

Abstract
PH domain Leucine-rich repeat protein phosphatase (PHLPP) directly dephosphorylates and inactivates Akt and protein kinase C, poising it as a prime target for pharmacological intervention of two major survival pathways. Here we report on the discovery of small molecule inhibitors of the phosphatase activity of PHLPP, a member of the PP2C family of phosphatases for which there are no general pharmacological inhibitors. First, the Diversity Set of the NCI was screened for inhibition of the purified phosphatase domain of PHLPP2 in vitro. Second, selected libraries from the open NCI database were docked into a virtual model of the phosphatase domain of PHLPP2, previously trained with our experimental data set, unveiling additional inhibitors. Biochemical and cellular assays resulted in the identification of two structurally diverse compounds that selectively inhibit PHLPP in vitro, increase Akt signaling in cells, and prevent apoptosis. Thus, chemical and virtual screening has resulted in the identification of small molecules that promote Akt signaling by inhibiting its negative regulator PHLPP.
Transient phosphorylation of proteins is a fundamental mechanism by which cells integrate and transduce signals. Kinases and phosphatases act in dynamic opposition to control the extent, duration, and intensity of signaling and to maintain cellular homeostasis. Dysregulation of the precisely tuned balance between phosphorylation and dephosphorylation results in pathophysiological states.
The phosphatidylinositol-3 kinase (PI3Ka)-Akt pathway is one of the major phosphorylation cascades that control cell fate.(1) Stimulation by growth factors, such as EGF or insulin, results in phosphorylation of receptor tyrosine kinases and recruitment of effector proteins, notably PI3K, to the receptors. PI3K phosphorylates the lipid phosphatidylinositol-4,5-bisphosphate (PIP2) to yield phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 recruits Akt to the plasma membrane where the protein is phosphorylated by its upstream kinase phosphoinositide-dependent kinase-1 (PDK-1) at the activation loop (Thr308 in Akt1). A subsequent phosphorylation occurs at the hydrophobic motif (Ser473 in Akt1) by a mechanism that depends on the TORC 2 complex.(2) Once phosphorylated, Akt is released from the membrane and phosphorylates diverse substrates throughout the cell, thus inducing a wide range of physiological effects, notably cell growth, proliferation, and survival. In addition, Akt is a master regulator of glucose metabolism, playing a key role in mediating the biological effects of insulin.(3) The activation of Akt is opposed by (1) lipid phosphatases that dephosphorylate, and thus remove, the lipid second messenger, and (2) protein phosphatases that dephosphorylate, and thus inactivate, Akt. Specifically, PTEN dephosphorylates PIP3(4) to terminate the activation of Akt. Activated Akt is dephosphorylated at the activation loop by okadaic acid sensitive phosphatases such as PP2A5,6 and at the hydrophobic motif by the recently discovered PH domain leucine-rich repeat protein phosphatase (PHLPP),7,8 resulting in inhibition of activity and promotion of apoptosis.
PHLPP was initially discovered as the phosphatase that dephosphorylates and inactivates Akt in cells, but it also dephosphorylates and regulates the levels of protein kinase C (PKC) isozymes,(9) another important class of kinases that control cell growth and survival. PHLPP is a family of three isoforms: the alternatively spliced PHLPP1α and PHLPP1β, and PHLPP2.(10) The phosphatase domains of the three enzymes are highly similar, with 58% amino acid identity. They belong to the PP2C family of phosphatases, which, in turn, belong to the larger PPM (protein phosphatase magnesium/manganese dependent) family of serine/threonine protein phosphatases, which require Mn2+ or Mg2+ for their activity. The primary known function of the PP2C family is to down-regulate stress responses in eukaryotes.11,12 PP2C phosphatases differ from those in the PPP family (which also require metallic cations for their activity) by their resistance to common serine/threonine phosphatase inhibitors such as okadaic acid and microcystin.(13) In fact, there are no general inhibitors of the PP2C family available, although cyclic peptide inhibitors for PP2Cδ(14) and small molecule inhibitors for PP2Cα, identified by virtual screening,(15) have been reported. Given the high therapeutic value of inhibitors for protein kinases to target disease,16,17 discovery of phosphatase inhibitors is likely to have a major impact in future therapeutics.
Because PHLPP dephosphorylates Akt and PKC, positioning it as a suppressor of two major survival pathways, PHLPP inhibition would be particularly relevant therapeutically in diseases where survival pathways are repressed, notably diabetes and heart disease. Indeed, Akt and PKC activities are repressed in both diabetes mellitus and cardiovascular conditions such as myocardial infarction and ischemia-reperfusion (I/R) injury. In diabetes mellitus, the Akt pathway is a therapeutic target for islet transplant and survival as well as in the treatment of associated vascular complications.(18) Akt activity is important for β-cell growth, survival, and insulin production.19,20 Studies have demonstrated that transgenic overexpression of Akt in islet β-cells gives rise to larger islets resulting from increases in the number and size of cells.21,22 This hypertrophy is combined with an increase in insulin production; mice are also resistant to streptozotocin-induced diabetes. Conversely, overexpression of kinase-dead mutants(23) or impaired PDK-1(24) in transgenic mice leads to defective insulin production and increased susceptibility to streptozotocin. Activation of Akt by different means has been used to improve transplantation success already.25,26 In cardiovascular diseases, activation of pro-survival pathways is key to protect the heart from damage because cardiovascular injuries are often linked to myocyte cell loss through apoptosis.27−29 Akt has a number of positive effects on I/R-mediated damage of the heart that are mediated by different substrates.30,31 For example, infarct size is reduced through inhibition of GSK3β and this effect is reversed by the PI3K inhibitors, LY 294002, and wortmannin. In the case of PKC, activation of PKC ε has been established to mediate cardiac protection from cardiac ischemia.32−36 Ischemic preconditioning(36) and many pharmacological agents,37,38 including insulin, adenosine A1/A2 agonist, bradykinin, natriuretic peptides, or erythropoietin, achieve their protective effect through activation of Akt and PKC. Thus, inhibition of PHLPP, a repressor of Akt and PKC activity, would provide a novel tool promoting the concomitant activation of the two key survival pathways.
Here we report on the discovery of small molecule inhibitors of PHLPP phosphatase activity. These molecules were identified by medium throughput chemical screening and virtual screening of the NCI repository. We identified molecules that inactivate PHLPP at low micromolar concentrations in vitro, increase basal and agonist-evoked Akt phosphorylation in cells, and suppress apoptosis.
Results
As there is no general inhibitor of PP2C, we started our search for inhibitory small molecules of PHLPP by screening the First Diversity Set of the National Cancer Institute. This set comprises 1990 compounds chosen among the 140000 compounds in the repository to encompass the largest chemical space possible. These molecules were assayed in a 96-well format, at concentrations of 100 μM, using the isolated phosphatase domain of PHLPP2 purified from Escherichia coli as the enzyme and pNPP as the substrate. Statistical analysis revealed a z value(39) of 0.5 and a signal-over-background ratio of almost 4, indicating the assay was statistically valid (Figure 1a). Dephosphorylation of pNPP results in an increase of the optical density of the solution, thus the slope of the change of OD over time served as a measure of the activity of the phosphatase (Figure 1b). In one assay, 80 compounds can be tested, as well as 12 controls of uninhibited activity (DMSO) and four controls for background (absence of enzyme). Activity in each well was measured and the value normalized to that in the absence of inhibitor; 88 compounds were identified that reduced activity to below 0.3 of the control value (Figure 1c), the criterion chosen to warrant further testing as an inhibitor. Colored compounds, which interfered with the colorimetric assay, were further tested at concentrations of either 10 or 20 μM depending on the intensity of the color. Finally, the IC50 values of the 50 most promising compounds were determined; these ranged from 1 to 100 μM, with 10% false positives for which the colorimetric change was unrelated to phosphatase activity. Structural analysis of inhibitory molecules led to the identification of 11 different chemical backbones (Figure 2 and Supporting Information Figure 1). We then retested other compounds of the Diversity Set with these backbones and uncovered 49 additional inhibitors.
Figure 1.

Screen of NCI Diversity Set. (a) Scatter plot of the raw data from 25 different plates showing control (DMSO) (gray solid circle) and background (absence of enzymes) values (open tilted square). The solid lines represent the mean of the control and background data and the broken lines display 3 standard deviations (SD) from the mean of each data set. The z-factor is 0.5, and the signal-overbackground ratio is 3.8. (b) Representative time course for the hydrolysis of pNPP by PHLPP2 catalytic domain. pNPP (8 mM) was incubated in 125 μL assay buffer at room temperature, without (open tilted square) or with the phosphatase domain of PHLPP2, in the presence of DMSO (gray solid circle) or benzylcarbamic acid (NCS 24515) (solid square). The absorbance of the solution was recorded at 405 nm and the data fit by linear regression. (c) Representative activity data from one 96-well plate. NCI plate 4238 was assayed for PHLPP2 activity as in (b). Each compound is represented by a black bar, DMSO controls are represented by gray bars (background controls were omitted on this graph). The values obtained for the DMSO controls were averaged and normalized to a relative activity of 1.0 (black line). Compounds that reduced PHLPP2 relative activity to lower than 0.3 (red line) were considered a hit (lane 86).
Figure 2.

Chemical structures of some inhibitors of PHLPP2. Different compounds that have been identified as inhibitors of PHLPP2 are presented along with the experimental IC50 determined at 8 mM substrate. The chemical backbones used to determine the structural families are highlighted.
We turned to virtual screening to expand the scope of our investigation. Docking programs have been successfully used to identify novel inhibitory compounds of crystallographically solved signaling phosphatases,40−42 including PP2Cα.(15) We employed the GLIDE algorithm because it is well-established in virtual drug discovery work and has performed well in comparative docking studies.43−45
Because of the lack of crystallographic data, a structure for PHLPP2 based on homology modeling was created that was capable of discerning inhibitory binding compounds from nonbinding compounds. The first step consisted of designing a model that would correlate best with our experimental data. The amino acid sequence of the PHLPP2 phosphatase domain was aligned with that of the phosphatase domain of PP2Cα, resulting in an alignment score of 23% (Figure 3a), and a homology model was then produced based on the crystal structure of PP2Cα.(46) Initial docking results of our control set of known inhibitors using GLIDE did not correlate well with aforementioned inhibitors of PHLPP2. Thus, we concluded that our homology model was not in an optimal conformation or that metal ions or water molecules played a critical role in inhibitor binding. Because many of our inhibitors contain negatively charged moieties, the interaction between these groups and the metal ions are predicted to contribute significantly to the overall binding energy. The crystal structure of PP2Cα coordinates 2 Mn2+ ions in the catalytic center. PP2C phosphatases in mycobacteria were found to accommodate a third metallic center in their catalytic core.47−49 Previous work has also shown that the PP2Cα active site metal ions are coordinated by six different water molecules. Because our structure is a homology model of the phosphatase domain of PP2Cα, it should include 1−3 Mn2+ ions and coordinated water molecules. We tested this by placing varying numbers of Mn2+ ions inside the active site near residues that could coordinate them and relaxed each structure to accommodate the ions. This resulted in a variety of structures, which we tested for the ability to recognize inhibitory compounds. All structures with 1 or more Mn2+ ions in the active site recognized inhibitors markedly better than the structure with no Mn2+ ions (Figure 3c).
Figure 3.

Virtual screen for inhibitors using modeled structure of PHLPP2 phosphatase domain. (a) Sequence alignment of PP2Cα and PHLPP2. Amino acids in red may be important for coordinating metal ions. * denotes a conserved residue. (b) Three-dimensional representation of model 4 of table (c) with D806, E989, and D1024 shown coordinating 2Mn2+ ions in the phosphatase active site. (c) Table indicating the functional relevance of modified homology models. Model 4 was chosen for further docking because of its high docking scores and good performance identifying true hits from the Diversity Set. The number of true hits is derived from docking 50 compounds with experimentally derived IC50s for PHLPP2 and determining how many compounds scored better than −7. (d) Structural similarity search to create libraries of compounds similar to known binders of PHLPP2. Selected compounds were docked with the GLIDE algorithm employing calculations at three levels of accuracy, removing lower scoring compounds at each stage.
Next, the entire Diversity Set was docked against our model. This served as a means to test the model for its ability to discriminate true inhibitors from a decoy set of ligands with no experimental activity. The docking protocol was modified so that only the top 4% of ligands were given final docking scores, as would be the case during virtual screening. From these studies, we determined that the model with two Mn2+ ions in the active site coordinated by D806, E989, and D1024 was most capable of discriminating true binders from decoys. In addition, this model had the highest range of G-scores for true hits (Figure 3c, model 4 and Figure 3b). Addition of water molecules did not improve detection of true inhibitors, although it is likely that they contribute to the coordination of ions in the active site. Forty new compounds were found to dock with G-scores better than −7 kcal/mol, in addition to some of the previously characterized inhibitors. These new virtual hits were tested experimentally and 14 of these new compounds were determined to have IC50 values below 100 μM. Rarely do docking studies serve as a means to identify false negatives in a chemical screen but, in this case, combining chemical testing and virtual testing prevented us from missing 14 inhibitors of PHLPP. Model 4 was chosen for further studies because of its ability to distinguish hits from decoys and value in identifying 14 false negatives in the chemical screen.
Armed with a substantial data set of inhibitory molecules, we hypothesized that finding similar structures and docking them might enlarge our pool of known binders and increase our hit rate over random virtual screening of the NCI repository. As previously mentioned, 11 structurally related compound families were identified from in vitro screening; these were used as the references for similarity searches performed on the NCI Open Compound Library (Figure 3d). In addition, seven of the highest affinity compounds were also used as reference compounds for similarity searches. A total of 43000 compounds were identified from these similarity searches and docked to model 4. Eighty compounds among the top ranked structurally similar compounds were tested experimentally, at concentrations of 50 μM, using the same protocol as described for the original screen. These 80 compounds were selected based on good docking scores, structural diversity, and availability from the NCI. Twenty-three compounds reduced the relative activity of the PHLPP2 phosphatase domain to below 0.5 of control and were considered hits. Of these, 20 compounds had an IC50 below 100 μM, with 15 of these having an IC50 value below 50 μM (Figure 4). Thus, we discovered a number of new, experimentally verified low-μM inhibitors by integrating chemical data into our virtual screening effort.
Figure 4.

PHLPP2 inhibitors from the virtual screening. (a) Table of experimentally confirmed inhibitors of PHLPP2 and their estimated free energy of binding (kcal/mol) as well as IC50. (b) Two-Dimensional representations of selected compounds from the docking study which inhibit PHLPP2 from the docking study.
We next undertook a kinetic analysis of select compounds to determine their mechanism of inhibition. Because the chemical and virtual screen focused on the isolated phosphatase domain, we expected inhibitors to be primarily active-site directed rather than allosteric modulators. Determination of the rate of substrate dephosphorylation in the presence of increasing concentrations of the inhibitors revealed three types of inhibition: competitive, uncompetitive, and noncompetitive (data not shown). We docked pNPP (Figure 5a) and a phosphorylated decapeptide based on the hydrophobic motif sequence of Akt (Figure 5b; HFPQFpSYSAS) into the active site of our best homology model, in the same manner as described for the inhibitors, to determine which substrate binding sites our inhibitor compounds could be blocking. Competitive inhibitors (e.g., 7 (NCS 134149); Figure 5c,e) were predicted to effectively block the binding site of pNPP, as expected for a competitive inhibitor. In contrast, uncompetitive inhibitors (e.g., 4 (NCS 13378); Figure 5d) and most of the compounds determined from our virtual screen (e.g., 13 (NCS 45586); Figure 5f) were predicted to bind the hydrophobic cleft near the active site and interact with one of the Mn2+ ions. Noncompetitive inhibitors (e.g., 2 (NCS 47924)) tended to dock poorly into our model, as expected if they bind sites distal to the substrate-binding cavity. Note that pNPP is a small molecule which, although it binds the active site and is effectively dephosphorylated, does not recreate the complex interactions of PHLPP with hydrophobic motifs and large peptides. Therefore, the type of inhibition we observe toward pNPP may not necessarily hold for peptides or full-length proteins. Importantly, we identified a number of inhibitors predicted to dock well in the active site and with kinetic parameters (competitive or uncompetitive inhibition) consistent with such docking.
Figure 5.

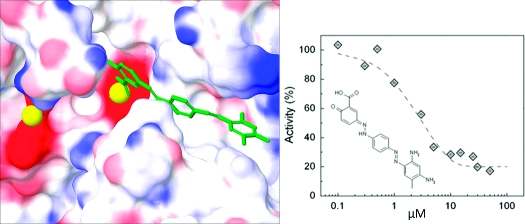
Model of active site of PHLPP2 docked with substrate or inhibitors. (a) pNPP, (b) a phosphopeptide based on the hydrophobic motif of Akt (HFPQFpSYSAS, where the phosphorylated Ser corresponds to Ser473); (c) compound 7, (d) compound 4, and (f) compound 13 were docked in the PHLPP2 model 4. Ligands are represented in tube format, Mn2+ ions are represented as yellow spheres, and surface representation of PHLPP2 shows the electrostatic potential where blue represents positive, white neutral, and red negative electrostatic potential. Docked substrates or inhibitors are in green, the phosphate groups are highlighted in purple. * denotes the carbonyl group of Gly 745 in all views. (e) Lineweaver−Burke analysis of inhibition mechanism of compound 7. The activity of PHLPP was measured at eight different concentrations of pNPP in the presence of DMSO (gray solid circle), 5 μM (solid square), 10 μM (gray solid square), or 30 μM (open square) compound 7.
We next tested whether the six most-promising compounds: (1) inhibited PHLPP in cells, and (2) were selective for PHLPP compared with other phosphatases in vitro. To investigate PHLPP inhibition in cells, HT29 cells (colon cancer cell line) were treated for 24 h with compounds at concentrations of either 100 or 250 μM, and the effect on Akt was assessed by examining the phosphorylation state of Akt on Ser 473 (the direct target of PHLPP) and, in addition, the phosphorylation state of two downstream targets of Akt, FoxO1, and GSK3 (Figure 6a,b). We chose to use HT29 cells for this study because the protein levels of PHLPP are not regulated by the level of Akt activity, as occurs in other cell lines via a recently described negative feedback loop.50,51 All compounds except 2 (Figures 6a,b, lane 7) caused an increase in the phosphorylation of Akt on Ser 473, with maximal increases of 4-fold caused by several of the compounds (see Figure 6b). Of these, four compounds (1 (NCS 117079), 24 (NCS 11404), 4, and 13) caused corresponding increases in the phosphorylation of the downstream substrates GSK3 α/β on Ser21/9 and FoxO1/3α on Thr 24/32. We have previously shown that knockdown of either PHLPP1 or PHLPP2 increases the phosphorylation of FoxO1 on Thr 24 and GSK3β on Ser9.(8) Some compounds selectively increased the phosphorylation of the downstream substrates but not Akt (e.g., lane 7), and others caused an increase in the phosphorylation of Akt but only one of the downstream substrates (e.g., lane 8). Compound 4 (lane 5) induced cells to detach from culture dishes, reflecting toxicity of the compound.
Figure 6.

Cellular potency and specificity of PHLPP inhibitors. (a,b) Effect of different inhibitors on Akt phosphorylation. HT29 cells in DMEM supplemented with 5% FBS were treated for 24 h with different compounds (see table). Blots were probed with antibodies specific to phospho-Akt (S473), phospho-GSK3α/β (S21/9), phospho-FoxO1/3a (T24/32), and actin. (b) Densitometric analysis was performed on blots from three separate experiments. The levels of phosphoproteins are normalized to actin. The phosphorylation of Akt at Ser 473 (white bars), FoxO 1/3 (light-gray bars), and GSK 3α/β (dark-gray bars) is calculated relative to control lanes. The graph represents mean values ± SEM from three separate experiments. (c) Selectivity toward other phosphatases. pNPP (8 mM) was incubated in 125 μL of the optimal assay buffer for each protein, in the presence of the PP2C domain of PHLPP2 (black bars), PP1 (dark-gray bars), PP2B (white bars), or PP2Cα (light-ray bars). Compounds (see table) were added. The activity of the enzyme is relative to the DMSO control. The graph represents mean values ± SEM of at least three different experiments. * denotes that compound 13 was tested at 100 and not 250 μM (d,e) in vitro inhibition curves for compound 1 (d) and 13 (e). The inhibitor, diluted in DMSO, was incubated in assay buffer with 8 mM pNPP in the presence of 1 μM enzyme. Activity was calculated relative to DMSO alone. The mean activity ± SEM for five different experiments are represented (gray solid circle, 1; gray tilted solid squares, 13) and were fit with eq 1.
In parallel with the cell study above, we tested the in vitro selectivity of the inhibitors by measuring their effect on the activity of the phosphatase domain of related and unrelated phosphatases. Figure 6c shows the effect of these inhibitors on the in vitro activity of the phosphatase domain of PHLPP2 (black bars), PP1 (dark-gray bars), PP2B (white), and PP2Cα (light-gray bars). A number of compounds proved to be selective for the PP2C domain of PHLPP2 over the other phosphatases tested, including the related family member, PP2Cα. We should point out that, among the 54 inhibitors for PHLPP2 tested against PHLPP1, none was specific; at best, IC50s were 5-fold different (data not shown), not unexpected given the high sequence homology of the phosphatase domains of the two isoforms. The most selective molecule for the PHLPP phosphatase domain was compound 1: a concentration of 10 μM resulted in 80% inhibition of PHLPP2, with no significant effect on the activity of the other phosphatases. A 10-fold higher concentration resulted in approximately 50% inhibition of PP1 and PP2Cα, indicating that the selectivity for PHLPP was over an order of magnitude. Importantly, compound 1 increased Akt phosphorylation and activity in cells. Compound 13 was also noteworthy: it selectively inhibited PHLPP2 compared to the other phosphatases tested (Figure 6c, lane 6) and was one of the compounds that induced a robust increase in the activity of Akt (Figure 6b, lane 6). Thus, compounds 1 and 13 were chosen for further studies. Their IC50 values for inhibition of pNPP dephosphorylation were 5.45 ± 0.05 μM and 3.70 ± 0.06 μM, respectively (Figure 6d,e).
The inhibitory potency of compounds 1(52) and 13 on PHLPP activity in cells was determined next. To discriminate between specific effects of the compounds on PHLPP activity vs nonspecific effects, we took advantage of the finding that PHLPP specifically and directly dephosphorylates Ser473 of Akt and does not dephosphorylate Thr308.(7) For these experiments, we examined the effect of the compounds on Akt phosphorylation in serum-starved cells in case PHLPP suppression is more dominant when Akt phosphorylation is maximally suppressed.
COS 7 cells, serum-starved for 24 h, were treated with increasing concentrations of the inhibitors for 35 min and the phosphorylation of Akt on Ser473 and Thr308 was determined; we also examined the activity of Akt by probing for the phosphorylation of downstream substrates with antibodies that recognize phosphorylated Akt substrates (Figure 7a−d). Treatment of cells with compound 1 resulted in an approximately 6-fold increase in the phosphorylation of Ser473 and a 4-fold increase in the phosphorylation of downstream substrates, with half-maximal increases caused by 29.1 ± 0.3 and 36.4 ± 0.3 μM inhibitor for Ser473 and substrate phosphorylation, respectively. Importantly, at the concentration of compound causing a half-maximal increase in phosphorylation of Ser473, there was little effect of compound 1 on the phosphorylation of Thr308 (Figure 7b, arrow). Higher concentrations did cause an increase in phosphorylation of Thr308; half-maximal increase in Thr308 phosphorylation was observed for 132 ± 1 μM inhibitor. Thus, the compound selectively increased Akt phosphorylation on Ser473 and this resulted in a commensurate increase in the activity of Akt. A similar increase in the phosphorylation on Ser473 and substrate phosphorylation was observed with compound 13. Specifically, this compound also caused an approximately 4-fold increase in the basal phosphorylation of Akt, with half-maximal increase caused by 70.6 ± 0.6 μM compound. The phosphorylation of the downstream substrates FoxO 1/3α and GSK3 β increased with comparable kinetics. For this compound, the sensitivity of phosphorylation of Akt on Thr308 was not significantly different from that of Ser473 (Figure 7d, lower panel). These data reveal that compounds 1 and 13 selectively inhibit the activity of PHLPP toward Akt in cells, with IC50 values of approximately 30 and 70 μM, respectively. Compound 1 has higher selectivity toward PHLPP as assessed by the uncoupling of phosphorylation at Ser473 and Thr308. At concentrations above 100 μM, this compound loses specificity as evidenced by the increase in Akt phosphorylation at both Ser473 and Thr308. Compound 13 was considerably less effective at modulating Ser473 phosphorylation in cells grown in serum (data not shown). In contrast, compound 1 increased Akt phosphorylation on Ser473 by 2-fold with comparable kinetics in the presence of serum. This acute treatment of cells with inhibitors did not change the levels of PKC isozymes (data not shown), as expected because PHLPP provides chronic control of PKC levels(9) (see Discussion).
Figure 7.

Effect of inhibitors on Akt-dependent signaling in cells. COS 7 cells were incubated in serum-free DMEM for 24 h and then treated for 35 min with compound 1 (a,b) or 13 (c,d) at different concentrations. The control corresponds to 35 min of treatment with DMSO alone. The graph represents mean values ± SEM from four separate experiments. (a,b) Blots were probed with antibodies specific to phospho-S473, phospho-T308, an antibody specific to phosphoserine or phosphothreonine in a consensus sequence for Akt, and actin. (b) The levels of phospho-Akt at Ser473 (open tilted square), at Thr308 (open circle) and phosphorylation of Akt substrates (gray tilted solid square) are normalized to total actin and relative to the level of phosphorylation in the control lanes. Arrow denotes IC50 value for inhibition of Ser473 phosphorylation. (c,d) Blots were probed with antibodies specific to phospho-S473, phospho-T308, phospho-GSK3 α/β, phospho-FoxO1/3a, and actin. (d) The levels of phospho-Akt at Ser473 (open tilted square), at Thr308 (open circle), and phosphorylation of Akt substrates (GSK 3: open square; FoxO, gray solid downward-pointing triangle) were normalized to total actin and relative to the level of phosphorylation in the control lane. Arrow denotes IC50 value for inhibition of Ser473 phosphorylation. (e,f) Effect of compound 1 on the agonist-evoked phosphorylation of Akt. COS 7 cells were incubated in serum-free DMEM for 24 h. The cells were pretreated with DMSO (dark symbols) or compound 1 (50 μM) (open symbols) for 35 min. EGF (1 mg·mL−1) was then added at various time. Blots were probed with the specified antibodies. (d) The levels of phospho-Akt and phospho-ERK were normalized to actin. The phosphorylation of Akt at S473 (solid tilted square, DMSO; open tilted square, 1), of Akt at T308 (solid circle, DMSO; open circle, 1) and ERK (solid upward-pointing triangle, DMSO; open upward-pointing triangle, 1) are relative to the levels of phosphoprotein in unstimulated cells treated with DMSO (lane 1). The graphs represent mean values ± SEM from five separate experiments.
PHLPP controls the basal phosphorylation state of Akt as well as the amplitude of the agonist-evoked increase in phosphorylation of Akt.(8) We therefore tested the effect of the inhibitors on agonist-evoked phosphorylation of Akt by pretreating serum-starved COS-7 cells with or without 50 μM of 1 and then stimulating with EGF (Figure 7e,f, open symbols (DMSO) and dark symbols (inhibitor)). As in previous experiments, the basal phosphorylation at Ser473 was significantly higher in cells treated with 1 compared with DMSO (Figure 7e, compare lanes 1 and 7). In cells treated with DMSO, addition of EGF caused an approximately 7-fold increase in the phosphorylation of Akt on Ser473 that peaked after 8 min (Figure 7f, top panel). In contrast, EGF had a smaller effect on the already elevated phosphorylation of Akt on Ser473 in cells treated with 1 (Figure 7f, top panel). Phosphorylation at Thr308 was slightly elevated under basal conditions in cells treated with the inhibitor compared to control cells (Figure 7e, lanes 1 and 7). EGF treatment resulted in an approximately 6-fold increase in p308 phosphorylation for both control and treated cells, which peaked earlier in inhibitor-treated cells (Figure 7f, middle panel). Thus, the magnitude of the increase in p308 and p473 phosphorylation was comparable in inhibitor vs DMSO-treated cells, but the rate of phosphorylation on p308 was significantly faster in inhibitor-treated cells and, most strikingly, the basal phosphorylation on Ser473 was highly elevated in inhibitor-treated cells. To discern whether this coupled phosphorylation of p473 and p308 resulted from off-target effects of the inhibitor or reflected the stabilization of phosphate on T308 when Ser473 is phosphorylated,(8) we examined the EGF-dependent phosphorylation of ERK 1/2: the kinetics and magnitude of the EGF-stimulated increase in ERK phosphorylation were the same for control cells and cells treated with the inhibitor (Figure 7f, bottom panel).
Because a major function of activated Akt is to promote cell survival, a function enhanced by loss of PHLPP,(7) we asked whether treatment of cells with compounds 1 or 13 suppressed etoposide-induced apoptosis. COS 7 cells were pretreated with DMSO, 1, or 13 (at 50 μM) for 30 min, then treated with DMSO or etoposide for 24 h (Figure 8). Etoposide treatment of control cells resulted in a (3.4 ± 0.4)-fold increase in apoptotic cells, as assessed by Trypan Blue exclusion. Pretreatment of cells with compound 1 reduced the magnitude of this increase by approximately 30%, to only (2.6 ± 0.3)-fold, and pretreatment with compound 13 essentially abolished the etoposide-induced increase in apoptotic cells. Note that the basal level of apoptotic cells was comparable in control cells and cells treated with compound 13 (approximately 3.4%) but elevated in cells treated with compound 1 (approximately 12%). These data reveal that the PHLPP inhibitors protect cells against etoposide-induced apoptosis.
Figure 8.

Effect of inhibitors on etoposide-induced cell death. COS 7 cells in DMEM supplemented with 0.1% FBS were treated for 30 min with DMSO, 1, or 13 at 50 μM followed by a 24 h treatment with DMSO or etoposide. Cells were then submitted to a Trypan Blue exclusion assay. The graph shows the etoposide-induced increase in cell death (compared to DMSO) and represents mean values ± SEM of three separate experiments .* corresponds to p values <0.0005 and ** corresponds to p values ≤2 × 10−5.
Discussion
By combining experimental and computational methods, we have identified the first set of inhibitors of the phosphatase PHLPP, a member of the PP2C family of phosphatases that has hitherto remained refractory to identification of general inhibitors. Specifically, we have identified small molecules that selectively inhibit PHLPP and show that treatment of cells with these inhibitors increases both the basal and agonist-evoked phosphorylation of Akt. Most relevant for therapeutic goals, these inhibitors selectively suppress cellular apoptosis.
We have specifically identified two molecules, with chemically distinct backbones that display selectivity for PHLPP both in vitro and in cells. Compound 1 (1-amino-9,10-dioxo-4-(3-sulfamoylanilino)anthracene-2-sulfonic acid, sodium salt) possesses an anthracene core, whereas compound 13 (1,3-[[4-(2,4-diamino-5-methylphenyl)diazenylphenyl]hydrazinylidene]-6-oxocyclohexa-1,4-diene-1-carboxylic acid) has aromatic groups linked by two diazene bonds. They inhibit PHLPP2 activity in vitro with IC50 values of 5.45 ± 0.05 and 3.70 ± 0.06 μM, respectively. Kinetic analyses reveal that both molecules are noncompetitive inhibitors with respect to pNPP. Both molecules are selective for PHLPP compared to other phosphatases tested: neither molecule affected the activity of PP2B at the highest concentration tested (100 μM) and inhibited PP1 and PP2Cα with IC50 values of approximately 100 μM (Figure 6c).
Both compound 1 and 13 show the potential for therapeutic development. Quikprop from the Schrodinger Suite was run to estimate properties that are potentially important to compound solubility, permeability, and drug development.(53) The Lipinski rules indicate that a potential drug compound should not contain more than 5 H-bond donors, 10 H-bond acceptors, a LogP (predicted octanol/water partition coefficient) greater than 5, or a molecular weight greater than 500 Da(54) (Table 1). There are no Lipinski violations for 13, and 1 contains one violation from extra H-bond acceptors. Virtual docking of 13 (Figure 5f) shows multiple interactions between the aromatic cycles of the compounds and residues composing the hydrophobic cleft as well as coordination of one Mn2+ by the acid moiety. Compound 1 was discovered by chemical screening and does not perform well in the virtual docking, so little information can be gained this way. Note that both compounds are a dark color and both tend to precipitate in the cell culture medium at high concentration (>300 μM).
Table 1. Calculated Chemical Properties of Compounds 13 and 1a.
| compd | mol weight (daltons) | donor H-bonds | acceptor H-bonds | LogP |
|---|---|---|---|---|
| 13 | 390 | 4 | 7.75 | 2.27 |
| 1 | 473 | 4 | 12 | 0.097 |
Quikprop was run to calculate the chemical properties that make up the Lipinski rules.
Cellular studies with compound 1 revealed that, at concentrations below 100 μM, it selectively inhibited the PHLPP-catalyzed dephosphorylation of Akt on Ser473 with little effect on the dephosphorylation on Thr308, a site that is not recognized by PHLPP. Indeed the IC50 value for inhibition of Ser473 dephosphorylation (29.1 ± 0.3 μM) was considerably lower than that for Thr308 dephosphorylation (132 ± 1 μM). At concentrations above 100 μM, the phosphorylation of Thr308 increased. This could result from off-target effects at higher concentrations, possibly by modulation of other phosphatases, or could reflect the stabilization of the phosphorylation on Thr308 by phosphorylation on Ser473.(8) Interestingly, elevation of the phosphorylation of Ser473 alone, and not Thr308, resulted in an accompanying increase in the phosphorylation of downstream substrates of Akt, including FoxO1/3 and GSK3 α/β. These data reveal that phosphorylation on only Ser473 activates cellular Akt sufficiently to mediate downstream signaling. Compound 13 was also an effective inhibitor of Akt dephosphorylation but displayed less selectively toward inhibiting the dephosphorylation of Ser473 compared to Thr308. Thus, both compounds are effective inhibitors of Akt dephosphorylation, with compound 1 displaying almost 1 order of magnitude selectivity for Ser473 compared to Thr308.
Akt plays a key role in controlling the balance between cell survival and cell death. Disruption of this balance results in severe pathological states. Most notably, in heart disease and diabetes, the balance is pushed toward cell death. Therapies are thus targeted toward enhancing cell survival. The ability of our compounds to selectively activate Akt, by inhibiting its negative regulator PHLPP, poises them as prime therapeutic molecules for diseases in which Akt activity is repressed. Indeed, both molecules effectively suppressed etoposide-induced apoptosis, with compound 13 abolishing etoposide-induced apoptosis under the conditions of our assays. The more dramatic effect of compound 13 may reflect its ability to modulate the phosphorylation of both Ser473 and Thr308. Thus, these molecules hold promise as the basis for novel therapeutics for diseases in which cell survival pathways are suppressed.
PHLPP also controls PKC isozymes: particularly in the case of the conventional isozymes, phosphorylation of the hydrophobic motif is constitutive and protects PKC from degradation. Dephosphorylation at this site triggers the down-regulation of the enzyme. Thus, PHLPP controls the amplitude of the PKC signal by controlling the levels of PKC. Genetic depletion of PHLPP results in elevated levels of PKC because phosphate on the hydrophobic motif is favored. However, PKC has a relatively long half-life (greater than 24 h), so chronic inhibition of PHLPP is required to modulate PKC levels. For this reason, the acute treatment of cells with inhibitors of PHLPP in this study did not modulate PKC levels.
In addition to uncovering new molecules to activate Akt signaling in cells, this work underscores both the value of computational work in drug discovery campaigns and the value of using chemical data to increase the efficiency of computational work. The efficiency of the experimental work was largely increased by the use and refinement of a virtual model in this drug development campaign. The primary screen of the Diversity Set identified 50 inhibitors out of 1990 tested, a random search yielding 2.5% hits. Overall, we identified 95 inhibitors of PHLPP in this set, representing 5% of the tested compounds. When the same set was first submitted to a virtual screen, 14 inhibitors were found out of 36 tested, increasing the experimental hit ratio to 16%. Finally, when the virtual screen was combined with a structural component, the experimental hit ratio reached 25%, a 10-fold increase, highlighting the value of computational methods for drug discovery.
Given the lack of structural information available for PHLPP, this work had to branch out from traditional docking studies. Although crystal structures are commonly preferred for docking studies, we have demonstrated that homology models, when carefully constructed and correlated to experimental results, can provide success in the search for new inhibitory ligands. The construction of our model included the modeling of metal ions and water molecules in the active site and some changes in conformations. As demonstrated by these data, placing metal ions in the active site can be extremely important in molecular modeling studies of this nature. Water molecules can also play a large role in inhibitor binding, but it is difficult to predict how the water molecules will be coordinated in the active site and which water molecules may be displaced by different inhibitors without crystallographic information. This may be the reason we did not observe an improvement in performance when adding water molecules to our homology structure. In addition to identifying new inhibitors by virtual docking, our docking studies also revealed the mechanism of binding of the various inhibitions (competitive, uncompetitive, or noncompetitive), with results validated by biochemical studies.
Compounds that inhibit PP2C phosphatases have been fairly refractory to identification, with few published studies.14,15 Here, we have identified a number of small molecules that not only inhibit this family of phosphatases but also selectively inhibit PHLPP compared to other phosphatases, including the highly related phosphatase PP2Cα. The combination of computational and chemical work allowed us to identify a variety of structurally distinct inhibitors for a phosphatase target without the need for a massive high-throughput chemical screen. It is noteworthy that these tests were performed without the use of robotics or highly automated methods, and the virtual screening was performed on a common desktop computer. Thus, collaboration between chemical and virtual screening provides an extraordinarily effective approach to drug discovery. Further refinement of these compounds to tune them to higher affinity and more specific inhibitors offers great therapeutic potential. Our identification of these new inhibitors for a PP2C family member is particularly relevant because these compounds could be potential therapeutics given the strategic position of PHLPP in cell survival pathways.
Experimental Section
The Diversity Set and compounds identified by virtual screen were obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis of the National Cancer Institute (http://dtp.nci.nih.gov/branches/dscb/repo_open.html). The compounds were used as provided in the in vitro assay. Purity of the compounds used to treat cells was verified by LC/MS using a Thermo LCQdeca mass spectrometer coupled with a Michrom BioResource HPLC at the UCSD Chemistry and Biochemistry Mass Spectroscopy Facility. Negative ion mode electrospray ionization was used. Purity was found to be >90% for compounds 24 and 4, 80% for compound 2, 60% for compound 7, and 55.5% for compound 13. See Supporting Information for compound 1.
The following phosphatases were purchased: PP1 (New England Biolabs), PP2B/calcineurin (Enzo). PP2Cα was purified from E. coli as previously described.(54)
The following polyclonal antibodies were purchased from Cell Signaling: phosphospecific to phosphorylated Akt at Ser473 (S473) (no. 9271), phosphospecific to phosphorylated Akt at Thr308 (T308) (no. 9275), phosphospecific to phosphorylated Ser/Thr Akt substrate (no. 9611), phosphospecific to phosphorylated GSK 3 α/β at Ser 21 and Ser 9, respectively (S21/9) (no. 9331), phosphospecific to phosphorylated FoxO1/3a at Thr 24 and Thr 32, respectively (T24/32) (no. 9464), phosphospecific to phosphorylated p44/42 MAPK (Erk 1/2) at Thr 202 and Tyr 204 (T202/Y204) (no. 9101), antibody against p44/42 MAPK (Erk 1/2) (no. 9102). Monoclonal antibody against actin was purchased from Sigma-Aldrich (no. A4700).
Experimental in Vitro Screen
In each well of a 96-well plate, 125 μL of a reaction mixture (100 mM tricine pH 7.5, 100 mM NaCl, 4 mM DTT, 0.02 mg·mL−1 BSA) containing 8 mM pNPP as the substrate, 1 μM enzyme and 100 μM compound were added. Reactions occurred at 23 °C. The optical density was monitored over time at 405 nm using an Emax Precision microplate reader (Molecular Devices). The absorbance was plotted against the time, and the slope was calculated. Background was averaged from four different reactions in the absence of enzyme and subtracted. Eight different controls (containing 5 μL of DMSO) were averaged and used to calculate the relative activity.
In Vitro Inhibition Concentration Assay
The reactions occurred in the same conditions as described above except that the inhibitor was added at seven different concentrations and DMSO served as a control. The relative activity was set at 100% for DMSO. The data were then fit to the eq 1:
The IC50 value is defined by −C0 ln(1/2).
Homology Modeling
The PP2C domain sequence of PHLPP2 (residues 745−1102) was used to make a homology model with the program MODELER using the PP2C domain of PP2Cα as the reference structure.19,20 The two sequences were aligned using ClustalW. Next a model of PHLPP2 was created from the reference structure using MODELER with default parameters. Further refinement of the model was performed by placing varying amounts of Mn2+ ions or water molecules in the active site and then relaxing the structure with Macromodel from the Schrodinger Suite.(49) The OPLS_2005 force field was used with 500 iterations of the gradient method.
Similarity Searches and Compound Library Generation
Accelrys software was used to search the NCI open repository, using PHLPP2 inhibitors determined previously in this study as reference compounds. Groups of inhibitors (families of compounds, or top inhibitors) were submitted as the reference compounds using the “Find Similar Molecules by Fingerprints” protocol provided with Accelrys Discovery Studio. Long range functional class fingerprint description 6 keys (FCFP 6 keys) were used with a Tanimoto distance coefficient to compute a similarity score. Top scoring compounds were selected for virtual screening.
Docking
The GLIDE virtual screening application in Schrodinger Molecular Modeling Suite was used to screen compounds using three levels of docking precision. A modified version of the Chemscore function is employed by GLIDE to assign a score to each ligand in all poses. Glide HTVS was run on all compounds to perform a complete conformational and positional search of three-dimensional space in the active site. For the compounds that scored in the highest 20%, GLIDE SP (standard precision) was run. Similarly for the top 20% high scoring GLIDE SP compounds, GLIDE XP (extra precision) was run too. Flexible docking was allowed in all stages and default parameters from the Virtual Screening Workflow were used in all docking studies, in addition to the aforementioned modifications to the percent of compounds entering each stage. All final scores and poses came from GLIDE XP. The grids were generated for each model in the phosphatase active site with the XYZ coordinates (−27.54, 26.44, 46.38). An inner box which must contain the center of each ligand docked was 14 Å in each direction, and the outer box in which all parts of the ligand must bind was 44 Å in each direction. The relatively large box allowed for a variety of docking poses and accepted large compounds. No other constraints were placed on the grids.
Homology Model Evaluation
Docking of the inhibitors found in the chemical screen into each modified homology model helped determine the best model to use for further docking studies. In addition, the experimentally validated binding compounds with a G-score below −7 were considered to be virtual hits in this study (Figure 2c). The docking protocol was modified slightly in the evaluation of models so that all known inhibitors were permitted to proceed through the three stages of GLIDE. Also, the entire diversity set was docked into the models with Mn2+ in the active site, these studies were performed with only the top 4% of compounds reaching the final GLIDE XP stage and receiving scores as described in the docking protocol. Models were evaluated based on the number of experimentally confirmed inhibitors receiving a G-score of better than −7.
Cell Culture and Immunoblotting
Cells are maintained in DMEM supplemented with 5% FBS and 1% penicillin/streptomycin, at 37 °C in 5% CO2. The medium was aspirated before addition of lysis buffer (50 mM Tris pH 7.5, 1 mM sodium pyrophosphate, 20 mM NaF, 2 mM EDTA, 1% Triton, 200 μM benzamidine, 40 μg mL−1 leupeptin, 1 mM PMSF, and 1 μM microcystin). Whole cell lysates were analyzed on 7.5% SDS-PAGE gels and Western blotting using the indicated antibodies. Chemiluminescent signals were imaged by an Alpha Innotech MultiImage III, and densitometric analysis was performed using AlphaView software (version 2.0.0.9).
Inhibitor Treatment of Cells
HT29 cells were plated in 12-well plates and maintained in 5% FBS in DMEM. Four μL of the different compounds diluted in DMSO were added to fresh medium in each well. Cells were lysed in 200 μL of lysis buffer, on ice, 24 h after addition of the inhibitors. For cellular IC50 studies, COS 7 cells were plated in 24-well plates. The cells were starved for serum 24 h prior to the assay by placing them in serum-free DMEM. The inhibitor (1 or 13) was added at various concentrations (4−400 or 3−300 μM, respectively) to each well, and the cells were allowed to sit for 35 min in the incubator before the plate was placed on ice. Cells were lysed in 100 μL of lysis buffer, and lysates were analyzed as described above. DMSO was used as a control. The relative activity was set to 1 for DMSO. The data were then fit to eq 2.
For time course studies, COS 7 cells were plated in a 24-wells plate. The cells were starved for serum 24 h prior to the assay by placing them in serum-free DMEM. Cells were pretreated with DMSO or 1 (50 μM) for 35 min. EGF was then added at different time to a final concentration of 1 μg mL−1. Between the different time points, cells were placed back in the incubator. The control was obtained in the absence of EGF. The plate was then placed on ice, and the cells were lysed in 100 μL of lysis buffer.
Apoptosis Assay
COS 7 cells were plated in a 6-well plates. When cells reached 80% confluency, medium was aspirated and replaced with 0.1% FBS in DMEM. DMSO, 1, or 13 were added to each well to a yield a final concentration of 50 μM. Following 30 min incubation at 37 °C, DMSO or etoposide (50 μM) was added. After 24 h, cells were detached from the plate using trypsin, centrifuged at 100g for 5 min, 4 °C, and stained with Trypan Blue. Cells were manually counted, and the percentage of dead cells was calculated.
Acknowledgments
We thank members of the Newton lab for critical reading of the manuscript, Chelsea Buma for help with the in vitro screen, Dr. Arneh Babakhani for guidance with the in silico screen, Drs. Nadia Fomina and Adah Almutairi for help with the synthesis, Drs. Li Xie and Philip E. Bourne for help with the homology modeling, and Drs. Yann Gambin and Edward Lemke for help with data analysis. This work was supported by a Juvenile Diabetes Research Foundation grant 3-2008-478 (E.S.), Molecular Biophysics Training grant GM08326 (W.S.), the National Science Foundation grant MCB-0506593, NBCR, CTBP, and Howard Hughes Medical Institute (J.A.M.), and National Institutes of Health grants GM31749 (J.A.M.) and GM067946 (A.C.N.).
Supporting Information Available
Table containing the NSC IDs of compounds that inhibit PHLPP 2 in vitro, grouped in clusters according to their chemical structures, best homology model in PDB format, and description of the synthesis of 1 and purification of compounds 1 and 13. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: DMEM, Dulbecco’s modification of Eagle’s medium ; DMSO, dimethyl sulfoxide; EGF, epidermial growth factor; ERK, external signal regulated protein; FBS, fetal bovine serum; FoxO, Forkhead box O; GSK-3, glycogen synthase kinase-3; MAPK, mitogen activated protein kinase; PDK-1, phosphoinositide-dependent kinase-1; PH, pleckstrin homology; PHLPP, PH domain leucine-rich repeat protein phosphatase; PI3K, phosphatidylinositol-3 kinase; PKC, protein kinase C; pNPP, para-nitrophenyl phosphate; PPM, protein phosphatase magnesium-dependent; PMSF, phenylmethyl sulfoxide; PP1, protein phosphatase 1; PP2A, protein phosphatase 2A; PP2B/calcineurin, protein phosphatase 2B; PP2C, protein phosphatase 2C; PPP, phosphoprotein phosphatase; PTEN, phosphatase and tensin homologue; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel.
Supplementary Material
References
- Cantley L. C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296 (5573), 1655–1657. [DOI] [PubMed] [Google Scholar]
- Newton A. C. Lipid activation of protein kinases. J. Lipid Res. 2009, 50 (Suppl), S266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning B. D.; Cantley L. C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129 (7), 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T.; Dixon J. E. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999, 9 (4), 125–128. [DOI] [PubMed] [Google Scholar]
- Trotman L. C.; Alimonti A.; Scaglioni P. P.; Koutcher J. A.; Cordon-Cardo C.; Pandolfi P. P. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 2006, 441 (7092), 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y.-C.; Huang K.-Y.; Yang C.-H.; Yang Y.-S.; Lee W.-Y.; Chiang C.-W. Regulation of Phosphorylation of Thr-308 of Akt, Cell Proliferation, and Survival by the B55α Regulatory Subunit Targeting of the Protein Phosphatase 2A Holoenzyme to Akt. J. Biol. Chem. 2008, 283 (4), 1882–1892. [DOI] [PubMed] [Google Scholar]
- Gao T.; Furnari F.; Newton A. C. PHLPP: A Phosphatase that Directly Dephosphorylates Akt, Promotes Apoptosis, and Suppresses Tumor Growth. Mol. Cell 2005, 18 (1), 13–24. [DOI] [PubMed] [Google Scholar]
- Brognard J.; Sierecki E.; Gao T.; Newton A. C. PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 2007, 25 (6), 917–931. [DOI] [PubMed] [Google Scholar]
- Gao T.; Brognard J.; Newton A. C. The Phosphatase PHLPP Controls the Cellular Levels of Protein Kinase C. J. Biol. Chem. 2008, 283 (10), 6300–6311. [DOI] [PubMed] [Google Scholar]
- Brognard J.; Newton A. C. PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 2008, 19 (6), 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweighofer A.; Hirt H.; Meskiene I. Plant PP2C phosphatases: emerging functions in stress signaling. Trends Plant Sci. 2004, 9 (5), 236–243. [DOI] [PubMed] [Google Scholar]
- Lammers T.; Lavi S. Role of Type 2C Protein Phosphatases in Growth Regulation and in Cellular Stress Signaling. Crit. Rev. Biochem. Mol. Biol. 2007, 42 (6), 437–461. [DOI] [PubMed] [Google Scholar]
- Mumby M. C.; Walter G. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 1993, 73 (4), 673–699. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H.; Durell S. R.; Feng H.; Bai Y.; Anderson C. W.; Appella E. Development of a Substrate-Based Cyclic Phosphopeptide Inhibitor of Protein Phosphatase 2Cδ, Wip1. Biochemistry 2006, 45 (44), 13193–13202. [DOI] [PubMed] [Google Scholar]
- Rogers J. P.; Beuscher; Flajolet M.; McAvoy T.; Nairn A. C.; Olson A. J.; Greengard P. Discovery of Protein Phosphatase 2C Inhibitors by Virtual Screening. J. Med. Chem. 2006, 49 (5), 1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J.; Plater L.; Elliott M.; Shpiro N. C.; Hastie J.; McLauchlan H.; Klevernic I.; Arthur J. S. C.; Alessi D. R.; Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007, 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26 (1), 127–132. [DOI] [PubMed] [Google Scholar]
- Zdychova J.; Komers R. Emerging role of Akt Kinase/Protein Kinase B signaling in Pathophysiology of Diabetes and Its Complications. Physiol. Rev 2005, 54, 1–16. [DOI] [PubMed] [Google Scholar]
- Elghazi L.; Bernal-Mizrachi E. Akt and PTEN: [beta]-cell mass and pancreas plasticity. Trends Endocrinol. Metab. 2009, 20 (5), 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elghazi L.; Rachdi L.; Weiss A. J.; Cras-Méneur C.; Bernal-Mizrachi E. Regulation of beta-cell mass and function by the Akt/protein kinase B signalling pathway. Diabetes, Obes. Metab. 2007, 9 (s2), 147–157. [DOI] [PubMed] [Google Scholar]
- Tuttle R. L.; Gill N. S.; Pugh W.; Lee J.-P.; Koeberlein B.; Furth E. E.; Polonsky K. S.; Naji A.; Birnbaum M. J. Regulation of pancreatic [beta]-cell growth and survival by the serine/threonine protein kinase Akt1/PKB[alpha]. Nature Med. 2001, 7 (10), 1133–1137. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi E.; Wen W.; Stahlhut S.; Welling C. M.; Permutt M. A. Islet beta cell expression of constitutively active Akt1/PKBalpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J. Clin. Invest. 2001, 108 (11), 1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal-Mizrachi E.; Fatrai S.; Johnson J. D.; Ohsugi M.; Otani K.; Han Z.; Polonsky K. S.; Permutt M. A. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet β cells. J. Clin. Invest. 2004, 114 (7), 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayascas J. R.; Wullschleger S.; Sakamoto K.; Garcia-Martinez J. M.; Clacher C.; Komander D.; van Aalten D. M. F.; Boini K. M.; Lang F.; Lipina C.; Logie L.; Sutherland C.; Chudek J. A.; van Diepen J. A.; Voshol P. J.; Lucocq J. M.; Alessi D. R. Mutation of the PDK1 PH Domain Inhibits Protein Kinase B/Akt, Leading to Small Size and Insulin Resistance. Mol. Cell. Biol. 2008, 28 (10), 3258–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ocana A.; Takane K. K.; Reddy V. T.; Lopez-Talavera J.-C.; Vasavada R. C.; Stewart A. F. Adenovirus-Mediated Hepatocyte Growth Factor Expression in Mouse Islets Improves Pancreatic Islet Transplant Performance and Reduces Beta Cell Death. J. Biol. Chem. 2003, 278 (1), 343–351. [DOI] [PubMed] [Google Scholar]
- Contreras J. L.; Smyth C. A.; Bilbao G.; Young C. J.; Anthony Thompson J.; Eckhoff D. E. Simvastatin induces activation of the serine−threonine protein kinase AKT and increases survival of isolated human pancreatic islets. Transplantation 2002, 74 (8), 1063–1069. [DOI] [PubMed] [Google Scholar]
- Armstrong S. C. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61 (3), 427–436. [DOI] [PubMed] [Google Scholar]
- Hausenloy D. J.; Tsang A.; Yellon D. M. The Reperfusion Injury Salvage Kinase Pathway: A Common Target for Both Ischemic Preconditioning and Postconditioning. Trends Cardiovasc. Med. 2005, 15 (2), 69–75. [DOI] [PubMed] [Google Scholar]
- Hausenloy D. J.; Yellon D. M. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc. Res. 2006, 70 (2), 240–253. [DOI] [PubMed] [Google Scholar]
- Mullonkal C. J.; Toledo-Pereyra L. H. Akt in Ischemia and Reperfusion. J. Invest. Surg. 2007, 20 (3), 195–203. [DOI] [PubMed] [Google Scholar]
- Zhu M.; Feng J.; Lucchinetti E.; Fischer G.; Xu L.; Pedrazzini T.; Schaub M. C.; Zaugg M. Ischemic postconditioning protects remodeled myocardium via the PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc. Res. 2006, 72 (1), 152–162. [DOI] [PubMed] [Google Scholar]
- Dorn G. W.; Souroujon M. C.; Liron T.; Chen C.-H.; Gray M. O.; Zhou H. Z.; Csukai M.; Wu G.; Lorenz J. N.; Mochly-Rosen D. Sustained in vivo cardiac protection by a rationally designed peptide that causes ε protein kinase C translocation. Proc. Natl. Acad. Sci. U.S.A. 1999, 96 (22), 12798–12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill E. N.; Mochly-Rosen D. The roles of PKC delta and epsilon isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem. Soc. Trans. 2007, 035 (5), 1040–1042. [DOI] [PubMed] [Google Scholar]
- Budas G. R.; Churchill E. N.; Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol. Res. 2007, 55 (6), 523–536. [DOI] [PubMed] [Google Scholar]
- Churchill E.; Budas G.; Vallentin A.; Koyanagi T.; Mochly-Rosen D. PKC Isozymes in Chronic Cardiac Disease: Possible Therapeutic Targets?. Annu. Rev. Pharmacol. Toxicol. 2008, 48 (1), 569–599. [DOI] [PubMed] [Google Scholar]
- Downey J.; Davis A.; Cohen M. Signaling pathways in ischemic preconditioning. Heart Failure Rev. 2007, 12 (3), 181–188. [DOI] [PubMed] [Google Scholar]
- Zhang X.-J.; Xiong Z.-B.; Tang A.-L.; Ma H.; Ma Y.-D.; Wu J.-G.; Dong Y.-G. Rosiglitazone-induced myocardial protection against ischemia/reperfusion injury is mediated through a PI3K/Akt-dependent pathway. Clin. Exp. Pharmacol. Physiol. 2009, 37, 156–161. [DOI] [PubMed] [Google Scholar]
- Przyklenk K.; Maynard M.; Whittaker P. Reduction of infarct size with d-myo-inositol trisphosphate: role of PI3-kinase and mitochondrial KATP channels. Am. J. Physiol. Heart Circ. Physiol. 2006, 290 (2), H830–H836. [DOI] [PubMed] [Google Scholar]
- Zhang J.-H.; Chung T. D. Y.; Oldenburg K. R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 1999, 4 (2), 67–73. [DOI] [PubMed] [Google Scholar]
- Park H.; Bahn Y. J.; Jung S.-K.; Jeong D. G.; Lee S.-H.; Seo I.; Yoon T.-S.; Kim S. J.; Ryu S. E. Discovery of Novel Cdc25 Phosphatase Inhibitors with Micromolar Activity Based on the Structure-Based Virtual Screening. J. Med. Chem. 2008, 51 (18), 5533–5541. [DOI] [PubMed] [Google Scholar]
- Vidal D.; Blobel J.; Pérez Y.; Thormann M.; Pons M. Structure-based discovery of new small molecule inhibitors of low molecular weight protein tyrosine phosphatase. Eur. J. Med. Chem. 2007, 42 (8), 1102–1108. [DOI] [PubMed] [Google Scholar]
- Yu W.-M.; Guvench O.; MacKerell A. D.; Qu C.-K. Identification of Small Molecular Weight Inhibitors of Src Homology 2 Domain-Containing Tyrosine Phosphatase 2 (SHP-2) via in Silico Database Screening Combined with Experimental Assay. J. Med. Chem. 2008, 51 (23), 7396–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47 (7), 1739–1749. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47 (7), 1750–1759. [DOI] [PubMed] [Google Scholar]
- Kontoyianni M.; McClellan L. M.; Sokol G. S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2003, 47 (3), 558–565. [DOI] [PubMed] [Google Scholar]
- Das A. H.; Helps N. R.; Cohen P. T.; Bradford D Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 A resolution. EMBO J. 1996, 15 (24), 6798–6809. [PMC free article] [PubMed] [Google Scholar]
- Wehenkel A.; Bellinzoni M.; Schaeffer F.; Villarino A.; Alzari P. M. Structural and Binding Studies of the Three-Metal Center in Two Mycobacterial PPM Ser/Thr Protein Phosphatases. J. Mol. Biol. 2007, 374 (4), 890–898. [DOI] [PubMed] [Google Scholar]
- Pullen K. E.; Ng H.-L.; Sung P.-Y.; Good M. C.; Smith S. M.; Alber T. An Alternate Conformation and a Third Metal in PstP/Ppp, the M. tuberculosis PP2C-Family Ser/Thr Protein Phosphatase. Structure 2004, 12 (11), 1947–1954. [DOI] [PubMed] [Google Scholar]
- Schlicker C.; Fokina O.; Kloft N.; Grüne T.; Becker S.; Sheldrick G. M.; Forchhammer K. Structural Analysis of the PP2C Phosphatase tPphA from Thermosynechococcus elongatus: A Flexible Flap Subdomain Controls Access to the Catalytic Site. J. Mol. Biol. 2008, 376 (2), 570–581. [DOI] [PubMed] [Google Scholar]
- Li X.; Liu J.; Gao T. {beta}-TrCP-Mediated Ubiquitination and Degradation of PHLPP1 Are Negatively Regulated by Akt. Mol. Cell. Biol. 2009, 29 (23), 6192–6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfel N.; Niederst M.; Newton A., unpublished results.
- HPLC analysis revealed that NCI compound 117079 contained a mix of chemicals. To obtain the pure product, we synthesized it (see Supporting Information) and purified it by flash chromatography to 90% purity. The purified compound 1 displayed similar inhibitory parameters in vitro and in cells as the original NCI 117079 mix.
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46 (1−3), 3–26. [DOI] [PubMed] [Google Scholar]
- Fjeld C. C.; Denu J. M. Kinetic Analysis of Human Serine/Threonine Protein Phosphatase 2Cα. J. Biol. Chem. 1999, 274 (29), 20336–20343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.