

Abstract
Inhibition of Bcr-Abl kinase activity by imatinib for the treatment of chronic myeloid leukemia (CML) currently serves as the paradigm for targeting dominant oncogenes with small molecules. We recently reported the discovery of GNF-2 (1) and GNF-5 (2) as selective non-ATP competitive inhibitors of cellular Bcr-Abl kinase activity that target the myristate binding site. Here, we used cell-based structure−activity relationships to guide the optimization and diversification of ligands that are capable of binding to the myristate binding site and rationalize the findings based upon an Abl−compound 1 cocrystal. We elucidate the structure−activity relationships required to obtain potent antiproliferative activity against Bcr-Abl transformed cells and report the discovery of new compounds (5g, 5h, 6a, 14d, and 21j-I) that display improved potency or pharmacological properties. This work demonstrates that a variety of structures can effectively target the Bcr-Abl myristate binding site and provides new leads for developing drugs that can target this binding site.
Introduction
Chronic myelogenous leukemia (CMLa) is a hematological disorder caused by a chromosomal rearrangement that generates a fusion protein, Bcr-Abl, with deregulated tyrosine kinase activity.(1) Although clinical remission has been achieved with the ATP-binding site targeting drug imatinib, many patients relapse because of emergence of clones expressing inhibitor-resistant forms of Bcr-Abl.(2) To address these relapses, two more potent ATP-site directed agents, nilotinib3,4 and dasatinib,5,6 have recently received clinical approval. Although both compounds inhibit most of the mutations that induce resistance to imatinib, neither compound is capable of inhibiting the “gatekeeper” T315I mutation, which is situated in the middle of the ATP-binding cleft.(7)
In an effort to find new pharmacological modalities of Bcr-Abl inhibition, we performed a differential cytotoxicity screen that resulted in the identification of 1, a non-ATP competitive inhibitor of cellular Bcr-Abl activity.(8) A major advantage of non-ATP competitive kinase inhibitors is that they can be highly selective for a particular kinase because they can exploit nonconserved kinase regulatory mechanisms. Indeed, 1 demonstrated exclusive cellular activity against Bcr-Abl transformed cells (EC50 = 140 nM) and did not inhibit the activity of 40 other tyrosine kinases in cellular assays or the biochemical activity of a panel of 80 diverse kinases.(8) Compound 1 was demonstrated to bind to the myristate binding site located near the C-terminus of the Abl kinase domain using protein crystallography and NMR spectroscopy.(9) Compound 2,(9) the hydroxylethylamide analogue of 1, with improved pharmacological properties is efficacious in Bcr-Abl-dependent xenograft and bone marrow transplantation models against wild-type Bcr-Abl and is capable of inhibiting T315I Bcr-Abl when used in combination with the ATP competitive inhibitor nilotinib. Binding of 1 or 2 to the myristate binding site induces changes to the conformational dynamics of the ATP-site as revealed by hydrogen−deuterium exchange studies, but the precise conformation induced by myristate-site binding remains to be elucidated.
Compound 1 was developed by iterative structure−activity guided optimization using Bcr-Abl transformed Ba/F3 cells. The original “hit” compound, GNF-1(4a),(8) was discovered by screening a combinatorial library of 50 000 heterocycles originally designed to target the ATP binding site. The library was synthesized on solid phase using the IRORI nanokan system.(10) The screen sought compounds that were differentially cytotoxic between murine 32D cells versus 32D cells transformed with Bcr-Abl. Compounds 1 and 4a have a 4,6-disubstituted pyrimidine core structure that has received some attention as an ATP-binding site directed scaffold11,12 but has not been as extensively investigated as the corresponding 2,4-disubstituted pyrimidines.13,14 Here, we describe the structure−activity relationships for the 4,6-disubsituted pyrimidine class of Bcr-Abl myristate binding site-targeted ligands. Using established medicinal chemistry approaches such as introduction of ring constraints to reduce entropic penalties upon ligand binding, we have successfully discovered additional heterocyclic ring systems such as thieno[2,3-d]pyrimidines, pyrrolo[2,3-d]pyrimidines, and pyrazolo[3,4-d]pyrimidines that can inhibit cellular Bcr-Abl activity by binding to the myristate-binding site.
Results and Discussion
Synthesis
4,6-Disubstituted pyrimidines were prepared from 4,6-dichloropyrimidine by base-promoted amination followed by palladium mediated coupling with boronic acids or esters, anilines, or phenols (Scheme 1).(15)
Scheme 1.

Reagents and conditions: (a) 4-trifluoromethoxylaniline (1.1 equiv), DIEA (1.1 equiv), EtOH, reflux; (b) primary or secondary amine, DIEA, 2-BuOH, reflux; (c) substituted phenylboronic acid (1.0 equiv), Pd(PPh3)4 (5%), Na2CO3 (4.0 equiv), CH3CN/H2O (v/v = 1/1), reflux; (d) amine, HATU, DIEA, DMF, room temp; (e) substituted pyrazoleboronic acid (1.0 equiv), Pd(PPh3)4 (5%), Na2CO3 (4.0 equiv), CH3CN/H2O (v/v = 1/1), reflux; (f) Cs2CO3, R6Br (1.1 equiv); (g) heteroaromatic boronic acid (1.0 equiv), Pd(PPh3)4 (5%), Na2CO3 (4.0 equiv), CH3CN/H2O (v/v = 1/1), reflux; (h) 2-morpholinoethanamine (1.0 equiv), DIEA (1.5 equiv), EtOH, reflux; (i) 4-(trifluoromethoxy)phenol, NaH, dioxane, room temp to reflux.
Further elaboration of the pyrimidine 6-substituent was accomplished by alkylation, sulfonamide or amide-bond formation. 2,4-Disubstituted pyrimidines, 2,6-disubstituted pyrazines, 2,4-disubstituted triazines, and 3,6-disubstituted pyridazines were prepared using methodology similar to that of 4,6-disubstituted pyrimidines preparation as described in the Supporting Information in order to explore similar core structures to the 4,6-disubstituted pyrimidines. Thieno[2,3-d]pyrimidines substituted at the 6-position were synthesized in three steps by condensation of methyl 2-mercaptoacetate with 4-chloro-6-(4-(trifluoromethoxy)phenylamino)pyrimidine-5-carbaldehyde under basic conditions, followed sequentially by ester hydrolysis and amide bond formation using HATU (Scheme 2).
Scheme 2.

Reagents and conditions: (a) methyl 2-mercaptoacetate, K2CO3, DMF, room temp to 90 °C; (b) LiAlH4, 0 °C ; (c) LiOH/THF; (d) primary or secondary amine, HATU, DMF, room temp.
Pyrrolo[2,3-d]pyrimidines were prepared by N9 alkylation on 4-chloro-7H-pyrrolo[2,3-d]pyrimidine with primary iodo- or bromoalkanes and sodium hydride, followed by thermal substitution of the 6-chloro group with the corresponding anilines. Further modification of the N9-substituent was accomplished by ester hydrolysis and amide bond formation using HATU (Scheme 3).
Scheme 3.

Reagents and conditions: (a) primary iodo- or bromoalkane, NaH, DMF, 0 °C to room temp; (b) 4-(trifluoromethoxy)aniline, 2-BuOH, 120 °C; (c) methyl 3-bromopropanoate, NaH, THF; (d) 4-(trifluoromethoxy)aniline, 2-BuOH, 120 °C; (e) Cs2CO3, THF/H2O; (f) R2NH2, HATU, DIEA, DMF, room temp.
Pyrazolo[3,4-d]pyrimidines were prepared through two different synthetic routes: either 4-chloro-6-(4-(trifluoromethoxy)phenylamino)pyrimidine-5-carbaldehyde was condensed with an appropriate alkylhydrazine or this condensation was performed using hydrazine followed by N1 alkylation with a primary iodo- or bromoalkane under cesium or potassium carbonate (Scheme 4). The regioisomeric products were subsequently separated by mass-triggered high-pressure liquid chromatography.
Scheme 4.

Reagents and conditions: (a) RNHNH2, Na2CO3, 2-BuOH, 60 °C; (b) NH2NH2, THF; (c) primary bromo- or chloroalkane, Cs2CO3 or K2CO3, DMF.
Bcr-Abl Structure−Activity Relationships for 4,6-Disubstituted Pyrimidines
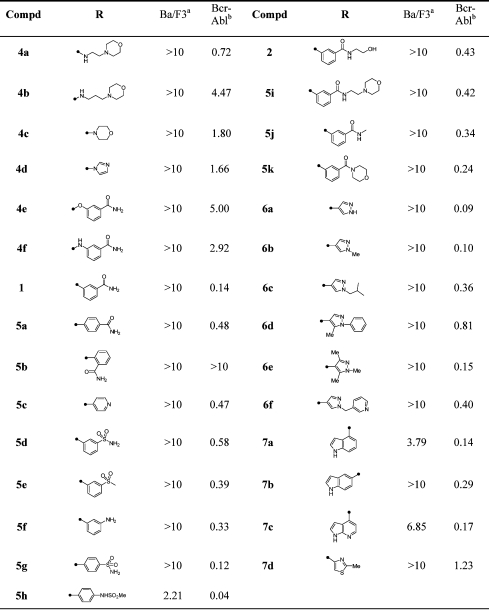
Following the identification of 4a as a screening “hit” that specifically inhibited Bcr-Abl transformed cells with an EC50 of 1 μM, a systematic evaluation of the structural features necessary to impart activity as a cellular Bcr-Abl inhibitor was investigated. We initiated our investigation by replacing the morpholinoethylamino substituent located at the pyrimidine C4-position with a range of functionalities including primary and secondary amines, anilines, phenols, and various aromatic and heteroaromatic substituents (Table 1). Compounds containing a direct aromatic linkage such as 1, 5a, and 5c displayed the most potent activity, while alkylamine (4b and 4c) derivatized compounds displayed reduced activity. The advantage of direct aromatic linkage at the pyrimidine C4-position (1) is evident by the loss of activity observed for the corresponding ether (4e) and amine-linked (4f) derivatives. In the aryl series, optimal activity was achieved with hydrogen bond donating or accepting functionalities (CONR1R2, SO2CH3, SO2NHR, NH2) at the 3- or 4-position of the phenyl ring as shown by 1, 2, 5a, and 5d−h. Substitution with a 2-carboxamide (5b) resulted in an inactive compound presumably as a result of steric hindrance or due to unfavorable torsional angle between the pyrimidine and 4-phenyl substituent. We next investigated additional analogues of 1 focusing on replacements of the C4-benzamide substitutent. Replacements of the amide with sulfone (5e), primary amine (5f), sulfonamide (5g), and homologated amides (5i−k) all resulted in compounds with cellular potency comparable to that of 1 and 5c (Table 1). These results suggested that this position maybe faces the solvent-exposed region of the enzyme and would provide a good location for modulating the physical and pharmacological properties of the compound. Indeed the hydroxyethylamide analogue 2 displayed similar cellular Bcr-Abl inhibitory activity as well as favorable pharmacokinetic properties and was chosen for evaluation in murine leukemia models.(9)
Table 1. SAR of 4,6-Disubstituted Pyrimidine.

 |
Cytotoxicity (EC50, μM) on parental Ba/F3.
Antiproliferative activity (EC50, μM) on wtBcr-Abl-Ba/F3.
We also introduced sulfonamide and “reverse” sulfonamide at the para position of the C6-phenyl. The unsubstituted sulfonamide 5g (EC50 = 120 nM) and reverse methylsulfonamide 5h (EC50 = 40 nM) were obtained with improved inhibitory activity. We installed a variety of heterocyclic ring systems at C4 and discovered that unsubstituted (6a) and alkyl substituted pyrazoles (6b, 6c, and 6e) displayed superior potency compared to 4a (Table 1). Introduction of 5,6-fused aromatic systems to the pyrimidine 4-position was also tolerated with potent activity being observed for indoles attached via the indole 4- or 5-position (7a, 7b) and azaindoles attached via the azaindole 4-position (7c).
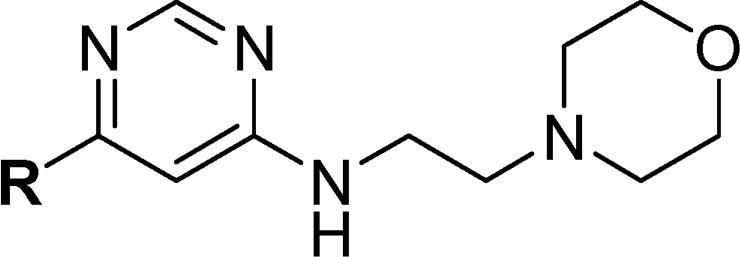
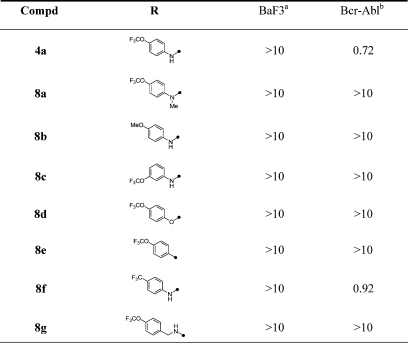
In sharp contrast to the range of functionality that was tolerated at the pyrimidine C4-position, very few replacements to the 4-trifluoromethoxyaniline were tolerated at the pyrimidine C6-position (Table 2). For example, replacement of the aniline NH with an N-methyl substitution (8a) or oxygen (8d) or direct phenyl ring connection (8e) resulted in a complete loss of cellular activity, suggesting that this group maybe responsible for a critical contact with the enzyme. N-Methyl substitution was chosen as the modification to prepare the negative control for both the affinity chromatography and NMR-binding studies.8,9 Similarly, regioisomeric m-trifluoromethoxyanilines (8c) or p-trifluoromethoxylbenzylamino-substituted compounds (8g) were completely inactive. To our surprise, replacement of the trifluoromethyl group with a methyl group also resulted in a complete loss of activity (8b). The only substitution that was tolerated at C6 was the conservative replacement of the trifluoromethoxy with a trifluoromethyl group (8f).
Table 2. SAR of Pyrimidine C6-position.
 |
Cytotoxicity (EC50, μM) on parental Ba/F3.
Antiproliferative activity (EC50, μM) on wtBcr-Abl-Ba/F3.
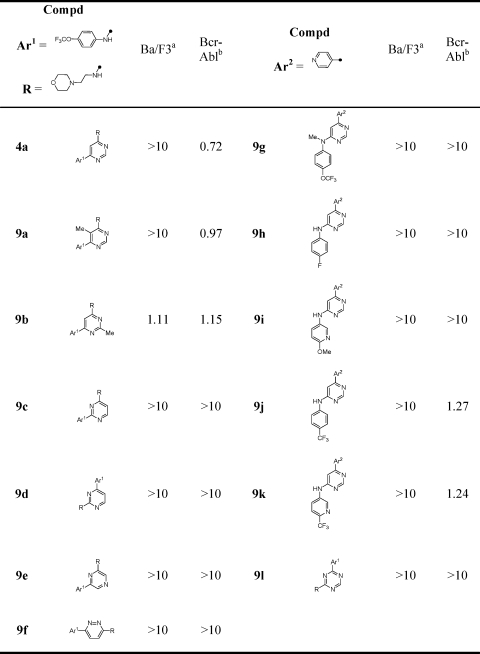
We next investigated the consequences of varying the positions of the nitrogens located on the central pyrimidine core (Table 3). Here, we discovered a strict requirement for the 4,6-pyrimidine; both of the 2,4-pyrimidine regioisomers (9c and 9d), pyrazine (9e), pyridazine (9f), and triazine (9l) were inactive. This result suggested that in the context of compound 4a, the 4,6-substitution pattern is strictly required to maintain cellular Bcr-Abl inhibitory activity.
Table 3. SAR of Central Pyrimidine Core.
 |
Cytotoxicity (EC50, μM) on parental Ba/F3.
Antiproliferative activity (EC50, μM) on wtBcr-Abl-Ba/F3.
We next focused our attention on using structure−activity relationships to deduce the bioactive conformation of improved leads such as 1. One interesting observation was that the 4,6-disubstitution requirement for the central pyrimidine was different from that of ATP-competitive kinase inhibitors where in general the 2,4-pyrimidine is favored as a motif that binds to the kinase hinge region. This is believed to be a consequence of 2,4-pyrimidines being able to assume the cis conformation with respect to the NH−C2 bond that is required to form a bidentate H-bond to the kinase hinge region. For example, the classical ATP-competitive 2,4-pyrimidine CDK inhibitors such as 3-[[4-[2-[(3-chlorophenyl)amino]pyrimidin-4-yl]pyridin-2-yl]amino]propan-1-ol (CGP60474) bind to CDK2 hinge residues using a cis conformation.(16) Considering that 1 was not able to function as an ATP-competitive Bcr-Abl inhibitor, this led us to speculate that the requirement for the 4,6-pyrimidine scaffold reflected a need for these compounds to adopt a “trans” conformation to be active as cellular Bcr-Abl inhibitors. To investigate this hypothesis, we created a series of compounds that would be structurally similar but exhibit different preferences for cis or trans conformations due to potential steric interactions between ortho position of the p-trifluoromethoxyaniline ring and the pyrimidine C5 in the cis-conformation (Figure 1). Biological evaluation of the resulting compounds demonstrated that only the inhibitors with a preference for the trans-conformation were able to inhibit cellular Bcr-Abl activity.
Figure 1.

SAR for analogues of 4a indicates that a trans conformation is favored.
Reasoning that the enhanced cellular potency of compound 5c might allow us to uncover subtleties regarding SAR at C6 that would not be apparent in the less potent 4a, we prepared further analogues at this position (Table 3). We again observed that methylation of the aniline NH (9g) resulted in a complete loss of cellular activity. Similar to 4a, substitution of the trifluoromethoxy group with fluoro (9h) and methoxy (9i) groups resulted in a complete loss in activity. Only removal of the oxygen of the trifluoromethoxy group (9j) and introduction of a meta nitrogen (9k) resulted in inhibitors that retained some activity.
Structural Biology of the Myristate Binding Site
The myristate binding site is a cylindrical shaped cavity located at the C-terminus of the Abl kinase domain. Before the availability of the 1−Abl cocrystal (PDB code 3K5V),(9) we initially used the docking program Glide to compute potential binding conformations of 1 to Abl.(17) The lowest energy pose places 1 in an extended trans conformation with the 4-trifluoromethoxyaniline pointed toward the bottom of the pocket. With the exception of the positioning of the benzamide, the Glide generated binding conformation for 1 was the same as in the subsequently determined crystal structure and could be used to rationalize the aforementioned structure−activity relationships. Specifically it explains the strict requirement for 4-substitution of the trifluoromethoxy group and the preference for 4,6- versus 2,4-disubstitution which favors the extended trans-conformation of the pyrimidine and the tolerance for diverse substitution at the pyrimidine 6-positions which point out toward the solvent exposed region. In the cocrystal (Figure 2), a water molecule forms a hydrogen bond bridge between the aniline NH and the main chain carbonyls of A433 (Abl 1a numbering) and E462 in the myristate binding site. This observation can explain the loss of potency observed when the aniline NH is replaced with oxygen (8d) or an N-methyl group (8a). Residues making contact with 1 at the base of the pocket are L341 and A344 from αE, I432 from αF, V468 from αH, F493 from αI, and I502 from αI′. The surface in the central part of the pocket is formed by A337 from αE, C464 and P465 from the start of αH, A433 from αF, and V506 from αI′. The fit in this region is quite snug, explaining the intolerance for substitution to the trifluoromethylphenyl ring. There are fewer interactions at the mouth of the pocket (Y435 from αF, E462 from the loop before αH, and L510 at the end of αI′), which is caused by the flexibility and hence slightly weaker electron density of the benzamide part of 1. This explains the diverse range of functionalities that can be incorporated into this position. A mutagenesis screen resulted in the identification of five mutations around the myristate binding pocket (C464Y, P465S, V506L, F497L, E505K) that cause resistance to 1 and 2.(9) On the basis of the crystal structure, these would be expected to cause resistance because of direct steric hindrance to binding or more indirectly because of the position of helix I.
Figure 2.

Cocrystal structure of 1 with Abl kinase (PDB entry 3K5V). (A) Binding conformation of 1 (cyan) in the myristate binding site of Abl (side chains from helix αE are green, αF orange, αI dark orange, α′I red). Hydrogen bonds to a water molecule (red sphere) are indicated by dashed yellow lines. (B) Solvent accessible surface of the myristate binding pocket colored as in panel A viewed from the mouth of the binding pocket.
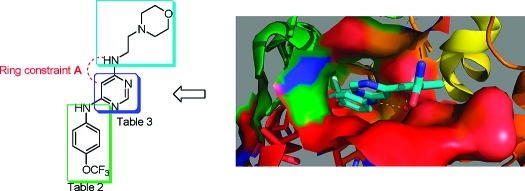
Ring Constrained Analogues of 1
We were interested in further optimizing the cellular potency of the 1 class of Bcr-Abl kinase inhibitors and exploring their potential to target a variety of Bcr-Abl mutants that are resistant to ATP-competitive inhibitors such as imatinib, nilotinib, and dasatinib. For example, 1 is active on wild-type Bcr-Abl, E255V, and Y253H mutants but is inactive on the T315I “gatekeeper” mutant which has proven to be the most recalcitrant to inhibit. To create further analogues of 1, we designed conformationally constrained compounds by introducing fused ring systems (Figure 3). We initially explored the potential to fuse a five-membered ring between the pyrimidine 5 and 6 positions to create four classes of 6,5-fused heterocyclic ring systems: 6-substituted thieno[2,3-d]pyrimidines, 7-substituted pyrrolo[2,3-d]pyrimidines, and 1- and 2-substituted pyrazolo[3,4-d]pyrimidines (Figure 3, constraint A). Collectively these scaffolds provide the possibility of directing one or two substituents from various positions of the five-membered ring system.
Figure 3.

Rationale for the evolution of structures from screening hit 4a to 1, 2, and a variety of 6,5-fused heterocyclic ring systems.
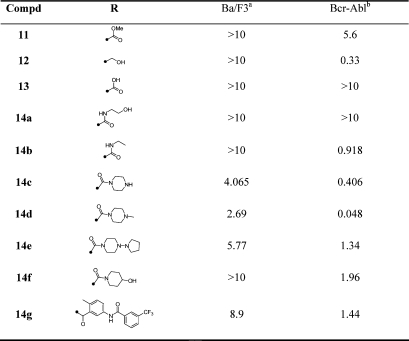
Substitution of the 6-position of the thieno[2,3-d]pyrimidines scaffold with a variety of carboxamides (Table 4) resulted in the identification of several compounds with EC50 values of approximate 1 μM for the inhibition of cellular Bcr-Abl activity. The optimal compound in this series was obtained by introduction of an N-methylpiperazine as exemplified by compound 14d, which possessed an EC50 of 48 nM (Table 4). Interestingly this compound was approximately 10-fold more potent than the compound lacking N-methylation (14c).
Table 4. SAR of the Thieno[2,3-d]pyrimidines.

 |
Cytotoxicity (EC50, μM) on parental Ba/F3.
Antiproliferative activity (EC50, μM) on wtBcr-Abl-Ba/F3.
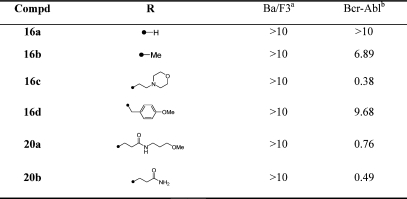
Preparation of six 7-substituted pyrrolo[2,3-d]pyrimidines resulted in the optimal activity being identified from morpholinoethyl (16c) substituent (Table 5). Inhibitor 16c, which can be viewed as the ring fused analogue of 4a, is approximately equipotent to 4a.
Table 5. SAR of 7-Substituted Pyrrolo[2,3-d]pyrimidines.

 |
Cytotoxicity (EC50, μM) on parental Ba/F3.
Antiproliferative activity (EC50, μM) on wtBcr-Abl-Ba/F3.
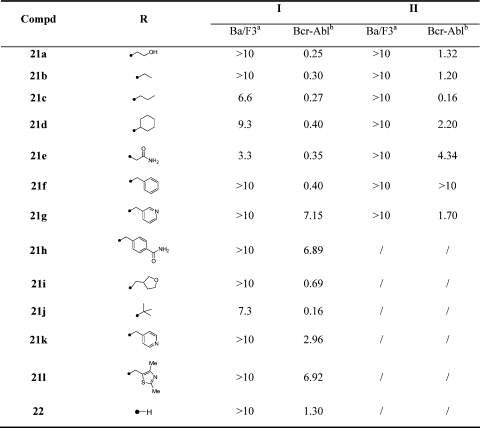
We prepared a 20-membered focused library for 1- and 2-substituted pyrazolo[3,4-d]pyrimidines (Table 6). The potency of the 1- or 2-substituted regioisomers was typically similar with the biggest difference being observed for the benzyl substituted compound 21f where the 2-substituted compound is completely inactive (Table 6). A variety of compounds were discovered that possessed potency comparable to that of 1 such as alkyl substituted inhibitors 21a−f. The most potent inhibitor in this series was tert-butyl substituted compound 21j-I which possessed an EC50 of 160 nM, even though compound 21j-I inhibited the growth of untransformed Ba/F3 cells to some degree (EC50 = 7.3 μM), suggesting additional cellular targets for this compound class.
Table 6. SAR of 1- and 2-Substituted Pyrazolo[3,4-d]pyrimidines.

 |
Cytotoxicity (EC50, μM) on parental Ba/F3.
Antiproliferative activity (EC50, μM) on wtBcr-Abl-Ba/F3.
Biological Profiling of Select 1 Analogues
We selected eight compounds that possessed optimal Bcr-Abl potency and that were representative of the different structure classes and tested them in a panel of biochemical and cellular assays to assess their mechanism of action relative to 1. Because several of the compounds are structurally similar to ATP-competitive kinase inhibitors, we sought to investigate their potential to interact with diverse kinase ATP-binding sites. This was assessed using KINOMEscan (Ambit Biosciences, San Diego, CA), a high-throughput method for screening kinase inhibitors against a panel of 353 kinases.(18) Compounds were screened at 10 μM; none of the compounds displayed the ability to target the ATP-binding site of any kinases in the panel. In order to demonstrate that these new compounds indeed function as myristate-site ligands, we tested their ability to compete for binding to Abl kinase domain with a fluorescein-linked 1 analogue using a fluorescence polarization assay (Figure 4).(19) By performing this assay at a range of inhibitor concentrations, we could measure a dissociation constant (Kd) for each compound (Figure 4B). We demonstrated that all compounds were competitive with respect to 1-FITC for binding to the myristate binding site. In addition, there was a good correlation between the EC50 for inhibition of cellular Bcr-Abl activity and Kd determined using the fluorescence polarization assay (Figure 4C). This provides evidence that changes in cellular potency that we observe for these analogues are likely to reflect the affinity with which they bind to the myristate binding site rather than differences in cell penetration and accumulation. All analogues were also unable to inhibit the proliferation of two point mutants located in the myristate binding site (A337N and A344L) that we had previously(8) demonstrated induced resistance to 1. This provides evidence that the cellular site of action of these analogues is also the Bcr-Abl myristate binding site. Cumulatively, these results are consistent with these new compounds exploiting the same mechanism of action as 1 to inhibit Bcr-Abl dependent proliferation.
Figure 4.

Dissociation constants (Kd) for a series of 1 analogues correlate with EC50 for inhibition of cellular Bcr-Abl activity. (A) Chemical structure of 1−FITC tracer. (B) Dissociation constants (Kd) were determined using a fluorescence polarization competition binding assay to Abl kinase domain with 1 analogues and fluorescein-linked 1 tracer. (C) Graph showing the correlation between compound dissociation constant (Kd) and its cellular EC50 value for Bcr-Abl inhibition.
To ascertain the ability of these compounds to inhibit clinically relevant Bcr-Abl mutants, they were tested against T315I, E255K, and E505K mutants (Table 7).(20)
Table 7. Cellular Activity of Select 1 Analogues against Some Clinically Relevant Bcr-Abl Mutantsa.
| cellular EC50 (μM) |
|||||
|---|---|---|---|---|---|
| compd | Wt-Bcr-Abl | T315I | E255K | E505K | Wt-Ba/F3 |
| 1 | 0.14 | >10 | 0.60 | >10 | >10 |
| 2 | 0.43 | >10 | 0.58 | >10 | >10 |
| 5g | 0.12 | >10 | 0.31 | >10 | >10 |
| 5h | 0.04 | >10 | 0.36 | >10 | >10 |
| 6a | 0.09 | >10 | 0.52 | >10 | >10 |
| 14d | 0.05 | >10 | 1.33 | >10 | >10 |
| 16c | 0.38 | >10 | 1.22 | >10 | >10 |
| 21a-I | 0.25 | >10 | 1.61 | >10 | >10 |
| 21j-I | 0.16 | >10 | 0.50 | >10 | >10 |
Antiproliferative activity (EC50, μM) on select cell lines.
The results revealed that most of the compounds have submicromolar potency against wild-type and E255K with the exception of 14d, 16c, and 21a-I. As expected, all the compounds are inactive on gate-keeper mutant T315I and myristate bind site mutant E505K. As we have previously reported that 2 is capable of synergizing with ATP-competitive inhibitors such as nilotinib to inhibit the proliferation of T315I Bcr-Abl, we tested the potential of selected compounds to exhibit synergistic inhibition with the type-I ATP competitive inhibitor dasatinib (Table 8).(9) Interestingly the compounds exhibited considerable differences in their ability to combine with dasatinib to inhibit T315I Bcr-Abl-dependent proliferation. In addition, the calculated combination indices (CI)(21) did not correlate with the ability of the compounds to inhibit wild-type Bcr-Abl as single agents. Understanding the mechanistic basis for this will require a structure of Abl ligated with both dasatinib and a myristate-site ligand.
Table 8. Synergistic Effect of Select 1 Analogues with Dasatinib against T315I Mutant.
| CI value at |
|||||
|---|---|---|---|---|---|
| compd (ratio | ED50 | ED75 | ED90 | Dm | r |
| dasatinib + 5g (1:1) | 0.4973 | 0.27007 | 0.48539 | 0.99256 | 0.94707 |
| dasatinib + 5h (1:1) | 0.61781 | 0.38123 | 0.25814 | 0.90849 | 0.55459 |
| dasatinib + 6a (1:1) | 0.54179 | 0.71033 | 0.95607 | 0.77202 | 0.94716 |
| dasatinib + 14c (1:1) | 0.75652 | 0.70471 | 0.65645 | 0.41015 | 0.90394 |
| dasatinib + 14d (1:1) | 0.30119 | 0.13126 | 0.11157 | 0.96888 | 0.50907 |
| dasatinib + 20a (1:1) | 0.40752 | 0.35515 | 0.47618 | 0.96398 | 2.23613 |
| dasatinib + 21a-I (1:1) | 0.70277 | 0.36185 | 0.48377 | 1.30175 | 0.76679 |
Conclusions
We have presented a comprehensive exploration of Bcr-Abl myristate targeted ligands derived from 1/4a lead structures. We demonstrate that the 4-trifluoromethoxyaniline is an obligate structural feature for achieving submicromolar potency as a cellular Bcr-Abl inhibitor but that the pyrimidine ring can be elaborated to include a variety of other heterocyclic ring systems. We identify several analogues with 5- to 10-fold improved potency against wild-type and some Bcr-Abl mutants such as E255K relative to 1. The most potent myristate-site targeted inhibitors we have discovered to date are sulfonamide 5g (EC50 = 120 nM), reverse methylsulfonamide 5h (EC50 = 40 nM), pyrazole-compound 6a (EC50 = 90 nM), 6-methylpiperazine amide thieno[2,3-d]pyrimidine 14d (EC50 = 48 nM), and pyrazolo[3,4-d]pyrimidines 21j-I (EC50 = 160 nM), which are still less potent than highly optimized ATP-competitive ligands such as dasatinib (EC50 = 20 nM) and nilotinib (EC50 = 40 nM) under our assay conditions. We demonstrate that a good correlation exists between the binding affinity for the myristate binding site and potency as a cellular Bcr-Abl inhibitor. Although further optimization of the binding affinity 1 class of ligand may be possible, because the mechanism of inhibition of Bcr-Abl is allosteric, it maybe limited by something other than myristate ligand affinity. We have also discovered that different myristate ligands display a differential ability to synergize with dasatinib to inhibit the T315I Bcr-Abl mutant. For example, potent wild-type inhibitor 6a (EC50 = 90 nM) does not exhibit synergy with dasatinib against T315I Bcr-Abl while the less potent inhibitor 21a-I does. The mechanistic basis for this effect is currently under investigation. The work presented here demonstrates that a variety of promising lead compounds can be identified that bind potently to the myristate binding site and inhibit cellular Bcr-Abl activity. The ability of these compounds to synergize with ATP-competitive inhibitors to inhibit the growth of cells transformed with Bcr-Abl mutants and likely to prevent or delay the emergence of resistance mutations should encourage this new therapeutic modality to be explored clinically. As a variety of kinases such as PKC,(22) mTor,(23) Arg,(24) and SK1(25) and Akt(26) have been demonstrated to be regulated through inter- or intramolecular interactions with lipids, screening this compound collection for allosteric inhibitors of lipid binding sites may prove valuable.
Experimental Section
Chemistry
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded at 400 MHz (Bruker XWIN-NMR) or 600 MHz (Varian AS600), and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), brs (broad singlet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Symmetry C18 column (19 mm × 50 mm, 5 μM) using a gradient of 5−95% acetonitrile in water containing 0.05% trifluoacetic acid (TFA) over 8 min (10 min run time) at a flow rate of 30 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis.
Cell Culture and Cell Proliferation Assay
Ba/F3.p210 cells were obtained by transfecting the IL-3-dependent murine hematopoietic Ba/F3 cell line with a pEYK vector containing p210BCR-ABL and Bcr-Abl mutations.9,27 All cell lines were cultured with 5% CO2 at 37 °C in RPMI 1640 (Invitrogen) with 10% fetal bovine serum (FBS) and supplemented with 1% l-glutamine. Parental Ba/F3 cells were similarly cultured with 10% WEHI-conditioned medium as a source of IL-3. Transfected cell lines were cultured in media supplemented with 25 μg/mL zeocin. The 48 h cell proliferation studies were obtained using the CellTiter-Glo assay (Promega) as previous described.(8)
For drug combination studies, compounds were added simultaneously at fixed ratios to cells, and cell viability was determined by CellTiter-Glo assay (Promega) and expressed as a function of growth affected (FA) drug-treated cells versus control cells. Synergy was assessed by Calcusyn software (Biosoft; Ferguson, MO, and Cambridge, U.K.), using the Chou−Talalay method.(21) The combination index CI indicates the strength of the synergy effect. Values less than 1 indicate synergy, whereas values greater than 1 indicate antagonism.
Procedure for the Synthesis of N4-(3-Morpholinopropyl)-N6-(4-(trifluoromethoxy)phenyl)pyrimidine-4,6-diamine (4b)
To a stirred solution of 4,6-dichloro-pyrimidine (298.0 mg, 2.0 mmol) and 4-trifluoromethoxyaniline (0.27 mL, 2.0 mmol) in 1.0 mL of EtOH was added DIEA (0.35 mL, 2.0 mmol). The reaction mixture was stirred for 16 h at 80 °C. Then the mixture was concentrated and purified by silica gel column chromatography to give the title compound 3 (521 mg, 90% yield). MS (ESI) m/z 290 (M + H)+.
To a stirred solution of compound 3 (29 mg, 0.10 mmol) and DIEA (35 μL, 0.20 mmol) in 1 mL of sec-BuOH was added 3-morpholinopropan-1-amine (29 μL, 0.20 mmol) at room temperature. Then the resulting mixture was stirred at 120 °C. After the reaction was complete as monitored by reverse phase analytical liquid chromatography−electrospray mass spectrometry (LC−MS), the solvent was removed and the title compound was purified by reverse-phase preparative HPLC using a water (0.05% TFA)/acetonitrile (0.05% TFA) gradient to afford the title compound 4b as the TFA salt (30 mg, 59%). 1H NMR (600 MHz, CD3OD) δ 8.26 (s, 1H), 7.50 (d, J = 7.2 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 5.92 (s, 1H), 4.03 (brs, 2H), 3.79 (brs, 2H), 3.47 (brs, 4H), 3.26−3.23 (m, 2H), 3.14 (brs, 2H), 2.12−2.08 (m, 2H). MS (ESI) m/z 398 (M + H)+. HRMS (ESI) calcd for C18H22F3N5O2 397.1726, found 398.1797 (M + H)+.
Procedure for the Synthesis of N-(2-Hydroxyethyl)-3-(6-(4-(trifluoromethoxy)phenylamino)pyrimidin-4-yl)benzamide (2)
A mixture of 6-chloro-N-(4-(trifluoromethoxy)phenyl)pyrimidin-4-amine (200 mg, 0.69 mmol), 3-carboxyphenylboronic acid (115 mg, 0.69 mmol), Pd(PPh3)4 (40 mg, 0.034 mmol), and sodium carbonate (292 mg, 2.76 mmol) in acetonitrile/water (v/v = 1/1, 10 mL) was heated to 90 °C under an argon atmosphere. After refluxing for 8 h, the hot reaction mixture was filtered. The filtrate was cooled to room temperature and treated with 6 N HCl (pH is around 4) to afford 3-(6-(4-(trifluoromethoxy)phenylamino)pyrimidin-4-yl)benzoic acid as a pale yellow precipitate, which was collected and washed with water and air-dried to give 202 mg (78% yield) of the title compound. MS (ESI) m/z 376 (M + H)+.
To a solution of 3-(6-(4-(trifluoromethoxy)phenylamino)pyrimidin-4-yl)benzoic acid (56.3 mg, 0.15 mmol), ethanolamine (27 μL, 0.45 mmol), and diisopropylethylamine (53 μL, 0.30 mmol) in 1.50 mL of dimethylformamide was added 1-hydroxy-1H-benzotriazole “HOBt” (30 mg, 0.22 mmol) at room temperature. Then the mixture was cooled to 0 °C and (3-(dimethylamino)propyl)ethylcarbodiimide hydrochloride “EDCI” (52 mg, 0.27 mmol) was added. And the mixture was warmed to room temperature gradually. After the reaction was completed, as monitored by LC−MS, the reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous Na2SO4, concentrated, and purified by silica gel column chromatography to afford 2 (50 mg, 80% yield). 1H NMR (600 MHz, DMSO-d6), δ 9.92 (s, 1H), 8.75 (s, 1H), 8.60 (t, J = 5.4 Hz, 1H), 8.51 (s, 1H), 8.17 (d, J = 7.8 Hz, 1H), 7.98 (d, J = 7.2 Hz, 1H), 7.84 (d, J = 9.0 Hz, 2H), 7.61 (t, J = 7.8 Hz, 1H), 7.34 (d, J = 9.0 Hz, 2H), 7.33 (s, 1H), 4.73 (t, J = 5.4 Hz, 1H), 3.54 (t, J = 6.0 Hz, 2H), 3.36 (t, J = 6.0 Hz, 2H). MS (ESI) m/z 419 (M + H)+.
Procedure for Synthesis of Methyl 4-(4-(Trifluoromethoxy)phenylamino)thieno[2,3-d]pyrimidine-6-carboxylate (11)
To a stirred solution of 4,6-dichloropyrimidine-5-carbaldehyde (1.0 g, 5.65 mmol) in 10 mL of THF was added 4-trifluoromethoxyaniline (0.72 mL, 5.36 mmol) at −10 °C. After 10 min, the mixture was stirred at room temperature for 5 h. Then the mixture was diluted by ethyl acetate and filtered through Celite. The filtrate was concentrated and purified by silica gel column chromatography to give compound 10 (1.13 g, 63% yield). MS m/z 318 (M + H)+.
4-Chloro-6-(4-trifluoromethoxyphenylamino)pyrimidine-5-carbaldehyde 10 (500 mg, 1.57 mmol) in DMF (2 mL) was added to a suspension of K2CO3 (620 mg, 4.5 mmol) in DMF (1 mL) at room temperature. After the mixture was stirred for 30 min, methyl thioglucolate (210 mg, 1.9 mmol) was added slowly to the reaction mixture. The resulting mixture was heated at 90 °C for 1 h. The reaction mixture was then cooled at room temperature and poured into ice−water. The precipitate was collected and dried to afford the title compound 11 (400 mg, 69% yield). 1H NMR (600 MHz, CDCl3) δ 8.64 (s, 1H), 8.16 (s, 1H), 7.78 (d, J = 9.0 Hz, 2H), 7.25 (d, J = 9.0 Hz, 2H), 3.92 (s, 3H). MS m/z 370 (M + H)+.
Procedure for the Synthesis of N-(2-Hydroxyethyl)-4-(4-(trifluoromethoxy)phenylamino)thieno[2,3-d]pyrimidine-6-carboxamide (14a)
A mixture of methyl 4-(4-(trifluoromethoxy)phenylamino)thieno[2,3-d]pyrimidine-6-carboxylate (152 mg, 0.41 mmol) and LiOH (25.5 mg, 1.07 mmol) was dissolved in THF (4 mL) and H2O (1 mL). The mixture was stirred at 60 °C for 3 h. Acidification with acetic acid resulted in a brown solid which was collected by filtration. Compound 13 was then dried in vacuo and used for the next step without any further purification (115 mg, 79% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.97 (brs, 1H), 8.61 (s, 1H), 8.00 (d, J = 9.2 Hz, 2H), 7.42 (d, J = 9.2 Hz, 2H). MS (ESI) m/z 356 (M + H)+.
4-(4-(Trifluoromethoxy)phenylamino)thieno[2,3-d]pyrimidine-6-carboxylic acid (30 mg, 0.08 mmol) was mixed with DIEA (30 μL, 0.172 mmol) and HATU (39 mg, 0.1 mmol) in 2 mL of DMF at room temperature. 2-Aminoethanol (6.1 mg, 0.1 mmol) was added to the reaction mixture 0.5 h later. After being stirred at room temperature for 2 h, the reaction mixture was concentrated and purified by preparative HPLC to afford the title compound 14a as a TFA salt (25 mg, 61% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 9.26 (s, 1H), 8.82 (s, 1H), 8.21 (d, J = 8.8 Hz, 2H), 7.85 (d, J = 8.8 Hz, 2H), 7.34 (s, 1H), 3.02 (t, J = 6.4 Hz, 2H), 2.65 (t, J = 6.4 Hz, 2H). MS m/z 399 (M + H)+.
Procedure for Synthesis of N-(4-(Trifluoromethoxy)phenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (16a)
To a stirred solution of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (46.1 mg, 0.3 mmol) and DIEA (78 μL, 0.45 mmol) in 2.0 mL of 2-BuOH was added 4-trifluoromethoxyaniline (79.7 mg, 0.45 mmol) at room temperature. Then the mixture was heated to 120 °C. After the reaction was complete, as monitored by LC−MS, the solvent was removed and purified by preparative HPLC to afford the title compound 16a (66.6 mg, 60% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.42 (s, 1H), 9.58 (s, 1H), 8.52 (s, 1H), 7.76 (d, J = 9.2 Hz, 2H), 7.24 (d, J = 9.2 Hz, 2H), 7.39 (d, J = 6.2 Hz, 1H), 6.52 (dd, J = 3.2, 1.6 Hz, 1H). MS (ESI) m/z 295 (M + H)+.
Procedure for Synthesis of N-(3-Methoxypropyl)-3-(4-(4-(trifluoromethoxy)phenylamino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)propanamide (20a)
To a stirred solution of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1.0 g, 6.51 mmol) in DMF (30 mL) was added NaH (60% in mineral oil) (391 mg, 9.76 mmol) at 0 °C. After the mixture was stirred for 30 min at room temperature, methyl 3-bromopropanoate (1.20 g, 7.18 mmol) was added and the resulting mixture was stirred for 16 h at 40 °C. Then the reaction was quenched with saturated NH4Cl solution and extracted with ethyl acetate. The organic extracts were washed with brine, dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography to afford compound 17 (1.2 g, 77% yield). MS m/z 240 (M + H)+.
To a solution of 3-(4-chloro-pyrrolo[2,3-d]pyrimidin-7-yl)propionic acid methyl ester (100 mg, 0.42 mmol) and DIEA (0.11 mL, 0.63 mmol) in sec-BuOH (3 mL) was added 4-trifluoromethoxyaniline (80 mg, 0.45 mmol). The resulting mixture was stirred at 120 °C for 2 h. Then the mixture was concentrated and purified by silica gel column chromatography to afford compound 18 (87.5 mg, 55% yield). MS m/z 381 (M + H)+.
To a solution of 3-[4-(4-trifluoromethoxyphenylamino)pyrrolo[2,3-d]pyrimidin-7-yl]propionic acid methyl ester (87.5 mg, 0.22 mmol) in 5 mL of mixed solvent THF/H2O (v/v = 3/1)) was added Cs2CO3 (78 mg, 0.40 mmol). The resulting mixture was stirred at 80 °C for 2 h. The mixture was concentrated and neutralized with acetic acid. The solid was filtered and dried to yield compound 19 as a brown solid (62 mg, 77%). MS m/z 367 (M + H)+.
3-[4-(4-Trifluoromethoxyphenylamino)pyrrolo[2,3-d]pyrimidin-7-yl]propionic acid (25 mg, 0.068 mmol) was mixed with DIEA (0.030 mL, 0.172 mmol) and HATU (28.5 mg, 0.075 mmol) in 1.0 mL of DMF at room temperature. 3-Methoxypropan-1-amine (12.1 mg, 0.14 mmol) was added into the reaction mixture 0.5 h later. After being stirred at room temperature for 2 h, the mixture was concentrated and purified by preparative HPLC to afford the title compound 20a (17.8 mg, 60% yield). 1H NMR (600 MHz, CDCl3) δ 11.53 (s, 1H), 8.29 (s, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 3.0 Hz, 1H), 6.23 (s, 1H), 5.46 (d, J = 2.4 Hz, 1H), 4.56 (t, J = 6.0 Hz, 4H), 3.43 (t, J = 6.0 Hz, 2H), 3.30 (s, 3H), 2.72 (t, J = 6.0 Hz, 2H), 1.70 (quintet, J = 6.0 Hz, 2H). MS (ESI) m/z 438 (M + H)+.
Procedure for Synthesis of 2-(4-(4-(Trifluoromethoxy)phenylamino)-1H-pyrazolo-[3,4-d]pyrimidin-1-yl)ethanol (21a-I) and 2-(4-(4-(Trifluoromethoxy)phenylamino)-2H-pyrazolo[3,4-d]pyrimidin-2-yl)ethanol (21a-II)(28)
To a solution of 4-chloro-6-(4-trifluoromethoxyphenylamino)pyrimidine-5-carbaldehyde (50 mg, 0.16 mmol) in 2.0 mL of sec-BuOH was added Na2CO3 (30 mg, 0.28 mmol) and (2-hydroxyethyl)hydrazine (20 mg, 0.26 mmol). The mixture was stirred at 60 °C for 8 h. The reaction mixture was partitioned in 50 mL of water and 50 mL of ethyl acetate. The aqueous layer was extracted with ethyl acetate three times. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude product was purified by silica gel column chromatography to give title compounds 21a-I (20 mg) and 21a-II (5.0 mg).
21a-I:1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 8.45 (s, 1H), 8.32 (s, 1H), 7.98 (d, J = 9.6 Hz, 2H), 7.41 (d, J = 9.6 Hz, 2H), 4.40 (t, J = 6.0 Hz, 2H), 3.83 (t, J = 6.0 Hz, 2H). MS (ESI) m/z 340 (M + H)+.
21a-II:1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.68 (s, 1H), 8.57 (s, 1H), 7.97 (d, J = 9.6 Hz, 2H), 7.45 (d, J = 9.6 Hz, 2H), 4.41 (t, J = 6.0 Hz, 2H), 3.85 (t, J = 6.0 Hz, 2H). MS (ESI) m/z 340 (M + H)+.
Procedure for Synthesis of 1-(Pyridin-3-ylmethyl)-N-(4-(trifluoromethoxy)phenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (21g-I) and 2-(Pyridin-3-ylmethyl)-N-(4-(trifluoromethoxy)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-amine (21g-II)
To a stirred solution of N-(4-(trifluoromethoxy)phenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (60 mg, 0.2 mmol) in 3.0 mL of DMF was added NaH (60% in mineral oil) (32 mg, 0.8 mmol) at 0 °C. After the mixture was stirred for 1 h, 3-(bromomethyl)pyridine hydrobromide (48 mg, 0.19 mmol) was added. The resulting mixture was warmed to room temperature gradually. After the reaction was complete, as monitored by LC−MS, the mixture was poured into ice−water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude product was purified by preparative TLC to give the title compounds.
21g-I:1H NMR (600 MHz, CD3OD) δ 8.54 (s, 1H), 8.44 (d, J = 4.8 Hz, 1H), 8.43 (s, 1H), 8.18 (brs, 1H), 7.89 (d, J = 9.0 Hz, 2H), 7.77 (dt, J = 1.8, 7.8 Hz, 1H), 7.38 (dd, J = 4.8, 7.8 Hz, 1H), 7.28 (d, J = 8.4 Hz, 2H), 5.64 (s, 2H). MS (ESI) m/z 387 (M + H)+.
21g-II:1H NMR (600 MHz, CD3OD) δ 8.62 (d, J = 1.8 Hz, 1H), 8.52 (dd, J = 1.2, 4.8 Hz, 1H), 8.47 (brs, 1H), 8.38 (s, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.86 (dt, J = 1.8, 7.8 Hz, 1H), 7.45 (ddd, J = 0.6, 4.8, 7.2 Hz, 1H), 7.28 (d, J = 8.4 Hz, 2H), 5.68 (s, 2H). MS (ESI) m/z 387 (M + H)+.
Fluorescence Polarization Assay
Abl kinases were prepared as previous described,(9) and fluorescent-labeled 1 compound (1-FITC) was used.(9)
Fluorescence polarization assays of the competitive binding of various concentrations of 1 analogues and fixed concentration of 50 nM 1−FITC to Abl were performed in the buffer 20 mM Tris-HCl, pH 8.0, 150 mM NaCl in 384-well plate with a total reaction volume of 40 μL. The samples were incubated at room temperature for 1 h, and the fluorescence polarization (FP) was then measured using a SpectraMax M5 microplate reader (Molecular Devices) equipped with a 530 nm emission filter and a 490 nm excitation filter. FP values were converted to percentage inhibition using the equation
where P0 is the FP value at 0%, P100 is the FP value when 100% of the 1−FITC has been competitively displaced from the kinase and 1−FITC complex, and P is the experimental FP value at each concentration of the competing 1 analogues being tested. The percentage inhibition versus competitor concentration curves were analyzed by nonlinear least-squares curve fitting, and the concentration of competing ligand required to displace half of the bound 1−FITC ligand was determined (IC50).
Acknowledgments
We thank the members from Nathanael S. Gray laboratory for experimental assistance and insightful comments throughout this study. Some of the cellular and biochemical assays were performed at the ICCB at Harvard Medical School with help from Andrew Daab, Sean M. Johnston, and Stewart Rudnicki. The Gray laboratory acknowledges the generous financial support from Novartis and NIH Grant R01 CA130876-02 and useful discussions with Michael Eck, Markus Warmuth, Paul Manley, Doriano Fabbro, and John Engen.
Supporting Information Available
Spectral data of 4a,c−e, 5a−k, 6a−f, 7a−d, 8a−g, 9a−l, 12, 14b−g, 16b−d, 20b, 21b−f, 21h−l, and 22; pharmacokinetic parameters of compounds 5g, 6a, 14d, and 21b-I; calculations of relative transformed energy from trans to cis (4a, 9a, 9c, 9l); fluorescence polarization assay results of additional compounds (5g, 7a, 12, 21a-I). This material is available free of charge via the Internet at http://pubs.acs.org.
PDB code: 3K5V.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: Akt, v-akt murine thymoma viral oncogene homologue 1; ATP, adenosine triphosphate; Bcr-Abl, breakpoint cluster region Abelson tyrosine kinase; CDK, cyclin-dependent kinases; DIEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; HATU, 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; mTor, mammalian target of rapamycin; PKC, protein kinase C; SK1, sphingosine kinase 1; TFA, trifluoroacetic acid; THF, tetrahydrofuran.
Supplementary Material
References
- Faderl S.; Kantarjian H. M.; Talpaz M.. Chronic myelogenous leukemia: update on biology and treatment. Oncology (Williston Park, N.Y.) 1999, 13, 169−80; discussion 181, 184. [PubMed] [Google Scholar]
- Cowan-Jacob S. W.; Guez V.; Fendrich G.; Griffin J. D.; Fabbro D.; Furet P.; Liebetanz J.; Mestan J.; Manley P. W. Imatinib (STI571) resistance in chronic myelogenous leukemia: molecular basis of the underlying mechanisms and potential strategies for treatment. Mini-Rev. Med. Chem. 2004, 4, 285–299. [DOI] [PubMed] [Google Scholar]
- Weisberg E.; Manley P. W.; Breitenstein W.; Bruggen J.; Cowan-Jacob S. W.; Ray A.; Huntly B.; Fabbro D.; Fendrich G.; Hall-Meyers E.; Kung A. L.; Mestan J.; Daley G. Q.; Callahan L.; Catley L.; Cavazza C.; Azam M.; Neuberg D.; Wright R. D.; Gilliland D. G.; Griffin J. D. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [DOI] [PubMed] [Google Scholar]
- Manley P. W.; Cowan-Jacob S. W.; Mestan J. Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochim. Biophys. Acta 2005, 1754, 3–13. [DOI] [PubMed] [Google Scholar]
- Lombardo L. J.; Lee F. Y.; Chen P.; Norris D.; Barrish J. C.; Behnia K.; Castaneda S.; Cornelius L. A.; Das J.; Doweyko A. M.; Fairchild C.; Hunt J. T.; Inigo I.; Johnston K.; Kamath A.; Kan D.; Klei H.; Marathe P.; Pang S.; Peterson R.; Pitt S.; Schieven G. L.; Schmidt R. J.; Tokarski J.; Wen M. L.; Wityak J.; Borzilleri R. M. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [DOI] [PubMed] [Google Scholar]
- Shah N. P.; Tran C.; Lee F. Y.; Chen P.; Norris D.; Sawyers C. L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004, 305, 399–401. [DOI] [PubMed] [Google Scholar]
- O’Hare T.; Walters D. K.; Stoffregen E. P.; Jia T.; Manley P. W.; Mestan J.; Cowan-Jacob S. W.; Lee F. Y.; Heinrich M. C.; Deininger M. W.; Druker B. J. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005, 65, 4500–4505. [DOI] [PubMed] [Google Scholar]
- Adrian F. J.; Ding Q.; Sim T.; Velentza A.; Sloan C.; Liu Y.; Zhang G.; Hur W.; Ding S.; Manley P.; Mestan J.; Fabbro D.; Gray N. S. Allosteric inhibitors of Bcr-Abl-dependent cell proliferation. Nat. Chem. Biol. 2006, 2, 95–102. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Adrian F. J.; Jahnke W.; Cowan-Jacob S. W.; Li A. G.; Iacob R. E.; Sim T.; Powers J.; Dierks C.; Sun F.; Guo G. R.; Ding Q.; Okram B.; Choi Y.; Wojciechowski A.; Deng X.; Liu G.; Fendrich G.; Strauss A.; Vajpai N.; Grzesiek S.; Tuntland T.; Liu Y.; Bursulaya B.; Azam M.; Manley P. W.; Engen J. R.; Daley G. Q.; Warmuth M.; Gray N. S. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010, 463, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S.; Gray N. S.; Wu X.; Ding Q.; Schultz P. G. A combinatorial scaffold approach toward kinase-directed heterocycle libraries. J. Am. Chem. Soc. 2002, 124, 1594–1596. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wu X.; Mitchell B.; Kintner C.; Ding S.; Schultz P. G. A small-molecule agonist of the Wnt signaling pathway. Angew. Chem., Int. Ed. 2005, 44, 1987–1990. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Liu Y.; Gao F.; Ding Q.; Cho C.; Hur W.; Jin Y.; Uno T.; Joazeiro C. A.; Gray N. Discovery of EGFR selective 4,6-disubstituted pyrimidines from a combinatorial kinase-directed heterocycle library. J. Am. Chem. Soc. 2006, 128, 2182–2183. [DOI] [PubMed] [Google Scholar]
- Manley P. J; Balitza A. E; Bilodeau M. T.; Coll K. E; Hartman G. D.; McFall R. C.; Rickert K. W.; Rodman L. D.; Thomas K. A. 2,4-Disubstituted pyrimidines: a novel class of KDR kinase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 1673–1677. [DOI] [PubMed] [Google Scholar]
- Beattie J. F.; Breault G. A.; Ellston R. P.; Green S.; Jewsbury P. J.; Midgley C. J.; Naven R. T.; Minshull C. A.; Pauptit R. A.; Tucker J. A.; Pease J. E. Cyclin-dependent kinase 4 inhibitors as a treatment for cancer. Part 1: Identification and optimisation of substituted 4,6-bis anilino pyrimidines. Bioorg. Med. Chem. Lett. 2003, 13, 2955–2960. [DOI] [PubMed] [Google Scholar]
- a Miyaura N. Top. Curr. Chem. 2002, 219, 11–59. [Google Scholar]; b Muci A. R.; Buchwald S. L. Top. Curr. Chem. 2002, 219, 131–209. [Google Scholar]; c Suzuki A.Modern Arene Chemistry; Astruc D., Ed.; Wiley-VCH: Weinheim, Germany, 2002; pp 53−106. [Google Scholar]; d Hartwig J. F.Modern Arene Chemistry; Astruc D., Ed.; Wiley-VCH: Weinheim, Germany, 2002; pp 107−168. [Google Scholar]
- Luesch H.; Wu T. Y.; Ren P.; Gray N. S.; Schultz P. G.; Supek F. A genome-wide overexpression screen in yeast for small-molecule target identification. Chem. Biol. 2005, 12, 55–63. [DOI] [PubMed] [Google Scholar]
- Glide, version 2.5; Schrödinger, L.L.C.: New York. The Glide 2.5 calculations used FirstDiscovery, version 2.5021, which was released in June 2003. [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Li Y.; Xie W.; Fang G. Fluorescence detection techniques for protein kinase assay. Anal. Bioanal. Chem. 2008, 390, 2049–2057. [DOI] [PubMed] [Google Scholar]
- Corbin A. S.; La Rosee P.; Stoffregen E. P.; Druker B. J.; Deininger M. W. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood 2003, 101, 4611–4614. [DOI] [PubMed] [Google Scholar]
- Chou T. C.; Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [DOI] [PubMed] [Google Scholar]
- Ziegler W. H.; Tigges U.; Zieseniss A.; Jockusch B. M. A lipid-regulated docking site on vinculin for protein kinase C. J. Biol. Chem. 2002, 277, 7396–7404. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Vilella-Bach M.; Bachmann R.; Flanigan A.; Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [DOI] [PubMed] [Google Scholar]
- Hantschel O.; Nagar B.; Guettler S.; Kretzschmar J.; Dorey K.; Kuriyan J.; Superti-Furga G. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 2003, 112, 845–857. [DOI] [PubMed] [Google Scholar]
- Delon C.; Manifava M.; Wood E.; Thompson D.; Krugmann S.; Pyne S.; Ktistakis N. T. Sphingosine kinase 1 is an intracellular effector of phosphatidic acid. J. Biol. Chem. 2004, 279, 44763–44774. [DOI] [PubMed] [Google Scholar]
- Castillo S. S.; Brognard J.; Petukhov P. A.; Zhang C.; Tsurutani J.; Granville C. A.; Li M.; Jung M.; West K. A.; Gills J. G.; Kozikowski A. P.; Dennis P. A. Preferential inhibition of Akt and killing of Akt-dependent cancer cells by rationally designed phosphatidylinositol ether lipid analogues. Cancer Res. 2004, 64, 2782–2792. [DOI] [PubMed] [Google Scholar]
- Azam M.; Latek R. R.; Daley G. Q. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 2003, 112, 831–843. [DOI] [PubMed] [Google Scholar]
- Structure assignment was according to the reported analogs. See the following: Da Settimo F.; Primofiore G.; La Motta C.; Taliani S.; Simorini F.; Marini A. M.; Mugnaini L.; Lavecchia A.; Novellino E.; Tuscano D.; Martini C. Novel, highly potent adenosine deaminase inhibitors containing the pyrazolo[3,4-d]pyrimidine ring system. Synthesis, structure−activity relationships, and molecular modeling studies. J. Med. Chem. 2005, 48, 5162–5174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.