

Abstract
Motivation: Traditional genomic prediction models based on individual genes suffer from low reproducibility across microarray studies due to the lack of robustness to expression measurement noise and gene missingness when they are matched across platforms. It is common that some of the genes in the prediction model established in a training study cannot be matched to another test study because a different platform is applied. The failure of inter-study predictions has severely hindered the clinical applications of microarray. To overcome the drawbacks of traditional gene-based prediction (GBP) models, we propose a module-based prediction (MBP) strategy via unsupervised gene clustering.
Results: K-means clustering is used to group genes sharing similar expression profiles into gene modules, and small modules are merged into their nearest neighbors. Conventional univariate or multivariate feature selection procedure is applied and a representative gene from each selected module is identified to construct the final prediction model. As a result, the prediction model is portable to any test study as long as partial genes in each module exist in the test study. We demonstrate that K-means cluster sizes generally follow a multinomial distribution and the failure probability of inter-study prediction due to missing genes is diminished by merging small clusters into their nearest neighbors. By simulation and applications of real datasets in inter-study predictions, we show that the proposed MBP provides slightly improved accuracy while is considerably more robust than traditional GBP.
Availability: http://www.biostat.pitt.edu/bioinfo/
Contact: ctseng@pitt.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Microarray technology is a promising methodology for predicting prognosis and response to treatment for cancer patients. However, a stable prediction model requires features selected from a large training dataset (Dobbin and Simon, 2005; Dobbin et al., 2008). Since microarray analyses and clinical trials are expensive as well as time and effort intensive, to validate information and to predict patient outcomes from individual studies, it is crucial to utilize accumulated inter-study data. For over a decade, microarray data have been accumulated from different array technologies or different versions within technologies performed on similar clinical samples. However, to use a dataset or integrated datasets from one platform to build a model that robustly and accurately predicts clinical characteristics of a new dataset or a new sample from another platform remains a challenge (Park et al., 2004; Tan et al., 2003).
Although current automated microarray assay systems have made microarray methodology straightforward, accurate use of genomic information from microarray analysis to classify patients or to predict patient prognosis is not trivial. To effectively predict clinical outcomes by genomic data requires careful data preprocessing, gene selection and model construction based on training data. The constructed model is then validated on independent test data. Currently, many prediction models have been developed and cross-validated within the single study used for the model construction (Pusztai and Leyland-Jones, 2008). As a result, the developed prediction models are usually underrepresented or over fitted due to a lack of heterogeneity of sampling and do not account for cross platform problems when training data and test data are generated from different microarray platforms and protocols.
Traditional prediction methods have used a gene-based (GBP) approach in which individual genes are selected as model components (Fig. 1A). A common cross platform problem encountered with such methods involves issues of gene missingness or gene mismatching. Frequently, genes in the prediction model based on training data cannot be found in the test data, which is termed as gene missingness in this article. Occasionally, genes matched across platforms may contain errors (i.e. gene mismatching). The need for inter-study prediction across different platforms is commonly encountered (Supplementary Fig. 1A). For example, suppose a pilot study has been performed in an old Affymetrix U95 platform and an effective prediction model has been constructed. The test site of another medical center may apply another commercial system (such as Agilent or Illumina platforms) or the original medical center may migrate to a newer U133 system. Many genes in the old training study may not be found in the new test study. Current cross-platform gene prediction methods use only those genes common to both training and test datasets (Irizarry et al., 2005; Shi et al., 2004, 2005). There are two main drawbacks to this GBP approach. One drawback is that the prediction model must be reconstructed with each new test dataset. Thus, the model cannot be created independently of the test data and the model elements must be adjusted every time different platforms of test data are used for prediction (Supplementary Fig. 1B). A second drawback involves the potential loss of important information. By ignoring the genes in the training dataset that are not found in the test dataset, important information from the training set may be lost. As a result, the prediction accuracy of GBP methods is unstable. Further instability arises because these methods are sensitive to noise in expression measurements.
Fig. 1.

Concept of GBP versue MBP methods: (A) the GBP method selects individual genes from training samples to construct a prediction model and uses the model to predict new samples. (B) The MBP method performs gene clustering a priori to form gene modules and uses these modules to construct a prediction model (color figure shown in Supplementary Figure 1A).
We propose a module-based prediction (MBP) strategy to overcome these aforementioned drawbacks. In MBP, groups of genes sharing similar expression patterns rather than individual genes are used as the basic elements of the prediction model (Fig. 1B). Such an approach borrows information from genes' similarity when genes are absent in test sets. By overcoming expression measurement noise and avoiding the problem of missing genes across platforms, the MBP method is hypothesized to yield robust predictions completely independent of information from the test data.
Recently, several similar approaches, such as metagene (Huang et al., 2003; Pittman et al., 2004; Potti et al., 2006; Spang et al., 2002; Tamayo et al., 2007; West et al., 2001, 2006), supergene (Park et al., 2007) and gene pathway module (Segal et al., 2004; van Vliet et al., 2007; Wong et al., 2008) methods, which used unsupervised gene cluster or supervised gene pathway information instead of individual gene information as predictors, have been reported and successfully applied in microarray prediction. However, unlike the MBP method proposed here, these methods mostly focus on improving prediction accuracy rather than on the robustness issue in inter-study gene prediction. Models constructed by most of these methods are still inevitably voided by gene missingness or expression measurement noise in the test study.
In this article, we will explore properties of the MBP strategy and compare them to those of the GBP approach. We will show, by simulation and applications to real microarray datasets, that MBP is more robust to gene missingness and expression measurement noise while not sacrificing prediction accuracy. We evaluate univariate gene/module filter selection in three popular classification methods, including k-nearest neighbor (KNN), linear discriminant analysis (LDA) and support vector machines (SVM). We also investigate an embedded method [prediction analysis of microarray (PAM)] and a multivariate gene/module selection method (R-SVM). Conceptually, the module-based approach can generally be applied to extend any existing classification method.
2 METHODS
2.1 Datasets and gene matching
Seven publicly available datasets were used to check the validity and adequacy of the MBP method (Supplementary Table 1). Four prostate cancer datasets, Luo (Luo et al., 2001), Yu (Yu et al., 2004), Welsh (Welsh et al., 2001) and Dhan (Dhanasekaran et al., 2001) were downloaded from the public domain. The malignant prostate cancer and its matched adjacent prostate tissue samples from Yu and Welsh datasets, and the malignant prostate cancer and its matched donor samples from Luo and Dhan datasets, were used for two sets of pairwise cross-platform analyses. Three lung cancer datasets, Beer (Beer et al., 2002), Bhat (Bhattacharjee et al., 2001) and Garber (Garber et al., 2001), were downloaded from publicly accessible information supporting the published manuscripts. Only the normal and the adenocarcinoma samples were used for analysis. All three datasets were from different platforms or different versions, and pairwise cross-platform analyses were performed. For matching genes across platforms, Entrez ID was used to match Affymetrix datasets using the R package ‘annotate’ (Kuhn et al., 2008), and a web-based match tool, MatchMiner, was used for cDNA datasets (Bussey et al., 2003). The genes sharing the same Entrez ID were averaged for their expression.
2.2 Module-based prediction
The MBP algorithm was developed under the rationale that genes sharing similar expression profiles could be grouped together and that a representative gene can be selected from the group of genes for prediction model construction. The disadvantages of GBP and motivation of MBP is illustrated in Supplementary Figure 1. An MBP schema compared with GBP is shown in Figure 1. The MBP algorithm involves five major steps: gene clustering, module merging, module selection, representative gene selection and model construction. More detailed mathematical notations and algorithm are shown in Supplementary Section A.
2.2.1 Data preprocessing
The training data are preprocessed using standard data filtering by eliminating genes with low expressions and genes not varying sufficiently across the training samples. The data are standardized using first column-wise and then row-wise standardization by subtracting column or row means and dividing by the corresponding column or row standard deviations.
2.2.2 Gene clustering
The processed data are clustered into K gene clusters by the classical K-means method (Hartigan and Wong, 1979). The clusters are defined as gene modules. Normally we choose K = 100 and we also tested K = 150 and 200 to show robustness of this selection. Many recent articles have discussed the issue of scattered genes that should not be clustered in gene clustering (Thalamuthu et al., 2006; Tseng and Wong, 2005). We have tested the penalized K-means approach (Tseng, 2007) and did not find improved performance over K-means, likely because the modules are already robust and including a small number of scattered genes in modules (clusters) does not deteriorate the prediction performance. We will use K-means algorithm to generate unsupervised modules throughout the article.
2.2.3 Module merging (δ-merge)
When the number of genes within a module is less than a given threshold δ, the small module is merged into its nearest neighboring module based on the minimum distance between module centroids. The selection of δ is determined by a probabilistic model described below to avoid missing genes of the entire module in the test study with high probability.
2.2.4 Module selection
Although the dimensionality has been reduced from several thousand genes to hundreds of modules in the MBP approach, the dimensionality is still high and proper feature (module) selection is needed to achieve better performance. In this article, we explore both univariate and multivariate feature selection methods. For univariate feature selection, the top M modules are selected according to their ranks of average absolute value of moderated t statistics (Tibshirani et al., 2002).
2.2.5 Representative gene selection
For each selected module, we use the ‘median gene’, that has the smallest sum of distances to other genes in the gene module, as the representative gene vector. These representative genes are used to construct the prediction model.
2.2.6 Model construction
For univariate feature selection methods, we examine three classical classification methods: LDA (Mardia et al., 1979), KNN (Dasarathy, 1991) and SVM (Cristianini and Shawe-Taylor, 2000). For embedded methods or multivariate feature selection, PAM (Tibshirani et al., 2002) and R-SVM (Zhang et al., 2006) are explored. Details of software and parameter setting of these methods are described in Supplementary Section B.
2.3 Estimate δ
One of the motivations to develop the MBP method is to build a prediction model solely on the training data, independent of test data and portable across studies with different microarray platforms. A necessary condition for the MBP procedure to succeed is that the test study should contain one or more genes in each gene cluster module in order to identify the representative genes in the prediction model. Below we provide a simplified probabilistic model to estimate the smallest δ needed to achieve the goal in the ‘module merging’ step. Assume π is the probability for a gene in the training study that is missing in the test study and the missingness of genes is independent of each other. The probability that the MBP method obtains no less than N matched genes in all of the K modules in the test study is
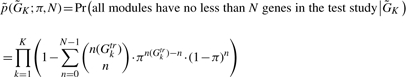 |
(1) |
where 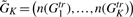 and n(Gktr) is the number of genes in cluster Gktr. In this article, we require N = 3. The probability calculation depends only on the gene missing probability π and the module sizes, n(Gktr).
and n(Gktr) is the number of genes in cluster Gktr. In this article, we require N = 3. The probability calculation depends only on the gene missing probability π and the module sizes, n(Gktr).
In our analysis of the seven datasets used in this article, we found that the cluster sizes generated by K-means clustering follow multinomial distributions very well (see Q–Q plots in Fig. 2); that is
| (2) |
Thus, the probability of each module in the test study to have no less than N genes (without δ-merge) becomes
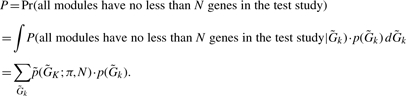 |
Fig. 2.

Q–Q plots: x-axis represents distribution of cluster sizes (i.e. number of genes in each cluster) generated by k-means clustering method and y-axis represents cluster sizes simulated by multinomial distribution. The linear trend in each plot shows good fitness of multinomial distribution.
With δ-merge algorithm, we require the probability of all modules containing no less than N genes in the test study to be greater than α = 99.9%. To estimate the minimal δ required in the module merge procedure, we perform the following simulation to calculate the probability after δ-merge:
Suppose n(Gtr), K, π, δ are given. Simulate
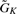 from
from 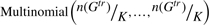 in Equation (2).
in Equation (2).Given δ, merge clusters with less than δ genes into a random cluster (since size of the nearest neighbor cluster in a real application is not known). Suppose the resulting cluster sizes become
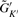 (K′ ≤ K).
(K′ ≤ K).Compute the conditional probability,
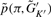 from Equation (1).
from Equation (1).- Repeat step 1–3 B times (B = 10 000 in this article). The probability of successful application to the test study can be estimated by
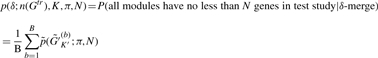
Repeat step 1–4. For different δ, increasingly from 1, 2, 3 … until finding the smallest δ such that p(δ; n(Gtr), K, π, N) ≥ 0.999.
The advantage of our probabilistic model is that the estimation of minimal δ only depends on the total number of genes in the training data (n(Gtr)), the number of clusters K used in K-means and the probability of gene missingness in the test study. It does not depend on the observed data and a table can be computed for a rapid decision in future applications (see Supplementary Table 2). For example, when we use a training data with 3000 genes to build a prediction model using 150 K-means modules and expect a gene missing probability to be 30% in the test data, we require δ = 13 in δ-merge to guarantee 99.9% success probability of application of MBP in the inter-study prediction.
2.4 Simulations
2.4.1 Simulation with varying gene measurement variability
To determine whether the MBP approach creates a model that is robust in the presence of expression measurement noise, we randomly added white noise to the Luo dataset. The added noises were randomly assigned across all data points in the expression intensity matrix and the noises followed a Gaussian distribution with mean μ = 0 and standard deviation σ = σ0 × average expression intensity. Variable magnitudes of noise (σ0 = 0, 0.1, 0.5, 1) were applied to various different proportions of the entire data (P = 10, 20, 50 and 70% of all data entries) and the leave-one-out cross-validation (LOOCV) prediction accuracies were compared between the MBP and GBP methods. In each iteration of LOOCV, a sample is left out as test data. Only the remaining samples, as training data, are used to perform the entire MBP model construction in Section 2.2. The exclusion of test sample from the entire model construction steps guarantees an unbiased estimate of prediction accuracy.
2.4.2 Simulation with gene missingness in cross-platform scenarios
The robustness for gene missingness across platforms was evaluated by randomly splitting the Luo dataset into a training dataset and a test dataset by a 1:1 ratio, and randomly deleting various proportions (π = 0.1–0.7) of genes from the test dataset to create missing genes. The prediction accuracies and prediction success rate (PSR), defined as the number of successful predictions (i.e. the inter-study prediction can be successfully implemented under the effect of missing genes) divided by the total number of prediction attempts, were compared between the MBP and GBP methods.
2.4.3 Simulation with gene mismatching
To examine robustness of prediction methods against erroneous gene matching, we randomly split the Luo dataset into equal size of training and testing datasets, selected a portion of genes (1–70%) and swapped their gene names in the test study. Although erroneous gene matching is not expected to be as high as 70%, we perform the simulation with this wide range to observe the empirical impacts.
2.5 Evaluations in real data
2.5.1 MBP versus GBP in within-study prediction
Prediction accuracies were assessed for every dataset using a LOOCV approach. For every dataset, the accuracy was calculated as the number of samples correctly predicted divided by the total number of samples in the dataset. Since there were random factors in K-means algorithm in the MBP method, the LOOCV procedure was run 30 times. The means and standard deviations of accuracies from the MBP methods were calculated and compared with the accuracies obtained from the traditional GBP method. The result demonstrates whether or not MBP provides better prediction accuracy than GBP in within-study cross validation.
2.5.2 MBP versus GBP in inter-study prediction
Cross-study prediction was performed by the standard MBP algorithm stated above. The test data used in the pairwise cross platform analyses were three lung cancer datasets (Bhat, Beer and Garber), two prostate cancer datasets matched with adjacent tissues as controls (Yu and Welsh) and two prostate cancer datasets using donors' samples as controls (Dhan and Luo). The results were compared with those of the GBP method, which only used genes common to both training and test datasets.
2.5.3 Comparison to metagene approach
We compared the traditional GBP and our proposed MBP to a popular metagene approach using non-negative matrix factorization (NMF) techniques (Tamayo et al., 2007). NMF is performed to identify gene clusters (modules) in the data and the prediction analysis is performed based on the gene modules by the factorized metagenes. Unlike MBP, the metagene approach adopts weighted averaging gene signatures similar to singular value decomposition and cannot address the issue of non-overlapping genes between training and testing studies.
3 RESULTS
3.1 Estimation of minimum cluster size
To estimate the minimum cluster size for δ-merge, one needs to know the distribution of the cluster sizes generated by K-means. The cluster sizes (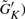 differ by datasets and can be viewed as a random vector. We examined the seven datasets used in this article by conditional Poisson distributions and multinomial distributions to fit the distribution of
differ by datasets and can be viewed as a random vector. We examined the seven datasets used in this article by conditional Poisson distributions and multinomial distributions to fit the distribution of 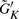 . The conditional Poisson distribution did not perform well (data not shown), while all datasets fitted well under multinomial distributions in Q–Q plots (Fig. 2). This finding justifies the first step of δ-merge by simulating
. The conditional Poisson distribution did not perform well (data not shown), while all datasets fitted well under multinomial distributions in Q–Q plots (Fig. 2). This finding justifies the first step of δ-merge by simulating 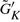 using Equation (2).
using Equation (2).
As a key parameter of the MBP model, δ was used to control or minimize prediction failure due to missing genes when genes in a model built on a training set did not exist in a test set. When a cluster size was smaller than δ, the cluster was merged into its nearest cluster to minimize the probability of prediction failure. The simulated results are listed in Supplementary Table 2. The threshold δ increased when K, π and N increased or when Gtr decreased.
3.2 Simulation: robustness to expression measurement noise, gene missingness and gene mismatching
3.2.1 Robustness to expression measurement noise
To determine the stability of the prediction accuracy in the MBP method, white noise were added to the Luo dataset and prediction accuracies were evaluated using LOOCV and the three classifiers: LDA, KNN and SVM. The added noise followed N(0, (σ0 × mean intensity)2) (with σ0 = 0, 0.1, 0.5 and 1) at serial proportions of 10, 20, 50 and 70% noise in the data. The results in Figure 3 show that prediction accuracies of the MBP approach were robust across varying amounts of noise added to the data (Fig. 3 top and middle panels; accuracies >90% for both K = 100 and K = 150), whereas the prediction accuracies of GBP dropped to around 80% when up to 1-fold of variation was added (Fig. 3 bottom panel).
Fig. 3.

Prediction accuracies in the simulation of added white noise in the Luo data. Top: MBP approach with K = 100 and δ = 20. Middle: MBP approach with K = 150 and δ = 20. Bottom: GBP approach.
3.2.2 Robustness to inter-study gene missingness
It is common that many genes appearing in one platform may be missing in another, causing difficulties in applying the GBP method to inter-platform predictions. To evaluate whether the MBP method is robust when genes are missing in the test data, the prediction accuracies were evaluated by splitting an array dataset, Luo, into a training set and a test set and randomly deleting genes from the test set at different proportions (π), from 10% to 70%. The procedure was repeated 100 times and the prediction accuracies were averaged. Among the 100 simulations, we also recorded the percentage of successful inter-study prediction implementation (PSR). For example, if any gene or module in the prediction model of a training study is missing in the test study, the inter-study prediction fails. In Figure 4A, the result shows that MBP was robust to gene missingness with PSR = 100% up to π = 70%. GBP was deemed to fail even when π = 10–20%. In Figure 4B, the prediction accuracy of MBP remained stable even when π increased to 60–70%.
Fig. 4.

(A): PSR of MBP versus GBP. MBP can be successfully applied in the presence of gene missingness across studies (up to π = 70%). GBP fails with >95% probability even when π = 10%. (B): Prediction accuracy of MBP averaged over successful inter−study prediction. (π = 10%,…, 70% on the x-axis).
3.2.3 Robustness to inter-study gene mismatching
MBP and GBP are evaluated for robustness by simulation when erroneous gene matching or annotation occurred in the training and test data. In 100 simulations, the results are shown in Supplementary Figure 2. As expected, MBP is generally more robust than GBP, particularly for high erroneous gene matching.
3.3 Prediction accuracies within studies
Although the motivation of MBP is to overcome the deficiency of the GBP approach in inter-study prediction, it is of interest to first compare their performances in LOOCV prediction within a study. Figure 5 shows the within-study prediction results. In the three lung cancer studies (Beer, Bhat and Garber), MBP and GBP had similarly high prediction accuracies. For the four prostate cancer studies, MBP usually had higher accuracy than GBP with a few exceptions. We applied a paired t-test and Wilcoxon signed rank test on the nine pairs of prediction accuracies (3 classifiers × 3 feature numbers) in each study and derived the two-sided test P-values. In two of the three lung cancer studies (Bhat and Garber), GBP's accuracy was better than MBP's with statistical significance but with a very small margin. In two out of the four prostate cancer studies, MBP outperformed GBP with statistical significance and with a large margin. Thus, MBP not only provides robustness for inter-study prediction (that will be shown below) but also performs similarly or outperforms GBP in cross-validated prediction within a study. Better performance by a multi-gene pooled decision has been reported previously (Park et al., 2007).
Fig. 5.

LOOCV within-study prediction accuracy of MBP (K = 100 and δ = 20) versus GBP for LDA, KNN and SVM classifiers with M = 10, 20 and 30 features in seven studies.
3.4 Prediction accuracies across studies
3.4.1 Lung and prostate cancer studies
Cross platform prediction was performed to evaluate MBP and GBP. Three sets of inter-study prediction analyses are outlined in Table 1 and the numbers of common genes across each pair of studies are shown. The minimal δ calculated from Section 2.3 for each pair of inter-study prediction is shown. The prediction results are shown in Figure 6 with similar presentation as in Figure 5. The prediction accuracies of MBP versus GBP for LDA, KNN and SVM classifiers with 10, 20 and 30 features in 10 inter-study predictions are demonstrated. In the inter-study prediction by MBP, the prediction model was built upon the entire gene list in the training study (i.e. the second column of Table 1). For GBP, it was, however, based on the common gene list of training and test studies (i.e. the fifth column of Table 1). In general, the MBP approach generated better inter-study prediction accuracy than did GBP (see two-sided test P-values in Beer => Garber, Bhat => Beer, Bhat => Garber, Luo => Dhan in Fig. 6).
Table 1.
Common genes across studies and required δ
| Training | Gtr (genes | Test | Gte | Gtr ∩ Gte (genes | π | δ |
|---|---|---|---|---|---|---|
| study | used by MBP) | study | used by GBP) | |||
| Beer | 4467 | Bhat | 4107 | 2493 | 0.44 | 1 |
| Bhat | 4107 | Beer | 4467 | 2493 | 0.39 | 1 |
| Garber | 3399 | Beer | 4467 | 1594 | 0.53 | 20 |
| Beer | 4467 | Garber | 3399 | 1594 | 0.64 | 29 |
| Bhat | 4107 | Garber | 3399 | 1493 | 0.64 | 33 |
| Garber | 3399 | Bhat | 4107 | 1493 | 0.56 | 25 |
| Welsh | 9494 | Yu | 9109 | 2521 | 0.73 | 1 |
| Yu | 9109 | Welsh | 9494 | 2521 | 0.72 | 1 |
| Dhan | 7784 | Luo | 3673 | 2352 | 0.70 | 1 |
| Luo | 3673 | Dhan | 7784 | 2352 | 0.36 | 1 |
Fig. 6.

Pairwise inter-study prediction accuracies of MBP and GBP. MBP used all genes in training data in model construction while GBP only used intersected genes across training and testing data. The evaluation was performed in three sets of studies (I: Beer, Bhat and Garber in the top three rows; II: Dhan and Luo in the fourth row; III: Welsh and Yu in the fifth row). LDA, KNN and SVM were evaluated and the top M = 10, 20, 30 features were used in the univariate feature selection (K = 100 and δ = 20).
One may argue that MBP's better performance may come from its accessibility to more genes than GBP. We performed an identical analysis of Figure 6 but forcing MBP to utilize only the intersected genes as in GBP. The result presented in Supplementary Figure 3 is similar to Figure 6, confirming that the improved accuracy of MBP does not come from availability of more genes.
3.4.2 Multivariate feature selection methods and penalized K-means
From the above simulation and application results, the MBP method showed an advantage over GBP in terms of accuracy and had a clear advantage over GBP with respect to prediction robustness, particularly in the presence of missing genes in inter-study prediction or increased measurement variability. The above evaluations were, however, based on univariate feature selection by selecting the top M features using moderated t-statistics. In the literature, there have been debates on whether multivariate feature selection improves upon univariate filtering-based feature selection in prediction accuracy (Guyon et al., 2002; Lai et al., 2006). Theoretically, multivariate feature selection considers interaction among genes and should perform better, while applications in some datasets have shown the opposite results as multivariate feature selection may add additional redundancy or may cause over-fitting. We tested a multivariate feature selection method in this category—R-SVM (Zhang et al., 2006) (an improved version of famous SVM-RFE)—and an embedded feature selection method, PAM (Tibshirani et al., 2002). We then compared them to their univariate filtering counterparts (i.e. SVM and nearest centroid method). The results are shown in Supplementary Figures 4.1 and 4.2. In general, multivariate feature selection or embedded methods did not necessarily improve univariate approaches. When comparing MBP and GBP, both results in PAM (Supplementary Fig. 4.1) and in SVM (Supplementary Fig. 4.2) did not show either method to have better accuracy than the other one when combining both univariate and multivariate feature selection methods.
An important factor that affects the performance of MBP is the quality of unsupervised modules (i.e. clusters). Given that the K-means method forces all genes into K clusters, many scattered genes are forced into clusters and may dilute the prediction power of the module. We examined a modified K-means method, penalized K-means (Tseng, 2007), and tested whether unsupervised modules generated by penalized K-means can outperform classical K-means. The results (shown in Supplementary Fig. 5) showed a negative conclusion. Better quality of clusters generated by penalized K-means does not further improve performance in MBP. The result suggests that robustness provided by MBP is probably strong enough to offset the effect of a small number of scattered genes in the modules.
3.4.3 Comparison to metagene approach
We compared GBP and MBP to a popular metagene approach. Although this approach is module based, its weighted averaging gene signatures do not address the non-overlapping gene issue between training and testing studies in inter-study prediction. The method has to be applied to the intersected gene set as GBP does. In Supplementary Figure 6, the performance of metagene approach is inferior to GBP and MBP in most of the inter-study predictions evaluated.
4 DISCUSSION
An ideal prediction model should be highly accurate, robust and simple for clinical utility. To pursue these standards, we developed the MBP method, which takes advantage of information from genes sharing similar expression patterns. The results of the current study show that the prediction accuracies of the MBP method are slightly better than those of the GBP method in both within-study and inter-study predictions. Furthermore, the MBP method is superior to the GBP method in being robust to gene missingness and to experimental noise. The results show great potential for MBP to improve inter-study prediction in microarray studies and enhance the application of this technology to clinical practice.
In the literature, it has been shown that multiple completely different prediction models may generate equally high prediction accuracy. For example, the well-known 70-gene signature to predict breast cancer patient survival was first proposed (van't Veer et al., 2002). Other investigators derived an additional six classifiers that performed as well as the 70-gene signature using the same dataset (Ein-Dor et al., 2005). Also, disparity in using different gene signatures to predict similar outcomes in different studies has been reported (Ramaswamy et al., 2003; Sorlie et al., 2001; van't Veer et al., 2002). It is important to allow reasonable inter-study prediction validations in relevant published studies. The stability of the MBP method observed in the present study is the result of grouping genes sharing a similar expression pattern and selecting a gene that can represent the group of genes. It has been postulated that using a cluster average would yield a higher prediction accuracy under certain conditions (Park et al., 2007). Although the MBP method only slightly outperforms the GBP method in prediction accuracy, the prediction robustness of MBP remains its major advantage.
The clinical utility of a genomic prediction model relies heavily on the model's simplicity and reproducibility. Recent cross-platform analyses used intersection genes across datasets (Bhanot et al., 2005; Bloom et al., 2004; Bosotti et al., 2007; Cheadle et al., 2007; Nilsson et al., 2006), an approach that required information from all datasets involved in the analysis. This approach is appropriate for meta-analysis of biomarker detection but is inadequate for cross-platform prediction. There are two elements needed for a prediction: (i) a selected gene signature and (ii) a prediction model. When the construction of a prediction model requires the common genes of training and test studies, the selected prediction signature must be readjusted whenever a different platform of the test study is applied, making it inconvenient to validate and for clinical use. Furthermore, loss of training data information by including only intersection genes to build the prediction model makes this approach less desirable. MBP is a natural solution to these hurdles.
A lack of reproducibility hinders the application of genomic prediction models. Many factors may affect model reproducibility. The MBP method focuses on two factors to increase model reproducibility: gene missingness and experimental noise. The robustness of the MBP method toward missing genes was provided by grouped decision in modules and the rare probability of model failure is controlled by merging small modules to nearest modules in our algorithm. The robustness of the method regarding expression measurement noise was assessed by testing on the Luo dataset. Although the MBP method was robust to added noise, the pattern of noise added may not adequately represent experimental variations in real data. Further study will focus on evaluating real data or introducing variation other than Gaussian noise.
In addition to demonstrating the clinical applicability of MBP, this study demonstrated some novel approaches in the algorithm. First, this is the first time that cluster sizes generated by K-means are demonstrated to consistently follow a multinomial distribution and a cluster merging procedure is proposed to avoid model prediction failure due to gene missingness. Second, we used a representative gene with the closest summed distance to all other genes within a module (similar to ‘sample median’ concept in estimating mean parameter) to summarize the module information, which is an actual gene with better annotation and interpretation rather than using a pseudo-gene such as eigen-gene or averaged gene vector used in many methods. Although we do not have enough evidence to prove or argue the superiority of adopting median representative genes, this procedure is conceptually more robust to accidental noises and has better interpretability. Third, MBP reduced redundant gene features by summarizing similar gene expression profiles within each module, diminishing gene collinearity and adding a novel technique for data reduction.
One limitation of the MBP method is the lack of correlation and interpretation of each module to known biological pathways. Further investigation will be made to integrate pathways from biological databases as supervised modules to improve the performance. Proper normalization across studies is another key to successful inter-study predictions. Our recent publication (Cheng et al., 2009) has discussed the issue of genewise normalization in addition to commonly practiced sample-wise normalization. MBP proposed in this article focuses on robust inter-study prediction from another angle and can potentially be combined with these advanced normalization methods to enhance prediction accuracy.
Recently, deep sequencing technology is emerging as an attractive alternative to microarrays for genotyping, analysis of methylation patterns, identification of transcription factor binding sites and quantification of gene expression. The digital quantification is far more precise than microarray although its widespread applicability is still now limited by its high cost. As the price goes down in the near future, we expect increased popularity of this technology. Our proposed MBP method can be extended to analyze deep sequencing data, where the feature dimensionality is even higher than microarray data. The fast algorithm of K-means clustering and the advantage of rapidly reducing dimensionality by gene modules make MBP a perfect tool for such type of extremely high-throughput technology.
Supplementary Material
ACKNOWLEDGEMENTS
The authors appreciate Janice Sabatine from Avanti Strategies for careful editing of the article.
Funding: National Institutes of Health (KL2 RR024154-04 to C.S., C.C. and G.C.T.); University of Pittsburgh (Central Research Development Fund; Competitive Medical Research Fund); Cooperative Studies Program, VA Maryland HCS to Z.M.; Precision Therapeutics Inc., Pittsburgh, PA to K.S. and N.S.
Conflict of Interest: none declared.
REFERENCES
- Beer DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- Bhanot G, et al. Robust diagnosis of non-Hodgkin lymphoma phenotypes validated on gene expression data from different laboratories. Genome Inform. 2005;16:233–244. [PubMed] [Google Scholar]
- Bhattacharjee A, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl Acad. Sci. USA. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom G, et al. Multi-platform, multi-site, microarray-based human tumor classification. Am. J. Pathol. 2004;164:9–16. doi: 10.1016/S0002-9440(10)63090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosotti R, et al. Cross platform microarray analysis for robust identification of differentially expressed genes. BMC Bioinformatics. 2007;8(Suppl. 1):S5. doi: 10.1186/1471-2105-8-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussey KJ, et al. MatchMiner: a tool for batch navigation among gene and gene product identifiers. Genome Biol. 2003;4:R27. doi: 10.1186/gb-2003-4-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle C, et al. A rapid method for microarray cross platform comparisons using gene expression signatures. Mol. Cell Probes. 2007;21:35–46. doi: 10.1016/j.mcp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Cheng C, et al. Ratio adjustment and calibration scheme for gene-wise normalization to enhance microarray inter-study prediction. Bioinformatics. 2009;25:1655–1661. doi: 10.1093/bioinformatics/btp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristianini N, Shawe-Taylor J. An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods. New York: Cambridge University Press; 2000. [Google Scholar]
- Dasarathy B. Nearest Neighbor (NN) Norms: Nn Pattern Classification Techniques (Unknown Binding). IEEE Computer Society Press Tutorial.; 1991. [Google Scholar]
- Dhanasekaran SM, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–826. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- Dobbin K, Simon R. Sample size determination in microarray experiments for class comparison and prognostic classification. Biostatistics. 2005;6:27–38. doi: 10.1093/biostatistics/kxh015. [DOI] [PubMed] [Google Scholar]
- Dobbin KK, et al. How large a training set is needed to develop a classifier for microarray data? Clin. Cancer Res. 2008;14:108–114. doi: 10.1158/1078-0432.CCR-07-0443. [DOI] [PubMed] [Google Scholar]
- Ein-Dor L, et al. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics. 2005;21:171–178. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- Garber ME, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl Acad. Sci. USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon I, et al. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002;46:389–422. [Google Scholar]
- Hartigan JA, Wong MA. A K-means clustering algorithm. Appl. Stat. 1979;28:100–108. [Google Scholar]
- Huang E, et al. Gene expression predictors of breast cancer outcomes. Lancet. 2003;361:1590–1596. doi: 10.1016/S0140-6736(03)13308-9. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, et al. Multiple-laboratory comparison of microarray platforms. Nat. Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- Kuhn A, et al. Cross-species and cross-platform gene expression studies with the Bioconductor-compliant R package annotationTools. BMC Bioinformatics. 2008;9:26. doi: 10.1186/1471-2105-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C, et al. A comparison of univariate and multivariate gene selection techniques for classification of cancer datasets. BMC Bioinformatics. 2006;7:235. doi: 10.1186/1471-2105-7-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, et al. Human prostate cancer and benign prostatic hyperplasia: molecular dissection by gene expression profiling. Cancer Res. 2001;61:4683–4688. [PubMed] [Google Scholar]
- Mardia K, et al. Multivariate Analysis. London: Academic Press; 1979. [Google Scholar]
- Nilsson B, et al. Cross-platform classification in microarray-based leukemia diagnostics. Haematologica. 2006;91:821–824. [PubMed] [Google Scholar]
- Park MY, et al. Averaged gene expressions for regression. Biostatistics. 2007;8:212–227. doi: 10.1093/biostatistics/kxl002. [DOI] [PubMed] [Google Scholar]
- Park PJ, et al. Current issues for DNA microarrays: platform comparison, double linear amplification, and universal RNA reference. J. Biotechnol. 2004;112:225–245. doi: 10.1016/j.jbiotec.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Pittman J, et al. Integrated modeling of clinical and gene expression information for personalized prediction of disease outcomes. Proc. Natl Acad. Sci. USA. 2004;101:8431–8436. doi: 10.1073/pnas.0401736101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potti A, et al. A genomic strategy to refine prognosis in early-stage non-small-cell lung cancer. N. Engl. J. Med. 2006;355:570–580. doi: 10.1056/NEJMoa060467. [DOI] [PubMed] [Google Scholar]
- Pusztai L, Leyland-Jones B. Promises and caveats of in silico biomarker discovery. Br. J. Cancer. 2008;99:385–386. doi: 10.1038/sj.bjc.6604495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, et al. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Segal E, et al. A module map showing conditional activity of expression modules in cancer. Nat. Genet. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- Shi L, et al. Cross-platform comparability of microarray technology: intra-platform consistency and appropriate data analysis procedures are essential. BMC Bioinformatics. 2005;6(Suppl. 2):S12. doi: 10.1186/1471-2105-6-S2-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, et al. QA/QC: challenges and pitfalls facing the microarray community and regulatory agencies. Expert Rev. Mol. Diagn. 2004;4:761–777. doi: 10.1586/14737159.4.6.761. [DOI] [PubMed] [Google Scholar]
- Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl Acad. Sci. USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang R, et al. Prediction and uncertainty in the analysis of gene expression profiles. In Silico Biol. 2002;2:369–381. [PubMed] [Google Scholar]
- Tamayo P, et al. Metagene projection for cross-platform, cross-species characterization of global transcriptional states. Proc. Natl Acad. Sci. USA. 2007;104:5959–5964. doi: 10.1073/pnas.0701068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PK, et al. Evaluation of gene expression measurements from commercial microarray platforms. Nucleic Acids Res. 2003;31:5676–5684. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalamuthu A, et al. Evaluation and comparison of gene clustering methods in microarray analysis. Bioinformatics. 2006;22:2405–2412. doi: 10.1093/bioinformatics/btl406. [DOI] [PubMed] [Google Scholar]
- Tibshirani R, et al. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl Acad. Sci. USA. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC. Penalized and weighted K-means for clustering with scattered objects and prior information in high-throughput biological data. Bioinformatics. 2007;23:2247–2255. doi: 10.1093/bioinformatics/btm320. [DOI] [PubMed] [Google Scholar]
- Tseng GC, Wong WH. Tight clustering: a resampling-based approach for identifying stable and tight patterns in data. Biometrics. 2005;61:10–16. doi: 10.1111/j.0006-341X.2005.031032.x. [DOI] [PubMed] [Google Scholar]
- van't Veer LJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- van Vliet MH, et al. Module-based outcome prediction using breast cancer compendia. PLoS ONE. 2007;2:e1047. doi: 10.1371/journal.pone.0001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JB, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- West M, et al. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc. Natl Acad. Sci. USA. 2001;98:11462–11467. doi: 10.1073/pnas.201162998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M, et al. Embracing the complexity of genomic data for personalized medicine. Genome Res. 2006;16:559–566. doi: 10.1101/gr.3851306. [DOI] [PubMed] [Google Scholar]
- Wong DJ, et al. Revealing targeted therapy for human cancer by gene module maps. Cancer Res. 2008;68:369–378. doi: 10.1158/0008-5472.CAN-07-0382. [DOI] [PubMed] [Google Scholar]
- Yu YP, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J. Clin. Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- Zhang X, et al. Recursive SVM feature selection and sample classification for mass-spectrometry and microarray data. BMC Bioinformatics. 2006;7:197. doi: 10.1186/1471-2105-7-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.