

Abstract
During vaccination or infection, adaptive and innate immune receptors of B cells are engaged by microbial antigens/ligands. A better understanding of how innate and adaptive signaling pathways interact could inform B lymphocyte biology as well as immunotherapy strategies and vaccine design. To address this goal, we examined the effects of TLR stimulation on BCR and CD40-induced B cell activation. Synergistic production of IL-6 was observed in both human and mouse primary B cells stimulated through B cell antigen receptors, CD40 and TLR7, and the latter two receptors also cooperated independently of BCR signals. The enhanced IL-6 production was dependent upon the activity of c-Jun kinase (JNK) and cFos. Dual stimulation through CD40 and TLR7 markedly enhanced JNK activity. The increased level of active JNK in dual stimulated cells was accompanied by an increase in the level of active AP-1 monomers cJun and cFos. The stimulation of B cells through both CD40 and TLR7 therefore enhanced the production of cytokines through increased JNK signaling and AP-1 activity. In addition, the dual stimulation increased cFos/AP-1 species in stimulated cells, effectively expanding the repertoire of AP-1 dimers as compared to singly stimulated B cells.
Keywords: B cells, AP-1, JNK, CD40, TLR7
Introduction
Natural immune responses to microbes and responses induced by vaccination involve the cooperation of innate and adaptive immunity. B lymphocytes express receptors allowing them to respond to both antigen-specific and innate immune stimuli [1, 2]. One of the key receptors involved in adaptive humoral responses is the TNF receptor superfamily member CD40. The interaction of CD40 on B cells with its ligand CD154, expressed by activated T cells, is integral to immunoglobulin production, isotype switching, and the formation of germinal centers [3], important events in the development of long-lived humoral memory. CD40 stimulation also contributes to B cell antigen presentation by the upregulation of T cell co-stimulatory molecules and production of acute phase cytokines such as TNF-α and IL-6 [4–6]. Previous studies have shown that dual stimulation of B cells through the BCR and CD40 leads to an enhancement of Ig and cytokine production [7]. However, the effects and mechanisms of interactions between innate immune receptors and BCR or CD40 expressed by B cells are not yet well defined.
B cells can be activated through engagement of a variety of innate immune receptors, including Toll-like receptors (TLRs). TLRs are related to the Drosophila receptor Toll, important for dorsal-ventral patterning in the developing fly as well as resistance to microbes [8]. The TLRs in higher organisms act as sensors of infection or `danger' by recognizing molecular patterns associated with microbes and/or cellular damage. As natural surveyors of environmental stresses, TLRs induce immune cell activity and direct the initial stages of immune responses [9]. Of the 13 TLR mammalian genes identified, B cells express TLRs 1, 4, 7, 8, 9, 10 and, under some circumstances TLR2, allowing detection of ligands that include LPS, double-stranded RNA, peptidoglycan, and hypomethylated DNA [10]. While activation of B cells through individual receptors has been the subject of much study, relatively little is known about the effects of endosomal TLR stimulation on signaling induced by adaptive receptors. These interactions are potentially important to effective immune responses as well as improved vaccine design.
Recent evidence suggests that the interaction of B cells with ligands for the BCR and TLR7/8 or TLR9 may enhance B cell effector functions during infection or autoimmune disease [11, 12]. TLRs 7 and 8 bind ssRNA as natural ligands, but also confer responsiveness to guanosine derivatives and the imidazolquinolines R848 (Resiquimod) and Imiquimod [13]. The latter experimental agonists have received considerable attention recently due to their effectiveness in treating genital warts and some skin malignancies [14–16]. Their mechanism of action is purported to be the activation of myeloid cells (primarily dendritic cells, DC) via interaction with TLRs 7 and 8 [17, 18]. However, B cells also express these TLRs and respond to their ligands, and unlike myeloid cells, also express adaptive immune receptors that may interact with TLR signals. These interactions could prove valuable in the use of TLR7/8 agonists as vaccine adjuvants [19–21].
B cells play important roles in protective immunity as both antibody producers and antigen presenting cells [22–25]. In an effort to design more effective vaccines and obtain a better understanding of B cell biology, it is important to study the effects of these potential adjuvants in the context of other immune receptor signaling events. Previous studies report that stimulation of B cells through TLRs enhances Ig production induced by either BCR or CD40 [26, 27]. However, these original findings did not address the effects of dual stimulation on other effector functions, such as cytokine production, nor were the molecular mechanisms of signal cooperation addressed.
In an effort to elucidate molecular mechanisms of cooperation between adaptive receptors and TLRs, we stimulated both mouse and human B cells with CD154 and the TLR7/8 agonist R848, which we previously demonstrated is a powerful activator of B cells [27, 28]. We found that this dual stimulation of B cells induced a synergistic production of specific cytokines, most dramatically IL-6. To elucidate the molecular mechanisms involved in the synergy, we focused on the enhancement of kinase pathways known to be important for IL-6 production by B cells. Our data indicate that the dual stimulation of B cells through CD40 and TLR7/8 cooperates in inducing the JNK signaling pathway and resulted in differential AP-1 monomer activation thereby increasing the amount of cfos/AP-1 dimers within the cell. These results reveal a novel molecular mechanism of innate/adaptive immune receptor interaction.
Results
Cooperation between CD40 and TLR7 signals in B cell cytokine production
Stimulation of human or mouse B cells with either TLR agonists or CD154 induces production of IL-6 [27, 29]. As peripheral B cells can be exposed to TLR agonists prior to CD40 stimulation, during infection or vaccination, we determined the effect of pre-treating human and mouse B cells with TLR7/8 agonists, with or without BCR engagement, on subsequent CD154 stimulation. Analysis of both species was done to ensure validity of the mouse as a model species for human B cell responses, as mouse and human B cells can respond differently to particular TLR agonists [30]. The pretreatment of B cells with TLR agonist or TLR + BCR stimuli enhanced the production of CD40-induced IL-6 (Fig. 1a–b). Pretreatment with BCR engagement, either alone or in tandem with the TLR7/8 agonist R848, modestly enhanced CD40 induced IL-6 production. However, the majority of the enhanced IL-6 production could be attributed to the effects of TLR pre-stimulation. Pre-stimulation with TLR agonists was not a prerequisite for enhancement of CD40 induced IL-6 production, as simultaneous co-stimulation of B cells with R848 and CD154 induced synergistic production of IL-6 in mouse and human primary cells as well as the mouse B cell line CH12.LX (Fig. 2). Human peripheral blood B cells also showed synergistic production of TNF-α and IL-12 (p40/p70) upon dual stimulation, but mouse splenic B cells did not (Fig. 2 and data not shown). We focused on IL-6 in subsequent experiments due to its role as a B cell differentiation factor and a `danger signal' cytokine in both mouse and human [31, 32].
Figure 1.

CD40 induced IL-6 production following pre-stimulation with TLR agonists. A) Human peripheral B cells and B) mouse high density B cells were treated with R-848 (0.2μg/ml), anti-BCR (Fab')2 (1μg/ml), both, or medium alone for 24 hours. Cells were then washed, rested for 1 hour, and stimulated with Hi5 cell controls or Hi5 cells expressing CD154 for 20 hours. Supernatants were then collected and assayed for IL-6 by ELISA. Each graph is representative of two individual experiments.
Figure 2.

Cytokine production following CD40 and TLR costimulation. A) human peripheral B cells, B) mouse high density (HD, resting) B cells, and C) the mouse B cell line CH12.LX were incubated with CD154-expressing Hi5 cells or control Hi5 cells alone (solid bars) or with R-848 (open bars) for 24 hours. Supernatants were assayed for IL-6 by ELISA. Each IL-6 graph is representative of five individual experiments, while the TNF-α and IL-12 graphs are representative of two individual experiments utilizing cells from two different donors. Hi5 to B cell ratios were 1:5, while R-848 was used at a final concentration of 0.2μg/ml for human and mouse primary cells and 10ng/ml for CH12 cells. * Statistical difference between CD154 and CD154 + R-848 (student's paired T-test P value < 0.01). ** Statistical difference between cells stimulated with R-848 alone and those receiving dual stimulation. Student's paired T-Test P value < 0.01).
Phosphorylation of JNK and its substrate cJun by dual CD40 and TLR signaling
TNFR associated factors (TRAFs) are cytoplasmic adaptor molecules that link receptors to downstream signaling events. The production of IL-6 following CD40 engagement is dependent upon the adapter protein 6 TRAF6, but not TRAFs 1, 2, or 3 [29]. TRAF6 is known to associate with CD40 [33], and is also utilized by TLR receptors in the activation of NF-κB and JNK [34]. Our previous work showed that basal nuclear NF-κB is necessary for IL-6 production but that CD40 induced IL-6 production is dependent upon factors downstream of JNK signaling [6]. Accordingly, the level of JNK activation in dual vs. singly-stimulated B cells was determined. The level of JNK activation was monitored by assaying the relative levels of JNK phosphorylation under suboptimal CD40 stimulatory conditions (1:10 ratio of B cells to Hi5 cells expressing CD154). The addition of both R848 and CD154 further increased the level of phosphorylated JNK in CH12.LX cells (~47% increase), resting mouse splenic B cells (~31% increase) and human peripheral B cells (~100% increase) above R848 alone (Fig. 3). Other MAP kinase family members, p38 and Erk1 and 2, were activated upon single stimulation, but did not display an increased level of phosphorylation, or alteration in kinetics, upon dual stimulation (data not shown).
Figure 3.


Phosphorylation of JNK following dual stimulation. A) CH12.LX cells and B) HD splenic B cells were stimulated with suboptimal doses of R-848 with or without CD154 and controls for times indicated. C) Human peripheral B cells were stimulated for 20 minutes with R-848 with or without CD154 and controls. Cells were then collected, lysed, and subjected to western blot analysis for phosphorylated JNK. Relative levels of p-JNK between treatments were compared by calculated fold increase over negative controls using normalized densitometry values (ratio of p-JNK value:internal control value / normalized negative control). Hi5 cells infected with wild type baculovirus were used as a negative control stimulus. Each western blot and corresponding graph are representative of two separate experiments.
Upon activation, JNK phosphorylates members of the Jun transcription factor family. After phosphorylation, Jun family members homo or heterodimerize with Fos, Fra, or ATF family members to produce the transcription factor AP-1 [35]. AP-1 composed of homodimers of cJun has been shown to be important for CD154 induced IL-6 production by B cells [6]. To determine the downstream consequences of enhanced JNK phosphorylation, the level of phosphorylated cJun (p-cJun) was measured in dual vs. singly stimulated cells. Cells stimulated through both CD40 and TLR7 demonstrated an enhanced level of p-cJun (Fig. 4). Both mouse and human B cells displayed enhanced p-cJun levels in dual vs. singly treated B cells. Therefore, the enhanced activity of JNK is reflected by enhanced phosphorylation of one of its primary targets, cJun, in both human and mouse dual stimulated B cells.
Figure 4.

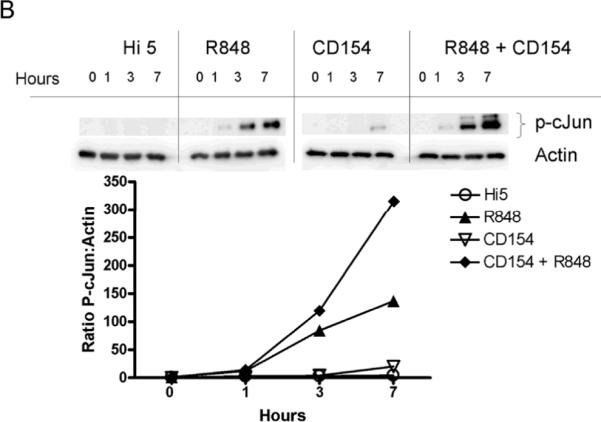
Enhancement of phosphorylated cJun by dual stimulation. A) CH12.LX cells (1 × 106 per treatment), B) mouse splenic B cells (1×106 cells), and C) human peripheral B cells (1×106 cells) were stimulated with R-848, CD154, or both for times indicated. Cells were then lysed and the presence of p-cJun and actin determined by western blot. Normalized densitometry values (ratio of p-cJun value:actin value) were used to compare p-cJun levels between treatments. Multiple bands present in p-cJun blot during dual stimulation are due to hyperphosphorylation of cJun. Data are representative of three individual experiments.
Differential AP-1 isoforms in dual stimulated B cells
We hypothesized that the enhanced p-cJun observed in dual stimulated B cells would enhance the amount of active AP-1 within the nucleus, contributing to synergistic IL-6 production. Nuclear extracts of dual stimulated mouse splenic B cells displayed an increase in active AP-1 monomers as determined by an EMSA based ELISA. Differences in total amounts of AP-1 complexes are difficult to quantitate by EMSA. We therefore used a commercial EMSA based ELISA to determine the relative amount of specific monomers in activated AP-1 complexes in dual compared to singly stimulated primary B cells. Nuclear extracts of stimulated B cells were collected and reacted with biotin-labeled AP-1 specific DNA probe. The AP-1 probe complex was then incubated on a streptavidin coated ELISA plate and monomer specific antibodies used to detect cJun, JunB, JunD, Fra1, Fra2, FosB and/or cFos. Using this approach, it was determined that the dual stimulated resting splenic B cells had an increased level of active cJun and cFos compared to those of singly treated cells (Fig. 5). To test the relevance of JNK activity in the increase in nuclear AP-1 subunits, dual stimulated cells were treated with or without JNK inhibitor and nuclear extracts probed for p-cJun and cFos. The increase in p-cJun and cFos within the nucleus of dual stimulated cells was abrogated by treatment of cells with a JNK inhibitor (Fig. 5c).
Figure 5.

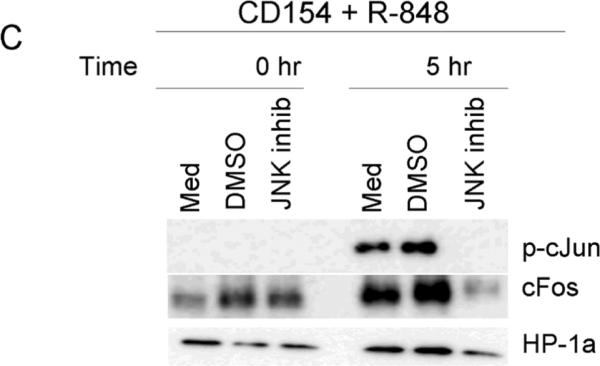

Differential activation of AP-1 monomers following dual signaling. Mouse cells were stimulated and nuclear extracts subjected to an EMSA based ELISA. Background normalized (described in materials and methods) absorbance values at 405nm are given for cJun (A) and cFos (B). There were no differences between treatments for other AP-1 monomers tested (Fra1, Fra2, JunB, JunD, and FosB, data not shown). Data are representative of two individual experiments. C) Nuclear extracts of B cells stimulated with both CD154 and R-848 in the presence or absence of JNK inhibitor. Inhibitor was added to cell cultures 30 minutes prior to stimulation. HP-1a (heterochromatin-associated protein 1a) was used as a nuclear extract loading control. D) Mouse B cells and E) human B cells were stimulated with R-848 with or without CD154 or controls. Dual stimulation increased the level of total cJun and cFos in both mouse and human B cells. Data are representative of 2 independent experiments.
Previous reports indicate that AP-1 positively auto-regulates production of AP-1 subunits [36, 37]. As the promoters for cjun and cfos contain AP-1 binding sites and are regulated, in part, by AP-1 activity, we determined the affect of TLR7/8 and CD40 stimulation on total levels of cJun and cFos [36, 37]. Stimulation of B cells increased the levels of total cJun and cFos protein in both mouse and human B cells (Figure 5d and e). While there may be species differences between levels of AP-1 subunit upregulation -- the human B cells appear to upregulate cJun in response to CD154 more readily than murine B cells - the overall trend of increased AP-1 subunits is conserved.
To further determine the relevance of cFos/AP-1 in the production of IL-6, previously characterized subclones of CH12.LX cells stably transfected with an IPTG inducible dominant negative form of AP-1 (A-Fos) [6] were used. A-Fos has been shown to inhibit AP-1 activity by forming nonfunctional dimmers with cJun [38]. The levels of IL-6 were then determined in both singly and dual stimulated cells in the presence and absence of IPTG. As shown in Fig 6, induced expression of A-Fos was able to abrogate the synergistic IL-6 production induced by dual stimulation. In addition, splenic B cells were tested for IL-6 production in the presence of JNK inhibitor VIII. The addition of JNK inhibitor reduced the amount of IL-6 production by ~55%, again indicating the importance of JNK in the synergistic cytokine production by dual stimulated B cells.
Figure 6.
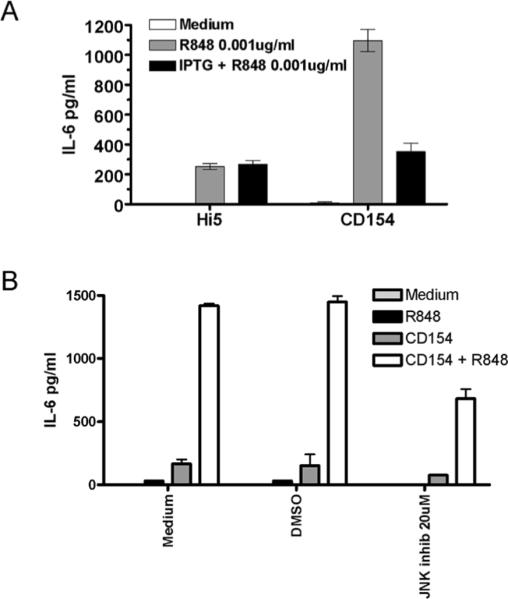
Inhibition of synergistic IL-6 production by inhibition of AP-1 and JNK. A) CH12.LX cells stably expressing an IPTG inducible dominant negative-Fos gene, FosA, were stimulated with R848, CD154 or both in the presence and absence of IPTG. IPTG at a final concentration of 1mM was added to cell cultures 24 hours prior to stimulation. B) Splenic B cells were stimulated as described in the presence or absence of JNK inhibitor for 24 hours. Supernatants were then collected and assayed for amounts of IL-6. Data are representative of 3 independent experiments.
Discussion
The present report demonstrates that stimulation of naïve B cells via TLR7 and CD40 induced the synergistic production of IL-6. This outcome required activation of the JNK pathway, resulting in heightened phosphorylation of the JNK substrate cJun, and increased activity of cJun and cFos-containing AP-1 complexes. Notably, the effects of dual stimulation were observed in both human and mouse B cells. Species differences between human and mouse TLR expression and response to TLR agonists have been described [30]. Human TLR8 recognizes R848 while mouse TLR8 is reported to be non-responsive to this compound [13]. The responses of human peripheral vs. mouse splenic B cells to the same TLR agonist can also vary. For example, while murine splenic B cells proliferate in response to R848, human peripheral B cells do not [28, 39]. Species-specific differences in TLR stimulation must therefore be taken into account when studying the effects of TLR-mediated activation.
Stimulation of B cells through CD40 induces JNK activation and subsequent IL-6 production that is driven by AP-1:cJun homodimers [6]. The addition of TLR7 signaling dramatically enhanced CD40-medaited IL-6 production via a mechanism involving heightened JNK activation and, in contrast to either stimulation alone, a marked increase in the level of active cFos/AP-1. Although the activity of JNK was enhanced in dual stimulated cells, this increase was transitory, and followed the same kinetics as that of activated JNK in cells treated with single stimuli. Despite the transient activity of JNK, enhanced amounts of phosphorylated cJun were observed for up to 16 hours (data not shown) after dual stimulation. The transitory JNK activation, followed by prolonged cJun activation, suggests that other kinases and/or phosphatases may be involved in the regulation/phosphorylation of cJun, as has been described [35]. The identification and role of these enzymes in cJun phosphorylation and IL-6 production by B cells is under current investigation.
Importantly, the inhibition of JNK in either dual or CD154 stimulated splenic B cells was not sufficient to completely abrogate IL-6 production. This suggests that other mechanisms leading to AP-1 formation may be induced in dual stimulated cells or other transcription factors could aid in driving IL-6 production under these circumstances. While both of these are possible, our data suggest the former, as AP-1 inhibition completely abrogated synergistic IL-6 production (Figure 6a). As mentioned above, previous studies have shown that other MAP kinases, such as p38 and ERK1/2, are capable of phosphorylating/activating cJun [35].
While cJun is known to be important for IL-6 production by B cells, other monomeric components of AP-1 are reported to have differential effects on cytokine production that are both cytokine, and potentially cell-type specific [35, 40, 41]. Our data indicate that cFos plays a positive regulatory role in IL-6 production. This appears to contradict previous reports that cFos acts as a negative regulator of IL-6, TNF-α, and IL-12 production [42, 43]. These studies, however, used macrophages isolated from a cFos−/− mouse stimulated through TLRs alone. The differences in results therefore, likely reflect cellular differences in the usage of cFos/AP-1 and/or differences in the stimulation of cFos/AP-1 (single vs. dual stimulation). Consistent with this hypothesis, cFos appears to have no negative effects on IL-6 production by TLR stimulated dendritic cells from cFos−/− mice [44]. Our data indicate that the role of cFos in B cell cytokine production may be more complex than previously thought, and worthy of further investigation.
The findings presented here indicate that stimulation of B cells through TLR7 and CD40 leads to a synergistic production of cytokines dependent upon the JNK signaling pathway and elevated p-cJun. In dual stimulated cells not only is the level of cJun activity increased, but the formation of potentially unique AP-1 complexes is also observed. Monomeric components of AP-1 such as cJun and cFos are transcriptionally regulated by, among other factors, AP-1. Changes therefore in AP-1 isoforms or percentages of monomeric proteins could have additional effects on the long-term or downstream AP-1 profile and overall activity. This positive feedback loop may, in turn, be important in the regulation of cytokine production by B lymphocytes.
The enhanced cytokine production by B cells stimulated through both adaptive and innate receptors could have positive physiological effects. Immune response efficacy could potentially be improved, either through the direct effects of IL-6 on B cell proliferation and survival, or inhibition of T regulatory cells [31]. Importantly, while enhancing cytokine production may be beneficial for combating infectious disease, it may have deleterious effects in the context of autoimmunity. Multiple cytokines have been associated with autoimmune disease severity. The blockade of IL-6 has been shown to alleviate clinical symptoms in multiple inflammatory diseases such as rheumatoid arthritis and SLE [45–47]. The regulation of IL-6 during immune activity is therefore of clinical relevance to both vaccine design and modulation of autoimmunity.
Our results demonstrate a conserved cooperation between innate and adaptive immune receptor signaling in B cells. The findings could be used to develop more effective vaccines by specifically designing optimally cooperative antigen/adjuvant combinations. In addition, AP-1 or its individual monomers could be targeted to improve clinical symptoms of autoimmune diseases that arise from aberrant IL-6 production such as SLE and rheumatoid arthritis [45–47].
Material and Methods
Reagents
Rabbit Anti-mouse IgM, goat anti-human IgM (BCR polyclonal Ab (Fab'2)), goat anti-mouse and goat anti-rabbit peroxidase-conjugated secondary antibodies were purchased from Jackson Immunologicals (West Grove, PA). Anti-mouse and anti-human IL-6 mAbs (MP5-20F3 and MP5-32C11 biotin-conjugated and MQ2-13A5 and MQ239C3 biotin-conjugated) were obtained from eBioscience (San Diego, CA). Multiplex assays (Ten-plex: IL-1b, TNF-a, IL-6, IL-2, IL-4, IL-5, IL-10, IL-12, IFN-g, GM-CSF) performed on human samples were obtained from Biosource (Carlsbad, CA). Abs specific for JNK, pJNK, HP-1a, cJun, and cFos were purchased from Cell Signaling Technologies (Beverly, MA). Antibodies specific for phospho-cJun were obtained from Upstate Biotechnologies (Lake Placid, NY). FITC-conjugated anti-CD19 Ab was purchased from Miltenyi (Auburn, CA). The TLR7/8 agonist R-848 was obtained from Alexis Biochemicals (San Diego, CA). JNK inhibitor VIII was obtained from Calbiochem Inc. (San Diego, CA). Hi5 insect cells expressing CD154 and controls have previously been described [48]. A B cell to Hi5 cell ratio of 1:10 was used for the suboptimal stimulatory conditions of B cells.
Cells
The mouse B cell line CH12.LX and subclones stably transfected with inducible dominant negative cFos/AP-1 (A-Fos) were maintained as previously described [6]. High density mouse splenic B cells were obtained from C57Bl/6 mice using complement directed lysis of T cells followed by Percoll density gradient separation [49]. Use of mice in this study followed a protocol approved by the University of Iowa Animal Care and Use Committee. Blood collected from healthy adult volunteers via veinipuncture was diluted 1:2 with warm PBS and layered onto a Ficoll density gradient. After centrifugation (600 × g for 25 minutes) the resulting PBMC band was removed and washed thoroughly with cold PBS. Recovered cells were subjected to magnetic separation using a negative selection B cell isolation kit as per manufacturer's instructions (StemCell Technologies Inc., Vancouver, Canada). B cell purity as determined by CD19 staining was > 94%. Cells were cultured in BCM10 medium (RPMI 1640 supplemented with 10uM 2ME, 10% FBS, glutamine, and antibiotics). The use of normal human B cells in this study was approved by The University of Iowa IRB.
Cytokine Assays
CH12.LX (1×105/ml), mouse splenic B cells (1×106/ml), or human B cells (1×106/ml) were incubated in 48 well plates at 0.5ml per well. Cells were treated as described with anti-BCR (Fab')2 antibodies, R848, CD154-expressing or Wt Hi5 insect cells, chemical inhibitors (JNK VIII), and/or IPTG for designated times. Chemical inhibitors were used at concentrations that did not decrease cell viability, as monitored by Trypan blue exclusion. The insect cells grow at 27°C and die forming membrane fragments at the 37°C temperature used for B cell cultures. Cell free supernatants were collected and quantitative ELISA for IL-6 (eBioscience) or Luminex based multiplex (human 10 plex - Biosource) was performed according to the manufacturer's recommended protocol.
Western blotting of cellular and nuclear lysates
Nuclear extracts were collected from purified splenic B cells as follows. 2 × 106 cells per treatment were stimulated as described in Figure legends. Upon completion of stimulation cells were collected by centrifugation (300 × g for 2 minutes at 4°C). Cells were resuspended in 400ul lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.2 mM Na Vanadate, and 1 mM DTT) for 15 minutes on ice. Following incubation, 25ul of 10% NP-40 detergent was added to each sample and vortexed for 10 seconds. Nuclei were collected by centrifugation (10,000 × g for 2 minutes at 4°C). Pelleted nuclei were then disrupted by the addition of 30ul nuclear lysis buffer (10 mM HEPES pH 7.9, 400 mM NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.2 mM Na Vanadate, and 1 mM DTT) and incubated with rocking at 4°C for 30 minutes. Nuclear debris was removed by centrifugation (10,000 × g for 5 minutes at 4°C) and the supernatant collected. SDS-treatment buffer was added to the supernatant samples and heated to 95°C for 7 minutes. Samples were subsequently used in Western blotting protocols below.
Both CH12.LX and splenic B cells were washed, resuspended at 1 × 106/ml in BCM-10 and rested for 1hr at 37°C. Cells were then treated as indicated and whole cell lysates prepared as previously described [50]. Cellular or nuclear lysates were separated using 10% polyacrylamide gels, transferred onto PVDF membranes, and analyzed by Western blot as previously described [50]. Membranes were incubated with designated Abs diluted in TBST (Tris buffered saline pH8.0 with 0.1% Tween 20) containing 1% BSA and 0.01% NaN3 at 4°C overnight. Blots were washed with TBST 3 times for 10 minutes each. Blots were then incubated in TBST containing peroxidase-conjugated secondary Ab for 1hr at RT with gentle rocking, followed by 5 washes in TBST for 5 minutes each. Peroxidase labeled secondary Abs were visualized using the chemiluminescent detection reagent West Pico peroxidase substrate (Pierce, Rockford, IL) and the luminescence measured using a Fujifilm LAS-1000 imaging system (Fujifilm Medical Systems, Ltd., Stanford, CT). Chemiluminescence was subsequently quantified using ImageGauge software (FujiFilm). Values were then used to calculate ratios of phosphorylated:total protein (phospho-protein:control protein). JNK inhibitor VIII was added to cell cultures 30 minutes prior to stimulatory treatments for Western blotting experiments.
AP-1 specific ELISA
Nuclear extracts were isolated as previously described [51]. In brief, 1×107 splenic B cells were stimulated as noted and collected by centrifugation (500 × g for 2 minutes). Cells were resuspended in 400uL suspension buffer (10mM HEPES, pH7.5, 10mM KCl, 0.1mM EDTA, 0.1mM EGTA, 0.5mM DTT, and 0.5mM PMSF) and placed on ice for 15 minutes. 100ul of lysis buffer (suspension buffer with 0.5% NP-40) was added to each sample, inverted 3 times and centrifuged (500 × g for 5 minutes). Nuclear pellets were then resuspended stepwise in three buffers: 4ul buffer B1 (20mM HEPES pH 7.5, 0.1 NaCl, 0.1mM EDTA, 0.1mM EGTA, 50mM NaF, 20% glycerol, 0.5mM DTT, 0.5mM PMSF, and 1× protease inhibitor cocktail - Roche), 4ul buffer B2 (20mM HEPES pH 7.5, 0.3 NaCl, 0.1mM EDTA, 0.1mM EGTA, 50mM NaF, 20% glycerol, 0.5mM DTT, 0.5mM PMSF, and 1× protease inhibitor cocktail - Roche), and 12ul buffer B3 (20mM HEPES pH 7.5, 0.6 NaCl, 0.1mM EDTA, 0.1mM EGTA, 50mM NaF, 20% glycerol, 0.5mM DTT, 0.5mM PMSF, and 1× protease inhibitor cocktail - Roche). The suspension was placed on orbital shaker for 1 hour at 4°C. Debris from nuclear extracts was removed by centrifugation (12,000 × g for 10 minutes at 4°C) and supernatant (nuclear extract) stored at −80°C until assay was performed. Protein content was determined by Bradford assay. AP-1 specific ELISA was purchased from Chemicon International (Temecula, CA) and performed as per manufacturer's instructions. 1ug of total nuclear protein (above) was used for each well of the AP-1 ELISA. Background corrected absorbance at 450nm, indicating the relative amount of individual AP-1 monomeric components, was compared between treatments. The non-biotinylated AP-1 specific probe supplied with the kit was used as the negative control. OD values obtained from the dual stimulated sample reacted with the AP-1 specific non-biotinylated competitor were used for background correction (OD treatment – OD cold competitor from dual stimulated samples).
Acknowledgements
We thank Drs. Laura Stunz, Bruce Hostager, and Jon Houtman for valuable discussions and critical reading of the manuscript. This work was supported by a career award from the VA to GAB, grants from the NIH to GAB (AI28847, AI49993, CA099997), and a postdoctoral fellowship from the American Cancer Society to TJV.
Abbreviations used
- cFos/AP-1
(AP-1 dimer containing cFos)
References
- 1.Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev. 2000;176:154–170. doi: 10.1034/j.1600-065x.2000.00607.x. [DOI] [PubMed] [Google Scholar]
- 2.Hodgkin PD, Basten A. B cell activation, tolerance and antigen-presenting function. Curr Opin Immunol. 1995;7:121–129. doi: 10.1016/0952-7915(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 3.Bishop GA, Hostager BS. The CD40–CD154 interaction in B cell-T cell liaisons. Cytokine Growth Factor Rev. 2003;14:297–309. doi: 10.1016/s1359-6101(03)00024-8. [DOI] [PubMed] [Google Scholar]
- 4.Urashima M, Chauhan D, Hatziyanni M, Ogata A, Hollenbaugh D, Aruffo A, Anderson KC. CD40 ligand triggers interleukin-6 mediated B cell differentiation. Leuk Res. 1996;20:507–515. doi: 10.1016/0145-2126(95)00098-4. [DOI] [PubMed] [Google Scholar]
- 5.Cerutti A, Zan H, Schaffer A, Bergsagel L, Harindranath N, Max EE, Casali P. CD40 ligand and appropriate cytokines induce switching to IgG, IgA, and IgE and coordinated germinal center and plasmacytoid phenotypic differentiation in a human monoclonal IgM+IgD+B cell line. J Immunol. 1998;160:2145–2157. [PMC free article] [PubMed] [Google Scholar]
- 6.Baccam M, Woo SY, Vinson C, Bishop GA. CD40-mediated transcriptional regulation of the IL-6 gene in B lymphocytes: involvement of NF-kappa B, AP-1, and C/EBP. J Immunol. 2003;170:3099–3108. doi: 10.4049/jimmunol.170.6.3099. [DOI] [PubMed] [Google Scholar]
- 7.Haxhinasto SA, Bishop GA. Synergistic B cell activation by CD40 and the B cell antigen receptor: role of B lymphocyte antigen receptor-mediated kinase activation and tumor necrosis factor receptor-associated factor regulation. J Biol Chem. 2004;279:2575–2582. doi: 10.1074/jbc.M310628200. [DOI] [PubMed] [Google Scholar]
- 8.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 9.Re F, Strominger JL. Heterogeneity of TLR-induced responses in dendritic cells: from innate to adaptive immunity. Immunobiology. 2004;209:191–198. doi: 10.1016/j.imbio.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Bourke E, Bosisio D, Golay J, Polentarutti N, Mantovani A. The toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood. 2003;102:956–963. doi: 10.1182/blood-2002-11-3355. [DOI] [PubMed] [Google Scholar]
- 11.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 13.Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, Lipford G, Bauer S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein DI, Harrison CJ, Tomai MA, Miller RL. Daily or weekly therapy with resiquimod (R-848) reduces genital recurrences in herpes simplex virus-infected guinea pigs during and after treatment. J Infect Dis. 2001;183:844–849. doi: 10.1086/319262. [DOI] [PubMed] [Google Scholar]
- 15.Garland SM. Imiquimod. Curr Opin Infect Dis. 2003;16:85–89. doi: 10.1097/00001432-200304000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Hengge UR, Benninghoff B, Ruzicka T, Goos M. Topical immunomodulators--progress towards treating inflammation, infection, and cancer. Lancet Infect Dis. 2001;1:189–198. doi: 10.1016/s1473-3099(01)00095-0. [DOI] [PubMed] [Google Scholar]
- 17.Ramakrishna V, Vasilakos JP, Tario JD, Jr., Berger MA, Wallace PK, Keler T. Toll-like receptor activation enhances cell-mediated immunity induced by an antibody vaccine targeting human dendritic cells. J Transl Med. 2007;5:5. doi: 10.1186/1479-5876-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller RL, Tomai MA, Harrison CJ, Bernstein DI. Immunomodulation as a treatment strategy for genital herpes: review of the evidence. Int Immunopharmacol. 2002;2:443–451. doi: 10.1016/s1567-5769(01)00184-9. [DOI] [PubMed] [Google Scholar]
- 19.McCluskie MJ, Cartier JL, Patrick AJ, Sajic D, Weeratna RD, Rosenthal KL, Davis HL. Treatment of intravaginal HSV-2 infection in mice: a comparison of CpG oligodeoxynucleotides and resiquimod (R-848) Antiviral Res. 2006;69:77–85. doi: 10.1016/j.antiviral.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Weeratna RD, Makinen SR, McCluskie MJ, Davis HL. TLR agonists as vaccine adjuvants: comparison of CpG ODN and Resiquimod (R-848) Vaccine. 2005;23:5263–5270. doi: 10.1016/j.vaccine.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 21.Vasilakos JP, Smith RM, Gibson SJ, Lindh JM, Pederson LK, Reiter MJ, Smith MH, Tomai MA. Adjuvant activities of immune response modifier R-848: comparison with CpG ODN. Cell Immunol. 2000;204:64–74. doi: 10.1006/cimm.2000.1689. [DOI] [PubMed] [Google Scholar]
- 22.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J Immunol. 1995;155:3734–3741. [PubMed] [Google Scholar]
- 23.Constant SL. B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J Immunol. 1999;162:5695–5703. [PubMed] [Google Scholar]
- 24.Schlaepfer E, Audige A, Joller H, Speck RF. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J Immunol. 2006;176:2888–2895. doi: 10.4049/jimmunol.176.5.2888. [DOI] [PubMed] [Google Scholar]
- 25.Casadevall A, Pirofski LA. A reappraisal of humoral immunity based on mechanisms of antibody-mediated protection against intracellular pathogens. Adv Immunol. 2006;91:1–44. doi: 10.1016/S0065-2776(06)91001-3. [DOI] [PubMed] [Google Scholar]
- 26.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 27.Bishop GA, Hsing Y, Hostager BS, Jalukar SV, Ramirez LM, Tomai MA. Molecular mechanisms of B lymphocyte activation by the immune response modifier R-848. J Immunol. 2000;165:5552–5557. doi: 10.4049/jimmunol.165.10.5552. [DOI] [PubMed] [Google Scholar]
- 28.Bishop GA, Ramirez LM, Baccam M, Busch LK, Pederson LK, Tomai MA. The immune response modifier resiquimod mimics CD40-induced B cell activation. Cell Immunol. 2001;208:9–17. doi: 10.1006/cimm.2001.1769. [DOI] [PubMed] [Google Scholar]
- 29.Baccam M, Bishop GA. Membrane-bound CD154, but not CD40-specific antibody, mediates NF-kappaB-independent IL-6 production in B cells. Eur J Immunol. 1999;29:3855–3866. doi: 10.1002/(SICI)1521-4141(199912)29:12<3855::AID-IMMU3855>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 30.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 31.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 32.Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther. 2006;8(Suppl 2):S2. doi: 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukundan L, Bishop GA, Head KZ, Zhang L, Wahl LM, Suttles J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J Immunol. 2005;174:1081–1090. doi: 10.4049/jimmunol.174.2.1081. [DOI] [PubMed] [Google Scholar]
- 34.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 35.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 36.Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 37.Sassone-Corsi P, Sisson JC, Verma IM. Transcriptional autoregulation of the proto-oncogene fos. Nature. 1988;334:314–319. doi: 10.1038/334314a0. [DOI] [PubMed] [Google Scholar]
- 38.Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C. A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J Biol Chem. 1997;272:18586–18594. doi: 10.1074/jbc.272.30.18586. [DOI] [PubMed] [Google Scholar]
- 39.Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, Hartmann G. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005;174:4043–4050. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 40.Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117:5965–5973. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- 41.Bakiri L, Matsuo K, Wisniewska M, Wagner EF, Yaniv M. Promoter specificity and biological activity of tethered AP-1 dimers. Mol Cell Biol. 2002;22:4952–4964. doi: 10.1128/MCB.22.13.4952-4964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ray N, Kuwahara M, Takada Y, Maruyama K, Kawaguchi T, Tsubone H, Ishikawa H, Matsuo K. c-Fos suppresses systemic inflammatory response to endotoxin. Int Immunol. 2006;18:671–677. doi: 10.1093/intimm/dxl004. [DOI] [PubMed] [Google Scholar]
- 43.Maruyama K, Sano G, Ray N, Takada Y, Matsuo K. c-Fos-deficient mice are susceptible to Salmonella enterica serovar Typhimurium infection. Infect Immun. 2007;75:1520–1523. doi: 10.1128/IAI.01316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, Maliszewski C, Akira S, Pulendran B. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. 2004;172:4733–4743. doi: 10.4049/jimmunol.172.8.4733. [DOI] [PubMed] [Google Scholar]
- 45.Anolik JH, Aringer M. New treatments for SLE: cell-depleting and anti-cytokine therapies. Best Pract Res Clin Rheumatol. 2005;19:859–878. doi: 10.1016/j.berh.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Smolen JS, Maini RN. Interleukin-6: a new therapeutic target. Arthritis Res Ther. 2006;8(Suppl 2):S5. doi: 10.1186/ar1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paul-Pletzer K. Tocilizumab: blockade of interleukin-6 signaling pathway as a therapeutic strategy for inflammatory disorders. Drugs Today (Barc) 2006;42:559–576. doi: 10.1358/dot.2006.42.9.1025692. [DOI] [PubMed] [Google Scholar]
- 48.Hostager BS, Haxhinasto SA, Rowland SL, Bishop GA. Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem. 2003;278:45382–45390. doi: 10.1074/jbc.M306708200. [DOI] [PubMed] [Google Scholar]
- 49.Bishop GA, Ramirez LM, Waldschmidt TJ. Differential responses to Ig and class II-mediated signals in splenic B cell subsets from normal and autoimmune mice. Int Immunol. 1994;6:1049–1059. doi: 10.1093/intimm/6.7.1049. [DOI] [PubMed] [Google Scholar]
- 50.Moore CR, Bishop GA. Differential regulation of CD40-mediated TNF receptor-associated factor degradation in B lymphocytes. J Immunol. 2005;175:3780–3789. doi: 10.4049/jimmunol.175.6.3780. [DOI] [PubMed] [Google Scholar]
- 51.Lenert P, Yi AK, Krieg AM, Stunz LL, Ashman RF. Inhibitory oligonucleotides block the induction of AP-1 transcription factor by stimulatory CpG oligonucleotides in B cells. Antisense Nucleic Acid Drug Dev. 2003;13:143–150. doi: 10.1089/108729003768247600. [DOI] [PubMed] [Google Scholar]