treated High and Low LG offspring showed that the methionine treatment significantly affected only 300 genes, representing 1% of the population of genes on the chip (Weaver et al., 2006). These findings suggest an impressive level of specificity. Several of the modified genes are relevant for the effects observed on the stress response, however it would appear that these results do not emerge as a function of a widespread alteration in hippocampal gene expression. Our findings suggest that alterations of cytosine methylation in the adult brain through global procedures are surprisingly specific. Because methionine alone does not methylate DNA but is converted to the methyl donor SAM in the DNA methylation reaction, the DNMTs must be poised to methylate GR exon 17 promoter. Taken together, the TSA and methionine experiments support the basic hypothesis that epigenetic programs in the brain are maintained by a dynamic equilibrium of methylating and demethylating enzymes, a balance which could be shifted by agents which either inhibit demethylases or stimulate DNMTs. Thus, despite the remarkable stability of epigenetic programs they are nevertheless reversible (Fig. 2).
Fig. 2.

Epigenetic reprogramming by maternal care; a model. Maternal licking and grooming (LG) in the rat triggers activation of 5-HT receptors in the hippocampus leading to increased intracellular cAMP, activation of the transcription factor NGFI-A and recruitment of the HAT CBP to the GR exon 17 promoter. Acetylation of histone tails facilitates demethylation. In offspring of Low LG mothers, this process is reduced in comparison with offspring of High LG mothers, leading to differential epigenetic programming of the GR promoter. In the adult rat, the epigenetic state is reversible. TSA, an HDAC inhibitor, increases histone acetylation and facilitates demethylation and epigenetic activation of the gene in the offspring of the Low LG mothers. Conversely, injection of methionine to adult offspring of the High LG mothers leads to increased SAM, inhibition of demethylation, increased DNA methylation, and reduced activity of the GR exon 17 promoter.
One open question regarding the role of maternal care and methionine concerns the regional specificity of these epigenetic effects. Although the hippocampus was the focus of the aforementioned studies, other brain regions are also likely targets of these effects. The particular neural pathways influenced, however, likely depend on the brain region in question. For example, variations in maternal care are associated with altered estrogen receptor alpha expression (Champagne et al., 2003) and altered methylation of estrogen receptor alpha promoter in the hypothalamus (Champagne et al., 2006). More studies are needed to determine whether epigenetic variations of the GR17 splice variant account for reported differences in GR expression in brain areas in addition to the hippocampus.
3.3. Mechanisms linking maternal care and epigenetic reprogramming
We have started to decipher the molecular events which link maternal licking and grooming and epigenetic changes at the GR gene locus. In vivo and in vitro studies suggest that maternal LG or postnatal handling, which increases maternal LG, increases GR gene expression in the offspring through a thyroid hormone-dependent increase in serotonin (5-HT) activity at 5-HT7 receptors, and the subsequent activation of cyclic adenosine 30, 50 monophosphate (cAMP) and cAMP-dependent protein kinase A (PKA) (Laplante et al., 2002; Meaney et al., 1987; 2000). Both the in vitro effects of 5-HT and the in vivo effects of maternal behavior on GR mRNA expression are accompanied by increased hippocampal expression of the NGFI-A transcription factor. The GR exon 17 promoter region contains at least one binding site for NGFI-A (McCormick et al., 2000). Interestingly, NGFI-A was previously shown to regulate transcription of the transcriptional coactivator and histone acetyl transferase CBP by both repression and activation under different cellular challenges (Yu et al., 2004). Signaling pathways that result in increased cAMP also activate CBP (Chawla et al., 1998). NGFI-A and CBP are recruited to the GR exon 17 promoter in response to maternal care, which explains the increased acetylation and demethylation observed in offspring of High LG mothers (Weaver et al., 2007). Tissue culture experiments demonstrate that recruitment of NGFI-A to the GR exon 17 promoter results in replication-independent DNA demethylation. The recruitment of NGFI-A to the promoter facilitates the interaction of MBD2, as mentioned above a protein proposed to be involved in replication-independent DNA demethylation, with the promoter (Weaver et al., 2007). Further experiments are required, including specific knock down of NGFI-A, CBP and MBD2 in vivo to fully demonstrate the pathway linking exposure to maternal care and demethylation of specific loci. Nevertheless, these experiments chart a feasible route leading from a behavioral exposure to a chemical change in chromatin (Fig. 2).
3.4. Dietary contributions to DNA methylation and histone modifications
The experiments described above involving infusion of methionine into the lateral ventricles of the brain raise the possibility that diet can affect the phenotype being studied. Because intracellular levels of methionine can be affected by both dietary intake and polymorphisms of enzymes involved in methionine metabolism, such as methylenetetrahydrofolate-reductase (Friso et al., 2002), it is tempting to consider the possibility that diet could modify epigenetic programming in the brain not only during early development but also in adult life.
Human epidemiological and animal model data indicate that susceptibility to adult-onset chronic disease is influenced by persistent adaptations to prenatal and early postnatal nutrition (Lucas, 1998). Rodent models have been particularly useful in elucidating the mechanisms involved in these developmental effects. For example, in rats, dietary L-methionine has been shown to be crucial for normal brain development, brain aging, and the pathogenesis of neurodegenerative disorders, playing an essential role in gene expression, protein synthesis, cell signaling, lipid transport/metabolism, and neuron survival (Slyshenkov et al., 2002; Van den Veyver, 2002). DNA methyltransferase requires SAM to establish or maintain DNA methylation patterns. Synthesis of SAM is dependent on the availability of dietary folates, vitamin B12, methionine, betaine, and choline (Cooney, 1993). Developmental choline deficiency alters SAM levels and global and gene-specific methylation (Kovacheva et al., 2007; Niculescu et al., 2006), and prenatal choline availability has been shown to impact neural cell proliferation and learning and memory in adulthood in rodents (Glenn et al., 2007; Meck et al., 1989; Meck and Williams, 2003). Several studies have shown that additional dietary factors, including zinc and alcohol, can influence the availability of methyl groups for SAM formation, and thereby influence CpG methylation (Davis and Uthus, 2004; Pogribny et al., 2006; Ross, 2003; Ross and Milner, 2007). Maternal methyl supplements affect epigenetic variation and DNA methylation and positively affect health and longevity of the offspring (Cooney et al., 2002; Waterland and Jirtle, 2003; Wolff et al., 1998). We hypothesize that reversal of epigenetic states in the brain, such as the remethylation of the exon 17 GR promoter, could be triggered not only by pharmacological agents but also by stable variations in environmental conditions.
Other studies have shown that certain dietary components may act as an HDACi, including diallyl disulfide, sulforaphane, and butyrate (Dashwood et al., 2006). For example, broccoli, which contains high levels of sulforaphane, has been associated with H3 and H4 acetylation in peripheral blood mononuclear cells in mice 3–6h after consumption (Dashwood and Ho, 2007). The long-term consequences of such epigenetic effects on human health remain to be studied, however HDACis are an active area of research as anti-inflammatory and neuroprotective agents in autoimmune diseases such as lupus and multiple sclerosis (Gray and Dangond, 2006), and sodium butyrate has been shown to have antidepressant effects in mice (Schroeder et al., 2007). Thus, it is conceivable that dietary compounds that influence histone acetylation may affect signaling mechanisms that regulate neural function. In light of the aforementioned link between histone modifications and DNA methylation, future studies are needed to address the possibility that sustained exposure to such compounds may affect DNA methylation at susceptible loci, with implications for mental health in humans. Further studies are required to map the effects of dietary components on epigenetic programming. The advent of whole-genome mapping methodologies will allow a detailed definition of the impact of dietary variations at different stages in life on long-term epigenetic programming.
4. Epigenetic contributions to mental health
The questions raised by evidence that epigenetic changes result in stable long-term changes in gene function that may nevertheless be reversible have broad ranging implications for our understanding of social, physiological and pathological processes and their interrelationships. In humans, several of the questions are similar to those raised by experimentation in non-human animals reviewed above, while others may have particular relevance to human populations. For example, what is the evidence for and the magnitude of inter-individual differences in the epigenetic profiles in humans, particularly in genomic loci involved in behavior? Could differences in early life adversity have long-term effects on epigenetic processes in humans, including increased risk for psychopathology? Finally, can behaviorally-mediated epigenetic reprogramming alter and be altered in response to diet? This list is by no means exhaustive and will serve in the following discussion only to illustrate particular ways in which these challenges are beginning to be addressed.
4.1. Interindividual differences in DNA methylation in humans
One line of evidence supporting the concept that there is a lifelong drift in DNA methylation in normal somatic tissue comes from the hypermethylation observed in aging tissue (Ahuja et al., 1998; Issa, 2000). Similarly, a recent study of monozygotic twins has revealed that a difference in DNA methylation emerges later in life, suggesting an environmental rather than a genetic basis for the lifelong DNA methylation drift (Fraga et al., 2005). The dynamic plasticity of the DNA methylation patterns revealed by these studies and its responsiveness to both the chemical and behavioral environment raises the possibility that errors in DNA methylation might emerge during adulthood and lead to changes in gene expression and the emergence of late onset pathologies (Feinberg, 2007).
4.2. Influence of DNA methylation on mental health
Genetic defects in genes encoding the DNA methylation and chromatin machinery exhibit profound effects on mental health. A classic example is RETT syndrome, a progressive neurodevelopmental disorder and one of the most common causes of mental retardation in females which is caused by mutations in the methylated DNA binding protein MeCP2 (Amir et al., 1999). Mutations in MeCP2 and reduced MeCp2 expression were also associated with autism (Ben Zeev Ghidoni, 2007; Herman et al., 2007; Lasalle, 2007; Nagarajan et al., 2006). ATRX a severe, X-linked form of syndromal mental retardation associated with alpha thalassaemia (ATR-X syndrome) is caused by a mutation in a gene which encodes a member of the SNF2 subgroup of a superfamily of proteins with similar ATPase and helicase domains which are involved in chromatin remodeling (Picketts et al., 1996). The ATRX mutation is associated with DNA methylation aberrations (Gibbons et al., 2000). Although these genetic lesions in the methylation machinery were present through development and are thus fundamentally different from methylation changes after birth, these data nevertheless support the hypothesis that DNA methylation defects could lead to mental pathologies as well. Thus, it is possible that environmental exposures which would affect the activity of the methylation machinery would also lead to behavioral and mental pathologies.
There are some data indicating aberrant methylation in late onset mental pathologies, although it is unclear whether these changes in DNA methylation originated during embryogenesis or later in life as a response to an environmental exposure. The gene encoding REELIN, a protein involved in neuronal development and synaptogenesis, which is implicated in long-term memory, was found to be hypermethylated in brains of schizophrenia patients. The methylation of REELIN was correlated with its reduced expression and increased DNMT1 expression in GABAergic neurons in the prefrontal cortex (Chen et al., 2002; Costa et al., 2002; 2003; Grayson et al., 2005; Veldic et al., 2007).
The promoters of the genes encoding rRNA were found to be heavily methylated in hippocampi from subjects who committed suicides relative to controls (McGowan et al., 2008). Methylation of rRNA defines the fraction of rRNA molecules which are active in a cell, and the output of rRNA transcription defines to a large extent the protein synthesis capacity of a cell (Brown and Szyf, 2007). Protein synthesis is critical for learning and memory. Thus, a reduced capacity for protein synthesis required for learning and memory in brains of suicide victims could be epigenetically determined. This might be involved in the pathology leading to suicide. Thus, evidence is emerging that aberrant DNA methylation is involved in psychopathologies. Because it remained unclear whether the documented epigenetic aberrations were present in the germ line or whether they were truly late onset changes, we examined the genomic and anatomical specificity of rRNA methylation. We found that the sequence of rRNA was identical in all subjects, and there was no difference in methylation between suicide victims and controls in the cerebellum, a brain region not normally associated with psychopathology, nor were there genome-wide differences in levels of methylation (McGowan et al., 2008). These data imply that epigenetic effects that influence psychopathology likely target particular neural pathways.
4.3. Chromatin modification and its role in mental health
The fact that histone methylation is reversible provides a wide platform for pharmacological and therapeutic manipulations of the state of histone methylation in both directions. Both histone demethylases and histone methyltransferase are excellent candidates for new drug discovery. Understanding the intricate details of their genomic targets will allow the design of targeted and specific therapeutics.
The epigenetic effects of current clinically used monoamine oxidase inhibitors provide leads for the further development of therapies targeting the epigenome. For example, H3-K4Me2 is a hallmark of active genes and the target of the histone demethylase LSD1 which demethylates H3-K4Me2. Interestingly, certain non-selective monoamine oxidase inhibitors used as antidepressants such as Tranylcypromine that were clinically used for some time and believed to be acting on monoamine oxidases also appear to inhibit LSD1 demethylase (Lee et al., 2006). It is tempting to speculate that inhibition of LSD1 is part of the mechanism of action of these antidepressants through activation of critical genes suppressed by the H3-K4Me2 demethylating activity of LSD1 in the brain (Shi et al., 2004) or by repressing genes activated by the H3-K9Me2 demethylation activity of LSD1 (Metzger et al., 2005). Thus, it is possible that LSD1 inhibition is involved in the mechanism of action of antidepressive agents. It is tempting to speculate that selective inhibitors of LSD1 might be effective as anti-depressants. This is an idea that might be pursued in the future.
Chromatin acetylation and memory were shown to be impaired in CBP knock out mice, suggesting a role for acetylation in memory formation (Alarcon et al., 2004). The fact that valproic acid, a long established antiepileptic and mood stabilizer, is also an HDACi (Phiel et al., 2001) alluded to a possible role for HDACi in treating mental disorders such as schizophrenia and bipolar disorder. Valproic acid has some effect in alleviating psychotic agitation as an adjunct to antipsychotics in schizophrenia (Bowden, 2007; Yoshimura et al., 2007). HDACi were shown to improve memory and induce dendritic sprouting in a transgenic mouse model of neurodegeneration, suggesting that HDACi might be of use in treating neurodegeneration and memory loss as well (Fischer et al., 2007). Although biological and behavioral effects of HDACi in the brain are somewhat characterized, the specific gene targets of HDACi in the brain and their function in mental pathologies are not well delineated. Nevertheless, the limited clinical and animal data suggest a potentially important role for HDACi in treatment of mental disorders. Experiments with a novel HDACi from the benzamide class N-(2-aminophenyl)-4-[N-(pyridin-3-yl-methoxycarbonyl)aminomethyl]benzamide derivative (MS-275) in mice resulted in brain region specific induction of acetylation in the frontal cortex at two genes involved with schizophrenia pathogenesis, REELIN and GAD(67) (Simonini et al., 2006). Valproic acid was shown to induce the expression of REELIN, which was silenced by methionine treatment in mice (Dong et al., 2007). These studies raise the possibility that treatment of schizophrenics with HDACi might cause activation of expression of critical genes such as REELIN and could reverse the course of this disease (Sharma et al., 2006). Several clinical trials have tested valproate as an adjunctive therapy to antipsychotics in schizophrenia (Basan and Leucht, 2004; Bowden, 2007; Citrome et al., 2007). There are indications that valproate might improve violent episodes in a subset of schizophrenia patients (Basan and Leucht, 2004) and might have an effect on euphoric mania in combination with antipsychotics (Bowden, 2007) as well as features of manic symptomatology in bipolar disorders (Bowden, 2007). It should be noted that many of these trials were of small size and that further clinical trials are needed with valproate and with more potent and selective HDACi to methodically test the therapeutic potential of HDACi in mental pathologies.
One question that needs to be addressed is whether the observed defects in histone acetylation in mental disease are a consequence of aberrant deregulation of the overall levels of certain HDAC isotypes or HATs, or whether it involves the aberrant targeting of HDAC to a selection of promoters. The fact that inhibition of a general enzyme such as HDAC results in exquisite positive changes in the brain implies some specificity, even for a general inhibitor of a specific class of HDACs as discussed above. How could such specificity be achieved by treatment with non-selective HDACi? It will be important to delineate the response of the transcriptomes of different brain regions to HDACi and to map the genes that are critically involved in the molecular pathology of the disease. It will also be important to characterize the critical isoforms of HDAC for regulation of these genes. The advent of isotypic specific HDACi might enhance the efficacy and potency of the treatment and reduce its toxicity.
4.4. Relevance of diet to the risk for psychopathology
The idea that epigenetic modifications play a role in cancer has gained wide acceptance over the last two decades (Szyf, 2008). There has been more recent acknowledgement that metabolic syndrome has an epigenetic component (Ross and Milner, 2007). Evidence that nutrition plays a role at the interface between the environment and the genome in cancer and metabolic syndrome is beginning to be recognized. However, there is as yet little evidence for the role of nutrition in the epigenetic regulation of mental health. As mentioned above, a wide range of epigenetic effects influence the epigenetic status of the brain, and some nutritional components such as SAM and sulfophrane can mitigate changes in DNA methylation and chromatin structure akin to those observed by classical drugs used to treat psychopathology, such as valproic acid and the monoamine oxidase inhibitors. It is interesting to speculate that nutritional components, especially those to which humans are exposed developmentally or via sustained exposure – particularly to those which act to modify chromatin – will have effects on mental health and risk for psychopathology.
The possible involvement of DNA methylation in schizophrenia implies that pharmacological and nutritional agents which increase SAM levels in the brain might aggravate schizophrenia. For example, methionine treatment was shown in the 60s to aggravate schizophrenia (Brune and Himwich, 1962; Israelstam et al., 1967). Similarly, there might be questions raised as to the impact of folate supplementation during pregnancy and beyond. Folates are required for the synthesis of tetrahydrofolate, which is required for methionine synthesis and consequently SAM levels. Research combining the identification of polymorphisms associated with folate metabolism with psychopathology may identify effects of methionine synthesis on risk for schizophrenia (Muntjewerff and Blom, 2005).
Another of the possible interactions between dietary components that modify the DNA methylation machinery and effects on mental health in humans may be found in the effects of SAM in mood disorders (McGowan and Kato, 2008). Many studies have found SAM to have antidepressive effects (Papakostas et al., 2003). Interestingly, in one study, nine of 11 patients with bipolar depression treated with SAM switched to mania, suggesting a specific effect of SAM on bipolar depression (Carney et al., 1989). As mentioned above, central infusion of L-methionine, a precursor of SAM, increases DNA methylation of the promoter of the GR gene. Methionine treatment was found to abolish the effect of a High LG mother on the offspring, leading to increased DNA methylation of GR and exacerbating a measure of behavioral despair (Weaver et al., 2005). The fact that SAM, which similarly enhances DNA methylation, is effective in the treatment of depression is apparently contradictory to this effect of methionine. However, SAM is a methyl residue donor not only for the DNA methylation reaction but also for other enzymatic reactions. For example, creatine is produced from SAM and guanidinoacetate, and SAM treatment increases phosphocreatine levels in the brain (Silveri et al., 2003). This effect may also contribute to the antidepressive effect of SAM because decreased phosphocreatine levels have been reported in bipolar depression (Kato et al., 1994). It is becoming clear that we need to consider these issues in the future when assessing the safety of drugs, nutraceuticals and dietary habits, as DNA methylation in the brain has both pharmacological and toxicological implications.
5. Summary and prospective
The realization that the genome is programmed by the epigenome and that this programming might be as important as the sequence itself in executing genome functionality offers a new approach to the long-standing mystery of gene-environment interactions. Epigenetic aberrations might have similar consequences to genetic damage, as far as gene expression and the resulting phenotype are considered. Epigenetic marks, though potentially reversible, are stable and could be long lasting. The differential epigenetic status of the GR exon 17 promoter in the offspring of High LG mothers is a possible mechanism for the maternal effect on hippocampal GR expression and HPA responses to stress. These findings provide a possible mechanism for the ‘environmental programming’ of gene expression and function during development and beyond. Studies on the reversal of maternal effects on DNA methylation using either TSA or methionine suggest that neurons express the enzymatic machinery necessary for methylation and demethylation in adulthood. DNA methylation, although a stable epigenetic mark maintained through carbon–carbon bonds, can be altered through sustained alterations of chromatin structure such as histone acetylation. These findings thus raise the fascinating question of the degree to which such processes might remain sensitive to environmental regulation throughout life. The emerging understanding that late onset diseases might have an epigenetic origin points to the importance of developing screens to identify epigenetic chemoprotective agents present in our diet.
Perhaps one of the finest examples of how the epigenome mediates the effects of the environment on our genome comes from studies of endocrine disruptors [for a review see Jirtle and Skinner (2007)]. Endocrine disruptors cause epigenetic changes by DNA methylation, which are heritable in rodents and can promote disease across subsequent generations (Anway et al., 2005). These observations put forward the thought-provoking notion that environmental exposures in one generation could have an impact on phenotype and disease susceptibility on generations to come. Interestingly, exposure to endocrine disruptors affect female mate preference in rodents three generation removed from the exposure, raising the possibility that epigenetics is a yet unappreciated force in evolution (Crews et al., 2007). In addition, dietary manipulations that affect the availability of the methyl donors during development had a protective effect against endocrine disruption (Dolinoy et al., 2007). New data from behavioral studies is shedding light on the relationship between the social environment and epigenetic programming. It has also illustrated the potential lifelong dynamic nature of the epigenome. The relationship between behavior and the epigenome is bilateral; behavior could result in epigenetic programming and epigenetic programming could affect behavior. Similarly behavior might affect susceptibility to dietary preferences while dietary preferences might have a long-term effect on behavior through affecting epigenetic reprogramming. In humans, such effects might contribute to the risk for and resilience to psychopathology.
Another important principle that is emerging from these studies is that behavioral parameters should be taken into consideration in our analysis of the environmental impact on the epigenome. The dynamic equilibrium of DNA methylation provides a template for diet to act upon. Dietary components could act through cellular signaling pathways, leading from cell surface receptors down to transacting factors, that deliver chromatin-modifying enzymes to specific sequences. The dynamic epigenome has obviously adaptive and physiological roles in the crosstalk between our environment and our inherited genome, but could at the same time serve as a target for dietary components (Figs. 1–2). Unraveling the conduits between our diet and our genomes should have an important impact on our health.
Acknowledgments
This work was supported by grants from the Human Frontiers Science Program (HFSP), the Maternal Adversity, Vulnerability and Neurodevelopment (MAVAN) project of the Canadian Institutes for Health Research (CIHR), and the National Institute of Child Health and Development (NICHD) to MJM and MS and the National Cancer Institute of Canada to MS.
Abbreviations
- HAT
Histone acetyltransferase
- HDAC
histone deacetylase
- HMT
histone methyltransferase
- DNMT
DNA methyltransferase
- SAM
S-adenosylmethionine
- HDACi
HDAC inhibitor
- CBP
CREB binding protein
- TSA
Trichostatin A
- MBD2
METHYLATED DOMAIN DNA BINDING PROTEIN 2
- NGFI-A
NERVE GROWTH FACTOR-INDUCIBLE PROTEIN A
- LG
Licking/Grooming
References
- Adams RL, et al. DNA methylation in nuclei and studies using a purified DNA methylase from ascites cells. In: Antoni F, Farago A, editors. Post-synthetic Modification of Macromolecules. North-Holland; Amsterdam: 1975. pp. 39–48. [Google Scholar]
- Ahuja N, et al. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–5494. [PubMed] [Google Scholar]
- Alarcon JM, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein–Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Anway MD, et al. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto G, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- Basan A, Leucht S. Valproate for schizophrenia. Cochrane Database Syst Rev. 2004;1:CD004028. doi: 10.1002/14651858.CD004028.pub2. [DOI] [PubMed] [Google Scholar]
- Ben Zeev Ghidoni B. Rett syndrome. Child Adolesc Psychiatr Clin N Am. 2007;16:723–743. doi: 10.1016/j.chc.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Bergmann A, Lane ME. HIDden targets of microRNAs for growth control. Trends Biochem Sci. 2003;28:461–463. doi: 10.1016/S0968-0004(03)00175-0. [DOI] [PubMed] [Google Scholar]
- Bhattacharya SK, et al. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397:579–583. doi: 10.1038/17533. [DOI] [PubMed] [Google Scholar]
- Bowden CL. Spectrum of effectiveness of valproate in neuropsychiatry. Expert Rev Neurotherapeutics. 2007;7:9–16. doi: 10.1586/14737175.7.1.9. [DOI] [PubMed] [Google Scholar]
- Braganca J, et al. Physical and functional interactions among AP-2 transcription factors, p300/CREB-binding protein, and CITED2. J Biol Chem. 2003;278:16021–16029. doi: 10.1074/jbc.M208144200. [DOI] [PubMed] [Google Scholar]
- Brandeis M, et al. Dynamics of DNA methylation during development. BioEssays. 1993;15:709–713. doi: 10.1002/bies.950151103. [DOI] [PubMed] [Google Scholar]
- Brenner C, et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. Embo J. 2005;24:336–346. doi: 10.1038/sj.emboj.7600509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SE, Szyf M. Epigenetic programming of the rRNA promoter by MBD3. Mol Cell Biol. 2007;27:4938–4952. doi: 10.1128/MCB.01880-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune GG, Himwich HE. Effects of methionine loading on the behavior of schizophrenic patients. J Nerv Ment Dis. 1962;134:447–450. doi: 10.1097/00005053-196205000-00007. [DOI] [PubMed] [Google Scholar]
- Burgers WA, et al. Viral oncoproteins target the DNA methyltransferases. Oncogene. 2006;26:1650–1655. doi: 10.1038/sj.onc.1209950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni GL. Biological methylation: selected aspects. Annu Rev Biochem. 1975;44:435–451. doi: 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
- Carney MW, et al. The switch mechanism and the bipolar/unipolar dichotomy. Br J Psychiatry. 1989;154:48–51. doi: 10.1192/bjp.154.1.48. [DOI] [PubMed] [Google Scholar]
- Cervoni N, et al. The oncoprotein Set/TAF-1beta, an inhibitor of histone acetyltransferase, inhibits active demethylation of DNA, integrating DNA methylation and transcriptional silencing. J Biol Chem. 2002;277:25026–25031. doi: 10.1074/jbc.M202256200. [DOI] [PubMed] [Google Scholar]
- Cervoni N, Szyf M. Demethylase activity is directed by histone acetylation. J Biol Chem. 2001;276:40778–40787. doi: 10.1074/jbc.M103921200. [DOI] [PubMed] [Google Scholar]
- Champagne FA, et al. Natural variations in maternal care are associated with estrogen receptor alpha expression and estrogen sensitivity in the medial preoptic area. Endocrinology. 2003;144:4720–4724. doi: 10.1210/en.2003-0564. [DOI] [PubMed] [Google Scholar]
- Champagne FA, et al. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. 2006;147:2909–2915. doi: 10.1210/en.2005-1119. [DOI] [PubMed] [Google Scholar]
- Chawla S, et al. CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science. 1998;281:1505–1509. doi: 10.1126/science.281.5382.1505. [DOI] [PubMed] [Google Scholar]
- Chen Y, et al. On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 2002;30:2930–2939. doi: 10.1093/nar/gkf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, et al. Crystal structure of the HhaI DNA methyltransferase. Cold Spring Harbor Symp Quant Biol. 1993;58:331–338. doi: 10.1101/sqb.1993.058.01.039. [DOI] [PubMed] [Google Scholar]
- Chuang JC, Jones PA. Epigenetics and MicroRNAs. Pediatr Res. 2007;61:24R–29R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- Citrome L, et al. Risperidone alone versus risperidone plus valproate in the treatment of patients with schizophrenia and hostility. Int Clin Psychopharmacol. 2007;22:356–362. doi: 10.1097/YIC.0b013e3281c61baf. [DOI] [PubMed] [Google Scholar]
- Comb M, Goodman HM. CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP-2. Nucleic Acids Res. 1990;18:3975–3982. doi: 10.1093/nar/18.13.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney CA. Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth Dev Aging. 1993;57:261–273. [PubMed] [Google Scholar]
- Cooney CA, et al. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393S–2400S. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- Costa E, et al. REELIN and schizophrenia: a disease at the interface of the genome and the epigenome. Mol Interv. 2002;2:47–57. doi: 10.1124/mi.2.1.47. [DOI] [PubMed] [Google Scholar]
- Costa E, et al. GABAergic cortical neuron chromatin as a putative target to treat schizophrenia vulnerability. Crit Rev Neurobiol. 2003;15:121–142. doi: 10.1615/critrevneurobiol.v15.i2.20. [DOI] [PubMed] [Google Scholar]
- Crews D, et al. Transgenerational epigenetic imprints on mate preference. Proc Natl Acad Sci U S A. 2007;104:5942–5946. doi: 10.1073/pnas.0610410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C, et al. Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci. 2003;116:4905–4914. doi: 10.1242/jcs.00804. [DOI] [PubMed] [Google Scholar]
- Dashwood RH, Ho E. Dietary histone deacetylase inhibitors: from cells to mice to man. Semin Cancer Biol. 2007;17:363–369. doi: 10.1016/j.semcancer.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashwood RH, et al. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27:344–349. doi: 10.1093/carcin/bgi253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CD, Uthus EO. DNA methylation, cancer susceptibility, and nutrient interactions. Exp Biol Med (Maywood) 2004;229:988–995. doi: 10.1177/153537020422901002. [DOI] [PubMed] [Google Scholar]
- Detich N, et al. Promoter-specific activation and demethylation by MBD2/demethylase. J Biol Chem. 2002;277:35791–35794. doi: 10.1074/jbc.C200408200. [DOI] [PubMed] [Google Scholar]
- Detich N, et al. Valproate induces replication-independent active DNA demethylation. J Biol Chem. 2003a;278:27586–27592. doi: 10.1074/jbc.M303740200. [DOI] [PubMed] [Google Scholar]
- Detich N, et al. The methyl donor S-adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S-adenosylmethionine. J Biol Chem. 2003b;278:20812–20820. doi: 10.1074/jbc.M211813200. [DOI] [PubMed] [Google Scholar]
- Di Croce L, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295:1079–1082. doi: 10.1126/science.1065173. [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, et al. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E, et al. Histone hyperacetylation induces demethylation of reelin and 67-kDa glutamic acid decarboxylase promoters. Proc Natl Acad Sci U S A. 2007;104:4676–4681. doi: 10.1073/pnas.0700529104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Finch JT, et al. Structure of nucleosome core particles of chromatin. Nature. 1977;269:29–36. doi: 10.1038/269029a0. [DOI] [PubMed] [Google Scholar]
- Fischer A, et al. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Fraga MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis D, et al. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286:1155–1158. doi: 10.1126/science.286.5442.1155. [DOI] [PubMed] [Google Scholar]
- Frank D, et al. Demethylation of genes in animal cells. Philos Trans R Soc Lond B, Biol Sci. 1990;326:241–251. doi: 10.1098/rstb.1990.0008. [DOI] [PubMed] [Google Scholar]
- Friso S, et al. A common mutation in the 5, 10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci U S A. 2002;99:5606–5611. doi: 10.1073/pnas.062066299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, et al. Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol Cell Biol. 1999;19:6415–6426. doi: 10.1128/mcb.19.9.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, et al. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24:88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- Fuks F, et al. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. Embo J. 2001;20:2536–2544. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, et al. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RJ, et al. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet. 2000;24:368–371. doi: 10.1038/74191. [DOI] [PubMed] [Google Scholar]
- Glenn MJ, et al. Prenatal choline availability modulates hippocampal neurogenesis and neurogenic responses to enriching experiences in adult female rats. Eur J Neurosci. 2007;25:2473–2482. doi: 10.1111/j.1460-9568.2007.05505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SG, Dangond F. Rationale for the use of histone deacetylase inhibitors as a dual therapeutic modality in multiple sclerosis. Epigenetics. 2006;1:67–75. doi: 10.4161/epi.1.2.2678. [DOI] [PubMed] [Google Scholar]
- Grayson DR, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Guidotti A, et al. S-adenosyl methionine and DNA methyltransferase-1 mRNA overexpression in psychosis. NeuroReport. 2007;18:57–60. doi: 10.1097/WNR.0b013e32800fefd7. [DOI] [PubMed] [Google Scholar]
- Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman GE, et al. Genetic testing in autism: how much is enough? Genet Med. 2007;9:268–274. doi: 10.1097/gim.0b013e31804d683b. [DOI] [PubMed] [Google Scholar]
- Ho DK, et al. Stereochemical studies of the C-methylation of deoxycytidine catalyzed by HhaI methylase and the N-methylation of deoxyadenosine catalyzed by EcoRI methylase. Arch Biochem Biophys. 1991;284:264–269. doi: 10.1016/0003-9861(91)90294-s. [DOI] [PubMed] [Google Scholar]
- Huang Y, et al. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci. 2002;22:8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamdar NM, et al. CpG methylation inhibits binding of several sequence-specific DNA-binding proteins from pea, wheat, soybean and cauliflower. Plant Mol Biol. 1991;17:111–123. doi: 10.1007/BF00036811. [DOI] [PubMed] [Google Scholar]
- Israelstam DM, et al. Methionine and schizophrenia. J Nucl Med. 1967;8:325–326. [PubMed] [Google Scholar]
- Issa JP. CpG-island methylation in aging and cancer. Curr Top Microbiol Immunol. 2000;249:101–118. doi: 10.1007/978-3-642-59696-4_7. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol. 2001;11:266–273. doi: 10.1016/s0962-8924(01)02001-3. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost JP. Nuclear extracts of chicken embryos promote an active demethylation of DNA by excision repair of 5-methyldeoxycytidine. Proc Natl Acad Sci U S A. 1993;90:4684–4688. doi: 10.1073/pnas.90.10.4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafri T, et al. Mechanistic aspects of genome-wide demethylation in the preimplantation mouse embryo. Proc Natl Acad Sci U S A. 1993;90:10558–10562. doi: 10.1073/pnas.90.22.10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, et al. Reduction of brain phosphocreatine in bipolar II disorder detected by phosphorus-31 magnetic resonance spectroscopy. J Affect Disord. 1994;31:125–133. doi: 10.1016/0165-0327(94)90116-3. [DOI] [PubMed] [Google Scholar]
- Kim GD, et al. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. Embo J. 2002;21:4183–4195. doi: 10.1093/emboj/cdf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirillov A, et al. A role for nuclear NF-kappaB in B-cell-specific demethylation of the Igkappa locus. Nat Genet. 1996;13:435–441. doi: 10.1038/ng0895-435. [DOI] [PubMed] [Google Scholar]
- Kovacheva VP, et al. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J Biol Chem. 2007;282:31777–31788. doi: 10.1074/jbc.M705539200. [DOI] [PubMed] [Google Scholar]
- Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Laplante P, et al. Serotonin regulates hippocampal glucocorticoid receptor expression via a 5-HT7 receptor. Brain Res Dev Brain Res. 2002;139:199–203. doi: 10.1016/s0165-3806(02)00550-3. [DOI] [PubMed] [Google Scholar]
- Lasalle JM. The odyssey of MeCP2 and parental imprinting. Epigenetics. 2007;2:5–10. doi: 10.4161/epi.2.1.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, et al. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Levenson JM, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Liang G, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22:480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, et al. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic–pituitary–adrenal responses to stress. Science. 1997;277:1659–1662. doi: 10.1126/science.277.5332.1659. [DOI] [PubMed] [Google Scholar]
- Lucas A. Programming by early nutrition: an experimental approach. J Nutr. 1998;128:401S–406S. doi: 10.1093/jn/128.2.401S. [DOI] [PubMed] [Google Scholar]
- McCormick JA, et al. 5¢-heterogeneity of glucocorticoid receptor messenger RNA is tissue specific: differential regulation of variant transcripts by early-life events. Mol Endocrinol. 2000;14:506–517. doi: 10.1210/mend.14.4.0438. [DOI] [PubMed] [Google Scholar]
- McGowan PO, Kato T. Epigenetics in mood disorders. Environ Health Prev Med. 2008;13:16–24. doi: 10.1007/s12199-007-0002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, et al. Promoter-wide hypermethylation of the ribosomal RNA gene promoter in the suicide brain. PLoS ONE. 2008;3:e2085. doi: 10.1371/journal.pone.0002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, et al. Thyroid hormones influence the development of hippocampal glucocorticoid receptors in the rat: a mechanism for the effects of postnatal handling on the development of the adrenocortical stress response. Neuroendocrinology. 1987;45:278–283. doi: 10.1159/000124741. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, et al. Postnatal handling increases the expression of cAMP-inducible transcription factors in the rat hippocampus: the effects of thyroid hormones and serotonin. J Neurosci. 2000;20:3926–3935. doi: 10.1523/JNEUROSCI.20-10-03926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005a;7:103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci. 2005b;28:456–463. doi: 10.1016/j.tins.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Meck WH, et al. Organizational changes in cholinergic activity and enhanced visuospatial memory as a function of choline administered prenatally or postnatally or both. Behav Neurosci. 1989;103:1234–1241. doi: 10.1037//0735-7044.103.6.1234. [DOI] [PubMed] [Google Scholar]
- Meck WH, Williams CL. Metabolic imprinting of choline by its availability during gestation: implications for memory and attentional processing across the lifespan. Neurosci Biobehav Rev. 2003;27:385–399. doi: 10.1016/s0149-7634(03)00069-1. [DOI] [PubMed] [Google Scholar]
- Metzger E, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Milutinovic S, et al. Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis. 2007;28:560–571. doi: 10.1093/carcin/bgl167. [DOI] [PubMed] [Google Scholar]
- Mudd SH, Cantoni GL. Activation of methionine for transmethylation. III The methionine-activating enzyme of Bakers’ yeast. J Biol Chem. 1958;231:481–492. [PubMed] [Google Scholar]
- Muntjewerff JW, Blom HJ. Aberrant folate status in schizophrenic patients: what is the evidence? Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1133–1139. doi: 10.1016/j.pnpbp.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Nagarajan RP, et al. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:172–182. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, et al. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Ng HH, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23:58–61. doi: 10.1038/12659. [DOI] [PubMed] [Google Scholar]
- Niculescu MD, et al. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. Faseb J. 2006;20:43–49. doi: 10.1096/fj.05-4707com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko VV, et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Ou JN, et al. Histone deacetylase inhibitor Trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem Pharmacol. 2007;73:1297–1307. doi: 10.1016/j.bcp.2006.12.032. [DOI] [PubMed] [Google Scholar]
- Papakostas GI, et al. S-adenosyl-methionine in depression: a comprehensive review of the literature. Curr Psychiatry Rep. 2003;5:460–466. doi: 10.1007/s11920-003-0085-2. [DOI] [PubMed] [Google Scholar]
- Pascale R, et al. Reversal by 5-azacytidine of the S-adenosyl-L-methionine-induced inhibition of the development of putative preneoplastic foci in rat liver carcinogenesis. Cancer Lett. 1991;56:259–265. doi: 10.1016/0304-3835(91)90011-6. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Picketts DJ, et al. ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum Mol Genet. 1996;5:1899–1907. doi: 10.1093/hmg/5.12.1899. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, et al. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat Res. 2006;593:80–87. doi: 10.1016/j.mrfmmm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Purucker M, et al. Structure and function of the enhancer 3¢ to the human A gamma globin gene. Nucleic Acids Res. 1990;18:7407–7415. doi: 10.1093/nar/18.24.7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A. CpG methylation, chromatin structure and gene silencing—a three-way connection. Embo J. 1998;17:4905–4908. doi: 10.1093/emboj/17.17.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A, Cedar H. Distribution of 5-methylcytosine in chromatin. Proc Natl Acad Sci U S A. 1977;74:2725–2728. doi: 10.1073/pnas.74.7.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A, Riggs AD. DNA methylation and gene function. Science. 1980;210:604–610. doi: 10.1126/science.6254144. [DOI] [PubMed] [Google Scholar]
- Razin A, Shemer R. DNA methylation in early development. Hum Mol Genet. 1995;4:1751–1755. doi: 10.1093/hmg/4.suppl_1.1751. [DOI] [PubMed] [Google Scholar]
- Razin A, Szyf M. DNA methylation patterns. Formation Funct Biochim Biophys Acta. 1984;782:331–342. doi: 10.1016/0167-4781(84)90043-5. [DOI] [PubMed] [Google Scholar]
- Razin A, et al. Variations in DNA methylation during mouse cell differentiation in vivo and in vitro. Proc Natl Acad Sci U S A. 1984;81:2275–2279. doi: 10.1073/pnas.81.8.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SA. Diet and DNA methylation interactions in cancer prevention. Ann N Y Acad Sci. 2003;983:197–207. doi: 10.1111/j.1749-6632.2003.tb05974.x. [DOI] [PubMed] [Google Scholar]
- Ross SA, Milner JA. Epigenetic modulation and cancer: effect of metabolic syndrome? Am J Clin Nutr. 2007;86:s872–s877. doi: 10.1093/ajcn/86.3.872S. [DOI] [PubMed] [Google Scholar]
- Rountree MR, et al. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- Saito Y, Jones PA. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle. 2006;5:2220–2222. doi: 10.4161/cc.5.19.3340. [DOI] [PubMed] [Google Scholar]
- Schlesinger Y, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- Schroeder FA, et al. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Sharma RP, et al. Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: preliminary results from a clinical population. Schizophr Res. 2006;88:227–231. doi: 10.1016/j.schres.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Shi Y, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A. 2003;100:13225–13230. doi: 10.1073/pnas.1735528100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Silveri MM, et al. S-adenosyl-L-methionine: effects on brain bioenergetic status and transverse relaxation time in healthy subjects. Biol Psychiatry. 2003;54:833–839. doi: 10.1016/s0006-3223(03)00064-7. [DOI] [PubMed] [Google Scholar]
- Simmons RA. Developmental origins of beta-cell failure in type 2 diabetes: the role of epigenetic mechanisms. Pediatr Res. 2007;61:64R–67R. doi: 10.1203/pdr.0b013e3180457623. [DOI] [PubMed] [Google Scholar]
- Simonini MV, et al. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc Natl Acad Sci U S A. 2006;103:1587–1592. doi: 10.1073/pnas.0510341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slyshenkov VS, et al. Protective role of L-methionine against free radical damage of rat brain synaptosomes. Acta Biochim Pol. 2002;49:907–916. [PubMed] [Google Scholar]
- Szyf M. Towards a pharmacology of DNA methylation. Trends Pharmacol Sci. 2001;22:350–354. doi: 10.1016/s0165-6147(00)01713-2. [DOI] [PubMed] [Google Scholar]
- Szyf M. The role of DNA hypermethylation and demethylation in cancer and cancer therapy. Curr Oncol. 2008;15:72–75. doi: 10.3747/co.v15i2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremolizzo L, et al. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci U S A. 2002;99:17095–17100. doi: 10.1073/pnas.262658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, et al. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Uchida C, et al. The role of Sp1 and AP-2 in basal and protein kinase A-induced expression of mitochondrial serine: pyruvate aminotransferase in hepatocytes. J Biol Chem. 2002;277:39082–39092. doi: 10.1074/jbc.M201380200. [DOI] [PubMed] [Google Scholar]
- Van den Veyver IB. Genetic effects of methylation diets. Annu Rev Nutr. 2002;22:255–282. doi: 10.1146/annurev.nutr.22.010402.102932. [DOI] [PubMed] [Google Scholar]
- Varga-Weisz PD, Becker PB. Regulation of higher-order chromatin structures by nucleosome-remodelling factors. Curr Opin Genet Dev. 2006;16:151–156. doi: 10.1016/j.gde.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Veldic M, et al. Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr Res. 2007;91:51–61. doi: 10.1016/j.schres.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vire E, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- Wade PA, et al. Histone acetylation: chromatin in action. Trends Biochem Sci. 1997;22:128–132. doi: 10.1016/s0968-0004(97)01016-5. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Weaver IC, et al. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci. 2005;25:11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, et al. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, et al. The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J Neurosci. 2007;27:1756–1768. doi: 10.1523/JNEUROSCI.4164-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff GL, et al. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. Faseb J. 1998;12:949–957. [PubMed] [Google Scholar]
- Wolffe AP, et al. DNA demethylation. Proc Natl Acad Sci U S A. 1999;96:5894–5896. doi: 10.1073/pnas.96.11.5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JC, Santi DV. On the mechanism and inhibition of DNA cytosine methyltransferases. Prog Clin Biol Res. 1985;198:119–129. [PubMed] [Google Scholar]
- Xue Y, et al. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- Yoshida M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- Yoshimura R, et al. Valproic acid improves psychotic agitation without influencing plasma risperidone levels in schizophrenic patients. Pharmacopsychiatry. 2007;40:9–13. doi: 10.1055/s-2007-958521. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. Coactivating factors p300 and CBP are transcriptionally crossregulated by Egr1 in prostate cells, leading to divergent responses. Mol Cell. 2004;15:83–94. doi: 10.1016/j.molcel.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Zhu B, et al. 5-methylcytosine-DNA glycosylase activity is present in a cloned G/T mismatch DNA glycosylase associated with the chicken embryo DNA demethylation complex. Proc Natl Acad Sci U S A. 2000;97:5135–5139. doi: 10.1073/pnas.100107597. [DOI] [PMC free article] [PubMed] [Google Scholar]