

Abstract
Nucleobindin 1 (NUCB1) is a widely expressed multidomain calcium-binding protein whose precise physiological and biochemical functions are not well understood. We engineered and heterologously expressed a soluble form of NUCB1 (sNUCB1) and characterized its biophysical and biochemical properties. We show that sNUCB1 exists as a dimer in solution and that each monomer binds two divalent calcium cations. Calcium binding causes conformational changes in sNUCB1 as judged by circular dichroism and fluorescence spectroscopy experiments. Earlier reports suggested that NUCB1 might interact with heterotrimeric G protein α subunits. We show that dimeric calcium-free sNUCB1 binds to expressed Gαi1 and that calcium binding inhibits the interaction. The binding of sNUCB1 to Gαi1 inhibits its basal rate of GDP release and slows its rate and extent of GTPγS uptake. Additionally, our tissue culture experiments show that sNUCB1 prevents receptor-mediated Gαi-dependent inhibition of adenylyl cyclase. Thus, we conclude that sNUCB1 is a calcium-dependent guanine nucleotide dissociation inhibitor (GDI) for Gαi1. To our knowledge, sNUCB1 is the first example of a calcium-dependent GDI for heterotrimeric G proteins. We also show that the mechanism of GDI activity of sNUCB1 is unique and does not arise from the consensus GoLoco motif found in RGS proteins. We propose that cytoplasmic NUCB1 might function to regulate heterotrimeric G protein trafficking and G protein-coupled receptor-mediated signal transduction pathways.
Keywords: Allosteric Regulation, Calcium-binding Proteins, G Protein-coupled Receptors (GPCR), G Proteins, Protein-Protein Interactions
Introduction
Heterotrimeric guanine nucleotide-binding proteins, G proteins, couple to heptahelical cell surface G protein-coupled receptors (GPCRs)3 and participate in intracellular signaling events. The G protein heterotrimer is composed of the Gα subunit and the Gβγ heterodimer. Upon ligand-mediated activation, GPCRs catalyze the exchange of GDP for GTP on Gα leading to dissociation of the heterotrimer into Gα·GTP and Gβγ subunits (1–3). These individual subunits then regulate downstream signaling cascades involving effector systems like adenylyl cyclases, Ca2+ and K+ channels, phospholipase C isozymes, and cyclic nucleotide phosphodiesterases (4, 5). Thereafter, the intrinsic GTPase activity of Gα reverts it back to the GDP-bound state, which can reassociate with Gβγ. This inhibits the interaction of G protein subunits with downstream effectors, which results in the turning-off of the signaling pathways. Hence, signaling by heterotrimeric G proteins is directly dependent on the lifetime of the GTP-bound state of Gα. This lifetime is regulated by GTPase-accelerating proteins (GAPs), which catalyze the rapid hydrolysis of the Gα-bound GTP to GDP and by guanine nucleotide dissociation inhibitors (GDIs), which inhibit the exchange of GDP for GTP in the catalytic pocket of Gα (6).
Together, GAPs and GDIs exert a regulatory control on G protein signaling. In recent years, novel interacting partners of heterotrimeric G proteins called the regulators of G protein signaling or RGS proteins have been discovered that possess GAP activity (7, 8). Interestingly, RGS12 and RGS14 in addition to functioning as GAPs can also act as GDIs (9, 10). In 1999, Lanier and co-workers (11) used yeast two-hybrid analysis to identify distinct receptor-independent activators of G protein signaling or AGS proteins. Several members of this AGS family (AGS3-6) have been shown to function as GDIs of Gαi subunits (12). The observed GDI activity of both AGS and RGS proteins toward Gαi/o is attributed to a 19-amino acid consensus sequence called the GoLoco motif (9, 13, 14). Earlier in 1995, in an independent yeast two-hybrid screen, Mochizuki et al. (15) established the interaction of a novel Golgi-resident Ca2+-binding protein Nucleobindin 1 or NUCB1, specifically with the heterotrimeric G protein α subunit, Gαi2. Subsequently, Lin et al. (16) demonstrated that NUCB1 interacts exclusively with the adenylyl cyclase inhibitory (Gαi) and stimulatory (Gαs) classes of Gα subunits. In a recent study, it was shown that overexpression of NUCB1 causes redistribution of only the Gαi1 subunits and not the Gβγ subunits to the plasma membrane and regulated secretion granules (17). However, a role for NUCB1 in modulating Gαi1 activation and the biochemistry of NUCB1-Gαi1 interaction has not been reported.
NUCB1 is a 55-kDa multidomain Ca2+-binding protein that was first identified as a novel B cell growth and differentiation factor associated with lupus syndrome (18). NUCB1 derives its trivial name, Calnuc, from its Ca2+-binding and DNA-binding ability (19). The DNA-binding domain of basic residues (172–218) lies at the N terminus following the signal sequence. The Ca2+-binding domain is at the core of the protein sequence consisting of two EF hand motifs with an intervening acidic region (residues 253–316). The Ca2+-binding domain is followed by a leucine zipper domain (residues 347–389), which has been postulated to induce NUCB1 dimerization (Fig. 1A) (20). The C-terminal (CT) region following the leucine zipper domain is predicted to be intrinsically disordered and unstructured. Intriguingly, NUCB1 is strongly conserved from flies to humans (21) and is widely distributed among Golgi (16, 22), nucleus (19, 23), endoplasmic reticulum (24, 25), and cytoplasm (17). The N-terminal signal sequence of NUCB1 targets it to different membrane compartments and its deletion renders NUCB1 cytosolic.
FIGURE 1.

Domain architecture of human NUCB1 and engineered NUCB1 variants and Ca2+-binding activity of a soluble NUCB1 variant. A, modular nature of the NUCB1 protein structure is depicted schematically with its N-terminal signal sequence (gray square), putative DNA binding domain (green square), two EF hand loops (magenta rectangles), acidic region (blue oval), and leucine zipper domain (yellow hexagon). The amino acid residues at domain borders are numbered. We engineered a construct for expression in E. coli of a soluble form of NUCB1 that included an N-terminal His6 tag and a PreScission protease recognition sequence with an intervening spacer sequence as shown. Endoprotease cleavage yields a soluble form of NUCB1 that begins with the amino acid sequence GPHMAS and continues with the remainder of the native sequence beginning at Gly-32. We refer to this expressed protein construct as soluble NUCB1 or sNUCB1 throughout the text. We also constructed a double replacement mutant sNUCB1(W232A/W333A) and a truncation mutant sNUCB1(W333Ter) as shown. B, we used isothermal titration calorimetry (ITC) to measure the Ca2+-binding affinity of sNUCB1. sNUCB1 presumably binds one Ca2+ cation at each of its two EF hand domains, and a nonlinear least squares fit of the calculated values using the two-set of sites model resulted in an excellent fit as shown in the inset. The calculated dissociation constants for Ca2+ binding to the two EF hand domains of sNUCB1 are 6.3 ± 0.18 and 73.5 ± 0.19 μm, respectively.
Recently, Brodeur et al. (26) reported the role of NUCB1 in LDL receptor-related protein 9 (LRP9) trafficking where the cytosolic NUCB1 fraction helps in LRP9 endosomal sorting and prevents its delivery to lysosomes. The ubiquitous expression of NUCB1 in various cell and tissue types results in a diverse interactome, including interacting partners like G protein α subunits, cyclooxygenases, and amyloid precursor protein (16, 25–27). Several classes of Gα subunits have been shown to interact with NUCB1 (16, 25–27). Yeast two-hybrid experiments performed with various deletion constructs of either Gαi3 or NUCB1 mapped the C-terminal α5 helix domain of the G protein and the acidic region of NUCB1 (residues 264–305) to be necessary for interaction (28). Immunofluorescence-based studies showed that NUCB1 and Gαi protein subunits co-localize on the Golgi lumen and regulated secretion granules (17). Because Gα subunits are ubiquitously involved in numerous signal transduction pathways in different tissue types, their interaction with NUCB1 might regulate downstream signaling events.
We are interested in understanding the role of the α5 helix of the G protein α subunit in regulating nucleotide exchange rates (29–31). Because the α5 helix of Gα is involved in interaction with NUCB1, we characterized and studied the interaction of NUCB1 with Gαi1 in detail. In this study, we present the detailed biophysical and biochemical characterization of the Ca2+-binding ability and the oligomeric state of a heterologously expressed N-terminally truncated soluble form of NUCB1, termed sNUCB1 (Fig. 1A). We show that sNUCB1 binds Ca2+ and exists as a dimer in solution. Ca2+-free sNUCB1 preferentially binds Gαi1 and inhibits its GDP release as demonstrated by several independent assays. We have shown that overexpression of sNUCB1 in HEK293 cells markedly decreases ligand-dependent receptor-mediated Gαi inhibition of adenylyl cyclase. We conclude that sNUCB1 serves as a novel Ca2+-regulated GDI of Gαi1. Our findings suggest that NUCB1 is involved in regulating cytoplasmic G protein signaling pathways.
EXPERIMENTAL PROCEDURES
Reagents
GDP sodium salt and GTPγS tetralithium salt were purchased from Sigma. The fluorescent GTPγS analogue, mant(2′,3′-O-(N-methyl-anthraniloyl)-GTPγS was purchased from Jena Biosciences (Jena, Germany), and BODIPY-FL-GTPγS was purchased from Molecular Probes (Invitrogen). The His6-tagged mutants of WT Gαi1 were generated by site-directed mutagenesis using the high fidelity thermostable DNA polymerase Pfu (Stratagene). The cAMP dynamic 2 kit was purchased from CisBio (Bedford, MA) for cell-based studies. All reagents and chemicals used were of highest available purity.
Heterologous Expression of sNUCB1 and Heterotrimeric G Protein α-Subunit Gαi1
The cDNA clones for human NUCB1 and rat Gαi1 were obtained from the ATCC. The DNA fragment for the soluble form of NUCB1 or sNUCB1, corresponding to residues 31–461 of the human NUCB1 protein without the N-terminal signal sequence (residues 1–31), was cloned into the pET28a(+) expression vector (Amersham Biosciences). Similarly, the DNA fragment corresponding to full-length WT rat Gαi1 subunit (residues 1–354) was cloned into the pET28a(+) expression vector. The vector was used to transform BL21 (DE3) cells to express an N-terminal His6 tag protein containing a PreScission protease site directly after the His6 tag. The expressed construct for sNUCB1 served as a template for introducing site-specific mutations using the QuikChange system (Stratagene, La Jolla, CA) to generate sNUCB1(W232A/W333A) or for introducing a missense mutation to generate the truncated version of the protein, namely sNUCB1(W333Ter). All proteins were expressed in BL21 (DE3) cells grown in the presence of 50 μg/ml kanamycin. Cells were grown at 37 °C to A600 nm of ∼0.7 and then induced with 500 μm isopropyl 1-thio-β-d-galactopyranoside (United States Biological). Post-induction, the culture was grown overnight at 17 °C and subsequently harvested. The resulting pellets were resuspended in a buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 50 mm β-mercaptoethanol, bovine lung aprotinin (20 mg/ml), 2 mm phenylmethanesulfonyl fluoride (PMSF), and complete EDTA-free protease inhibitor mixture tablets (Roche Applied Science). Thereafter, the cells were lysed, and the expressed His6 tag protein was purified from crude extract by affinity chromatography using nickel-nitrilotriacetic acid column pre-equilibrated with buffer A (50 mm Tris, pH 8.0, 150 mm NaCl, 50 mm β-mercaptoethanol) for sNUCB1 and its variants and buffer A′ (50 mm Tris, pH 8.0, 150 mm NaCl, 2 mm MgCl2·6H2O, 10 μm GDP, 50 mm β-mercaptoethanol) for WT Gαi1. The bound His6 tag protein was eluted from the column by using buffer A or buffer A′ supplemented with 500 mm imidazole. The His6 tag was cleaved by PreScissionTM protease, which was subsequently removed. To remove Ca2+ from sNUCB1 or its variants, the proteins were extensively dialyzed against buffer A supplemented with 5 mm EGTA and 1 mm EDTA, which are also eventually removed through dialysis. Finally, SEC was done using a Superdex200 26/60 HR column equilibrated with buffer S (50 mm Tris, pH 8.0, 150 mm NaCl) for sNUCB1 and its variants and equilibrated in buffer S′ (50 mm Tris, pH 8.0, 150 mm NaCl, 2 mm MgCl2·6H2O, 10 μm GDP, 1 mm DTT) for WT Gαi1 to obtain homogeneously pure protein. The typical yield for sNUCB1 and its variants was ∼5 mg/liter and for WT Gαi1 ∼12–15 mg/liter. The purity of the proteins was assessed by Coomassie Brilliant Blue staining after the proteins were separated through SDS-PAGE. The purity of all proteins was greater than 95%.
Isothermal Titration Calorimetry (ITC)
ITC was performed at 25 °C (298 K) using a MicroCal VP-ITC (MicroCal, Northhampton, MA) calorimeter. To measure quantitatively the binding of Ca2+ or WT Gαi1·GDP to sNUCB1, 2 ml of 50 μm sNUCB1 (Ca2+-free) and 700 μl of either 500 μm CaCl2·6H2O (prepared in 50 mm Tris, pH 8.0, 150 mm NaCl) or 600 μm WT Gαi1·GDP (prepared in 50 mm Tris, pH 8.0, 150 mm NaCl, 2 mm MgCl2·6H2O, 10 μm GDP, 1 mm tris(2-carboxyethyl)phosphine) was used and thoroughly degassed. Contents of the sample cell were stirred continuously at 280 rpm during the experiment. A typical titration of sNUCB1 involved 50 injections each of 5 μl of CaCl2·6H2O (500 μm) or WT Gαi1·GDP (600 μm) into a sample cell containing 1.4 ml of sNUCB1 (50 μm). The heat of dilution of the titrant (CaCl2·6H2O or WT Gαi1·GDP) was subtracted from the titration data for base-line correction. The base line-corrected data were analyzed with MicroCal OriginTM 6.0 software to determine the change in enthalpy (ΔH) and association constant (Ka). Thermal titration data fit showed two sets of binding sites for Ca2+ to sNUBC1. The parameters were calculated based on three or more independent titration experiments. Each numerical value reported is the mean ± S.E. among the independent data sets.
Size-exclusion Chromatography
Complex Formation between sNUCB1 and WT Gαi1
Complex formation between sNUCB1 and Gαi1 was monitored using SEC. 150 μm sNUCB1 (−Ca2+/+Ca2+) was incubated with 256 μm Gαi1·(GDP/GTPγS) in 20 mm Tris, pH 8.0, 100 mm NaCl, 10 mm MgCl2, 10 μm GDP, and 1 mm DTT at 30 °C for 20 min with minimal shaking to enable saturation binding. WT Gαi1 in the GTPγS-bound state was generated by incubating WT Gαi1·GDP with 5 mm GTPγS for 30 min at 30 °C to ensure complete exchange of nucleotide. Furthermore, the complete exchange was confirmed by the AlF4− exchange assay. About 200 μl of the reaction mixture was injected onto Superdex200 10/30 GL column attached to AKTA FPLC (GE Healthcare) and run at 4 °C. Elution was done with the incubation buffer at a flow rate of 0.5 ml/min with 0.35-ml fractions collected. Peak fractions were analyzed through SDS-PAGE and Coomassie-stained to analyze the complex formation.
Estimation of sNUCB1 Molecular Mass Using Heavy Molecular Weight Standards
Purified sNUCB1 or the heavy molecular weight standards (GE Healthcare) were injected onto a Superdex200 10/30 GL column pre-equilibrated with buffer S, and the normalized absorbance of the eluting peak was plotted. A calibration curve of Kav (Ve − Vo/Vc − Vo) versus log10(molecular weight) was plotted, where Ve is the elution volume; Vo is the column void volume corresponding to the elution of blue dextran, and Vc is the geometric column volume. The data were fit to a straight line curve like Y = A + Bx, where A = 1.915 and B = 0.311 were obtained from the fit. Therefore, for a Y = Kav value, the corresponding value of log10 (molecular weight) was obtained to estimate the molecular mass of sNUCB1 and sNUCB1(W333Ter).
Light Scattering
Multiangle Light Scattering (MALS) and Dynamic Light Scattering (DLS)
The light scattering data were collected using a Superdex200 10/30, HR SEC column (GE Healthcare), connected to an HPLC system (Agilent 1200, Agilent Technologies, Wilmington, DE) equipped with an autosampler. The elution from SEC was monitored by a photodiode array UV-visible detector (Agilent Technologies), differential refractometer (OPT-Lab rEx, Wyatt Corp., Santa Barbara, CA), static and dynamic, multiangle laser light scattering detector (HELEOS II with QELS capability, Wyatt Corp.). The SEC-UV/LS/RI system was equilibrated in buffer (50 mm Tris, pH 8.0, 150 mm NaCl) for sNUCB1 alone and in buffer (50 mm Tris, pH 8.0, 100 mm NaCl, 10 mm MgCl2, 10 μm GDP, 1 mm DTT) for WT and truncated sNUCB1/G protein complex at the flow rate of 1.0 ml/min. Two software packages were used for data collection and analysis; the Chemstation software (Agilent Technologies) controlled the HPLC operation and data collection from the multiwavelength UV-visible detector, and the ASTRA software (Wyatt Corp., Santa Barbara, CA) collected data from the RI detector and the LS detectors and recorded the UV trace at 280 nm sent from the photodiode array detector. The weight average molecular masses, Mw, were determined across the entire elution profile in the intervals of 1 s from static LS measurement using ASTRA software as described previously (32). The approach uses a Rayleigh-Debye-Gans light scattering model (Equation 1), which relates the amount of scattered light to the concentration and weight average molecular weight of solute and second virial coefficient,
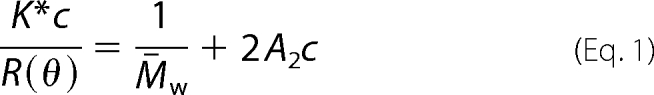 |
where R(θ) is the intensity of excess scattered light at angle θ; c is the concentration of the solute, M̄w is the weight average molecular weight of the solute; A2 is the second virial coefficient; K* is an optical parameter equal to 4π2n2 (dn/dc)2/(λ04NA), where n is the refractive index; dn/dc is the refractive index increment for the solute; NA is Avogadro's number, and λ0 is the wavelength of the scattered light.
DLS measurements were made “on line” at an angle of 100° with a 2-s collection time. Time-resolved scatter intensity fluctuations were analyzed using Astra software (Wyatt Corp.), which implements the cumulants method (33) to determine the time dependence of diffusive motion also referred to as the intensity correlation function, G(T), as shown in Equation 2 (34),
where B is the average base-line intensity; α is an instrument-specific correction factor; DT is the concentration-dependent translational diffusion constant of the solute; t is a delay time, and q is the scattering vector equal to (4πn)/λsinθ/2, where n is the refractive index of the solvent; λ is the wavelength of the scattered light, and θ is the scattering angle. Equation 2 describes the relationship between the time dependence of fluctuation in scatter intensity and the translational diffusion coefficient. The value of DT can be used to estimate the apparent hydrodynamic radius of an equivalent sphere using Stokes-Einstein relationship shown in Equation 3,
 |
where k is the Boltzmann constant; T is the absolute temperature, and η is the temperature-corrected viscosity of the solution.
Determination of Dimerization Equilibrium Constant from SEC-UV/LS/RI
The concentration-dependent changes in the weight average molecular mass determined for the apex of the eluting peak for sNUBC1 were used to determine the dimerization constant. The weight average molecular weights were plotted as a function of protein concentration for sNUCB1; error bars represent ±3% of M̄w for concentrations above 100 μg/ml, ±5% of M̄w for concentrations between 1 and 100 μg/ml, and ±10% of the M̄w for concentrations below 1 μg/ml. Lines represent the nonlinear least square fits to a monomer-dimer association model for sNUCB1 dimerization.
Determination of Kd of Complex Formation from SEC-UV/LS/RI
The concentration-dependent changes in the M̄w determined for the apex of the eluting peak were used to determine the complexation of Gαi1 with sNUCB1/sNUCB1(W333Ter). The M̄w values were plotted as a function of total protein concentration for complexation; error bars represent ±3% of M̄w for concentrations above 100 μg/ml, ±5% of M̄w for concentrations between 1 and 100 μg/ml, and ±10% of M̄w for concentrations below 1 μg/ml. Lines represent the nonlinear least square fits to a stimulated complexation model using Origin 6.0.
Fluorescence Spectroscopy
Steady-state Trp Fluorescence upon Ca2+ Binding
Steady-state fluorescence measurements for sNUCB1 were carried out in a SPEX τ3 spectrofluorimeter (Jobin-Yvon Instruments) at 25 °C. The excitation wavelength was set at 295 nm, although emission spectra were collected from 310 to 460 nm. The excitation and emission slit widths were 2.5 and 5.0 nm, respectively. 178 nm Ca2+-free sNUCB1 equilibrated in buffer containing 50 mm Tris, pH 8.0, 100 mm NaCl was incubated with increasing concentrations of Ca2+ with constant stirring for 5 min. Thereafter, Trp emission fluorescence spectra were collected for each concentration of Ca2+ added.
Quenching of Tryptophan Fluorescence by Small Molecule Quenchers
The excitation wavelength was set at 295 nm, and the emission spectra were collected from 315 to 460 nm with the excitation slit and emission slit width being 2.5 and 5 nm, respectively. 333 nm of protein was equilibrated in buffer containing 50 mm Tris, pH 8.0, and 100 mm NaCl. To study the interaction between the protein and the quenchers, small aliquots of the quenchers were added incrementally from a concentrated stock solution. In the case of potassium iodide, a small quantity of Na2S2O3 (0.1 mm) was added to prevent it from forming free triiodide (I3−). The accessibility of Trp was monitored by analyzing the quenching data using the Stern-Volmer Equation 4,
where F0 and F are the fluorescence intensities in the absence and presence of quenchers, respectively; [Q] is the concentration of the quenching molecules, and KSV is the Stern-Volmer quenching constant. In the case of purely collisional quenching mechanism, the Stern-Volmer plot of F0/F versus [Q] is linear with a slope equal to KSV.
Fluorescence-based mant-GTPγS (mGTPγS) Binding Assays
Steady-state fluorescence measurements experiments were carried out in a SPEX τ3 spectrofluorimeter (Jobin-Yvon Instruments) at 25 °C. 15 μm WT Gαi1 was incubated with 50 μm sNUCB1(W232A/W333A) in the reaction buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 2 mm MgCl2, and 1 mm DTT) for 5 min with constant stirring. Thereafter, mGTPγS (15 μm) was added to the reaction mixture. FRET was monitored by exciting the intrinsic Trp fluorescence at 295 nm and measuring the fluorescence emission from 310 to 540 nm. In all cases, blank spectra containing buffer with sNUCB1(W232A/W333A) alone were subtracted from the final spectra.
Time-based Fluorescence Assays
Time-based fluorescence measurements were performed in a SPEX Fluorolog-2 spectrofluorometer (SPEX Industries, Edison, NJ) at 25 °C. The excitation wavelength was set at 295 nm specifically for Trp residues, although emission wavelength was set at 340 nm. The excitation and emission slit width were 5 nm each.
AlF4− Uptake Assay
WT Gαi1 was incubated at 25 °C in reaction buffer K (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 2 mm MgCl2, and 1 mm DTT) with increasing amounts of sNUCB1(W232A/W333A) for 10 min with constant stirring. Thereafter, for AlF4−-dependent activation of Gαi1, a premixed solution of AlCl3− (20 mm) and NaF (1 m), respectively, was injected at 180 s. A time course of enhancement in Trp fluorescence coupled to activation was monitored in the presence of increasing concentrations of sNUCB1(W232A/W333A) in the reaction mixture.
GTPγS Nucleotide Exchange Assay
WT Gαi1 was incubated at 25 °C with increasing concentrations of sNUCB1(W232A/W333A) in reaction buffer K for 10 min with constant stirring. The base-line fluorescence was monitored for 200 s after which GTPγS (20 μm) was injected into the reaction mixture, and the relative increase in intrinsic Trp fluorescence (λex = 295 nm, λem = 340 nm) was measured as a function of time for each concentration of sNUCB1(W232A/W333A).
BODIPY-FL-GTPγS Nucleotide Exchange Assay
WT Gαi1 (20 μm) was incubated at 25 °C in reaction buffer K with increasing concentrations of Ca2+-free sNUCB1 for 10 min with constant stirring. BODIPY-FL-GTPγS (20 μm) was added to the reaction mixture, and the relative increase in intrinsic fluorescence (λex = 485 nm, λem = 512 nm) was measured for each concentration of Ca2+-free sNUCB1 as a function of time.
BODIPY-FL-GTPγS Nucleotide Binding Assay
WT Gαi1 (20 μm) was incubated alone and with Ca2+-free sNUCB1 (100 μm) at room temperature in reaction buffer K. 10-μl samples were withdrawn from each reaction mixture at various time points and loaded onto a pre-equilibrated ZebaTM micro-desalt spin column to remove the unbound nucleotide. The columns were spun at 1000 × g for 1 min, and the flow-through was collected. The amount of bound nucleotide in each flow-through was measured by using BODIPY absorbance at 504 nm (ϵ = 80,000 m−1 cm−1). The absorbance at 280 nm was also monitored to ensure that the same amount of protein was being collected in each flow-through sample.
Radioligand [35S]GTPγS-based Nucleotide Exchange Assay
The effect of Ca2+-free sNUCB1 or Ca2+-free sNUCB1(W333Ter) binding to WT Gαi1·GDP on the nucleotide exchange was monitored using [35S]GTPγS exchange assay. 20 μm WT Gαi1·GDP alone or with 100 μm Ca2+-free sNUCB1/sNUCB1(W333Ter) was incubated in buffer (10 mm Tris, pH 7.5, 100 mm NaCl, 2 mm MgCl2, 2 mm DTT) for 20 min at 30 °C. After incubation, [35S]GTPγS solution diluted with GTPγS was added to the reaction mixtures such that the final GTPγS concentration in each reaction mixture was 100 μm with an associated radioactivity of 50 nCi/8 μl of reaction volume. Thereafter, 8-μl samples (50 nCi) were withdrawn from each reaction mixture and added to buffer-equilibrated nitrocellulose filters. Subsequently, the filters were extensively washed with Wash buffer (50 mm Tris, pH 7.5, 5 mm MgCl2) to remove excess radioligand. For data analysis, the counts were recorded, and the value for buffer alone was subtracted from each reading. Each individual data point was scaled to the value of radioactivity within 8 μl of reaction volume to get the picomoles of radioligand bound to WT Gαi1 in each reaction. As a positive control, the C-terminal GoLoco motif peptide from RGS14, namely R14GL(496–531), was used in an identical experimental setup.
Circular Dichroism Spectroscopy
Wavelength Scan
All CD experiments were performed using an Aviv 62A DS CD spectrophotometer. Far-UV CD spectra were recorded at the end of each kinetic run. Spectra were recorded over the wavelength range of 190–250 nm at 1-nm intervals with an averaging time of 3 s using a 0.1-cm path length cell. Background spectrum was subtracted from each of the collected data sets. Each spectrum obtained was an average of three scans. The recorded spectra in millidegrees of ellipticity was converted to mean residue ellipticity in degrees·cm2 dmol−1 using Equation 5,
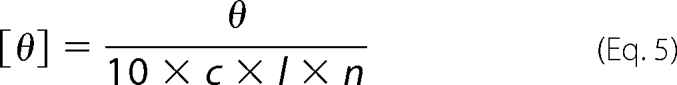 |
where [θ] is the mean residue ellipticity in degrees cm2 dmol−1; θ is the ellipticity in millidegrees; c is the concentration in m; l is the path length in cm, and n is the number of peptide bonds.
Thermal Unfolding
The unfolding of the protein (8 μm) with temperature was monitored using CD at a wavelength of 222 nm, which is characteristic of an α-helix. The data points were averaged over 30 s for every unit increment in temperature. A plot of CD signal versus temperature was fit to Equations 6 and 7,
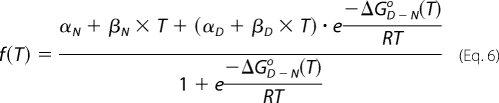 |
where,
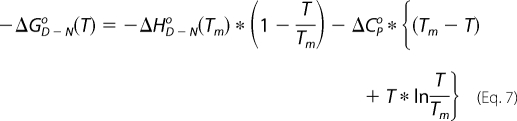 |
where f(T) is the signal as a function of temperature; T is the temperature; R is the gas constant; αN defines the intercept; βN is the slope of the post-transition region of the curve; αD defines the intercept; βD is the slope of the pre-transition region of the curve; ΔG0 is the free energy change for the unfolding reaction; ΔHD-No is the change in enthalpy for unfolding at the Tm, and ΔCpo is the change in heat capacity. Using the above expressions, Tm, the mid-point transition temperature, was estimated along with the change in enthalpy ΔHD-No, for the unfolding reaction at that Tm.
Analytical Ultracentrifugation (AUC)
Sedimentation equilibrium studies were carried out at different centrifugal speeds and protein concentrations for sNUCB1 and sNUCB1(W333Ter). AUC experiments for sNUCB1 (−/+Ca2+) were done at 25 °C with protein concentrations of 50, 100, and 150 μm each at speeds of 20,000, 30,000, and 40,000 rpm. Similarly for sNUCB1(W333Ter), AUC experiments were done with 32, 70, and 150 μm protein concentrations each at speeds of 25,000, 30,000, and 40,000 rpm. The individual protein samples were run for sufficiently long periods of time to allow for the equilibrium to generate a time invariant concentration gradient balanced by diffusion of the macromolecular species. The speeds were decided based on the molecular weight of the protein. Under no net transport conditions, the following correlation (Equations 8 and 9) should be observed between concentration and the radial distance (35),
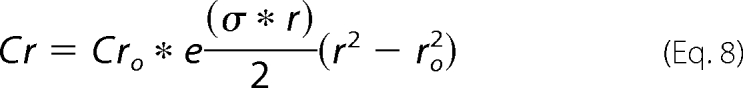 |
where,
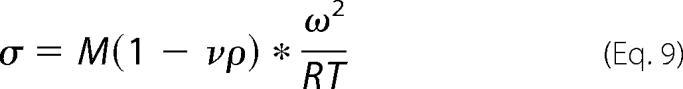 |
Cr is the concentration of macrosolute at any radial distance r; Cro is the concentration of the macrosolute at the reference radial distance ro; ν is the partial specific volume; ω is the angular velocity; ρ is the density; R is the gas constant; T is the absolute temperature; and M is the molecular mass. The data were analyzed using OptimaTM XL-A/XL-I data analysis software (Beckman, 2001).
Western Blot
HEK293 cells were maintained in Dulbecco's modified Eagle medium (Invitrogen) with 10% (v/v) fetal bovine serum (FBS) at 37 °C and 5% CO2. For Western blot analysis, cells were plated in 6-well plates and transiently transfected with 4 μg of total DNA (3.0 μg of pcDNA3.1(+) + 1.0 μg of WT Gαi1/sNUCB1/CXCR4), and cells were harvested after 48 h. Thereafter, cells were lysed, and the heat-denatured supernatant was subjected to SDS-PAGE analysis. Subsequently, the separated protein fractions were transferred to a PVDF membrane using semi-dry transfer procedures and probed with either 1D4 antibody to detect CXCR4, which has a C-terminal 1D4 tag, or with the respective primary antibodies for NUCB1 or Gαi1. Finally, the excess antibody was washed away, and the membrane was probed with a horseradish peroxidase-conjugated secondary antibody, and the chemiluminescence upon addition of the substrate was recorded.
cAMP Assay
cAMP dynamic2 assay kit was purchased from CisBio. HEK293 cells (ATCC) were maintained in Dulbecco's modified Eagle's medium (Invitrogen) with 10% (v/v) fetal calf serum (FCS) at 37 °C and 5% CO2. For cAMP assay, 800,000 cells were plated in a 6-well plate and transiently transfected with pcDNA3.1(+) control, WT Gαi1, sNUCB1, or CXCR4 expression constructs using Lipofectamine 2000 (Invitrogen). Briefly, 4.0 μg of total DNA (3.0 μg of pcDNA3.1(+) + 1.0 μg of WT Gαi1/sNUCB1/CXCR4) was complexed with 12 μl of Lipofectamine 2000 reagent and incubated for 20 min at room temperature before addition of the DNA/lipid mixture to the cells. Cells were plated 24 h after transfection at a density of 8000 cells/well in a poly-d-lysine-coated 384-well plate. 48 h after transfection, cells were left untreated or stimulated with forskolin alone or forskolin (10 μm) + SDF1α in media containing 100 μm inhibitor of phosphodiesterase, namely isobutylmethylxanthine (3,7-dihydro-1-methyl-3-(2-methylpropyl)-1H-purine-2,6-dione), for 15 min at 37 °C and 45 min at room temperature. After an hour of stimulation, lysis buffer alone was added to the untreated cells and lysis buffer with cAMP-d2 was added to the stimulated cells. Thereafter, cryptate-conjugated cAMP antibody was added to all cells and incubated for 1–2 h. Homogeneous time-resolved fluorescence was recorded with excitation at 320 nm and emission at 620 and 665 nm. A standard curve was prepared by varying the amount of cAMP-d2 and probing it with cryptate-conjugated cAMP antibody using homogeneous time-resolved fluorescence. The normalized signal from treated cells with respect to untreated cells was converted to the cAMP concentration using the standard curve.
RESULTS
Ca2+ Binding to EF Hand Domains of sNUCB1 Induces Conformational Changes
A soluble form of NUCB1 was engineered, expressed in Escherichia coli, and purified to homogeneity (Fig. 1A and supplemental Fig. S1A). The domain architecture of sNUCB1 shows two EF hand Ca2+-binding regions sandwiched between an N-terminal DNA binding domain and a C-terminal leucine zipper (20). The thermodynamics of Ca2+ binding to sNUCB1 were studied using ITC. The Ca2+ binding was exothermic with ΔH1o = −2.1 ± 0.06 kcal mol−1 and ΔS1o = 16.5 cal mol−1 K−1 for the first EF hand and ΔH2o = −0.62 ± 0.31 kcal mol−1 and ΔS2o = 17.1 cal mol−1 K−1 for Ca2+ binding to the second EF hand, suggesting a spontaneous uptake of Ca2+ by Ca2+-free sNUCB1. The recorded data were best fit to a “two-set binding sites” model giving Kd values of 6.3 and 73.5 μm for Ca2+ binding (Fig. 1B). The difference in binding affinities arises due to the presence of a noncanonical Arg residue substituting for the canonical Gly at the sixth position of the 12-residue Ca2+ binding loop of the second EF hand. Our results show that the two EF hands in sNUCB1 bind to Ca2+ differentially with an order of magnitude difference in their Kd values. In 2004, de Alba and Tjandra (36) heterologously expressed and purified only the EF hand domain (residues 228–326) of NUCB1 and also reported two Ca2+-binding sites with different affinities. The EF hand regions undergo conformational changes upon Ca2+ binding that can be monitored through change in sNUCB1 Trp fluorescence. sNUCB1 has two Trp residues at positions 232 and 333 lying sequentially before and after the EF hand domain, respectively. Steady-state fluorescence measurements show a blue shift of 15 nm and an enhancement in fluorescence intensity as more Ca2+ was added to Ca2+-free sNUCB1 (supplemental Fig. S1B). Thus, Ca2+ binding causes a conformational change in sNUCB1 causing reorientation of the Trp residues from a solvent-exposed polar to a hydrophobic environment. Fluorescence quenching experiments with acrylamide and iodide were performed to probe the conformational changes around the Trp residues upon Ca2+ binding. Acrylamide is a diffusible neutral quencher of Trp fluorescence unaffected by the charges surrounding the fluorophore. I−, on the other hand, is an anionic quencher for surface-exposed fluorophores only (37). Quenching experiments show that Trp residues are more accessible to acrylamide than I− for quenching. On binding to Ca2+, acrylamide quenching decreases much more than I− quenching (supplemental Fig. S1C). F0/F versus quencher concentration gave a straight line indicative of a dynamic quenching rather than static quenching. These observations suggest that conformational changes associated with Ca2+ binding make Trp residues less accessible by reorienting them into a hydrophobic environment.
The binding of Ca2+ to an EF hand motif forms an octahedral coordination sphere, which can cause structural rearrangement in the protein (38). This secondary structural change can in principle be followed by CD spectroscopy. Ca2+ binding to sNUCB1 causes only a slight increase in helicity. Intriguingly, the far-UV CD spectra of sNUCB1 show a smooth structural transition from an α-helical secondary structure at lower concentrations to a predominant β-sheet structure at higher concentrations. These concentration-dependent structural changes were observed both in the absence and presence of Ca2+ (supplemental Fig. S2A). Furthermore, the thermal unfolding data for sNUCB1 showed an increase in apparent Tm of 4 °C on binding to Ca2+. The folding of sNUCB1 was observed to be noncooperative and irreversible both in the absence and presence of Ca2+ (supplemental Fig. S2B). The increase in stabilization on binding to Ca2+ suggests a structural ordering of sNUCB1.
NUCB1 Is a Dimer
The domain diagram of NUCB1 reveals the presence of a C-terminal domain leucine zipper, which is a canonical structural motif known to cause dimerization (39). The oligomeric state of NUCB1 was evaluated through sedimentation equilibrium experiments using AUC. This experiment is insensitive to the shape of the molecule and directly reports on the molar mass of the sedimenting species characterized by the Boltzmann distribution. Sedimentation equilibrium measurements were performed with Ca2+-free sNUCB1 for concentrations varying from 50 to 150 μm. The data could be well fit to a monomer-n-mer model, which gave a molecular mass of ∼100 kDa corresponding to a dimer of sNUCB1 in solution (Fig. 2A). AUC analysis of Ca2+-bound sNUCB1 indicated that it is also primarily dimeric in solution (Fig. 2A, inset). To confirm that the dimerization of sNUCB1 is caused by the leucine zipper domain, a C-terminal truncation mutant, sNUCB1(W333Ter), lacking the leucine zipper was designed and heterologously expressed. The far-UV CD spectrum of sNUCB1(W333Ter) showed an α-helical secondary structure even at high protein concentrations in contrast to the concentration-dependent transition observed for sNUCB1 (supplemental Fig. S2C). AUC analysis of sNUCB1(W333Ter) revealed a monomeric species in solution over the concentration range of 32 to 150 μm (Fig. 2B). Similar to sNUCB1, the association state of sNUCB1(W333Ter) was unaffected upon binding to Ca2+ (data not shown). The experiments with the truncation mutant confirm that the leucine zipper domain of sNUCB1 is essential for the dimeric state of the protein in solution.
FIGURE 2.

sNUCB1 forms a stable dimer mediated by its leucine zipper domain. A, we carried out AUC experiments to determine the oligomeric state of sNUCB1 in solution. The sedimentation equilibrium plot for Ca2+-free sNUCB1 (50 μm monomeric concentration) shows the evolution of a sample concentration curve resulting from the applied centrifugal force. The data were fit to a monomer-n-mer equilibrium model, which gave a measured molecular mass of 98.9 ± 0.41 kDa, consistent with a dimeric structure for sNUCB1 in solution (the theoretical molecular mass of monomeric sNUCB1 is ∼51 kDa). The inset shows the sedimentation equilibrium plot for Ca2+-bound sNUCB1, which gave essentially an identical molecular mass of 99.4 ± 0.78 kDa. B, AUC experiments were carried out on the truncation mutant, sNUCB1(W333Ter), which lacked the leucine zipper domain in the C-terminal region of the protein. The AUC sedimentation equilibrium plot for Ca2+-free sNUCB1(W333Ter) (70 μm monomeric concentration) gave a molecular mass of 35.2 ± 0.05 kDa, consistent with a monomeric species in solution (the theoretical molecular mass of sNUCB1(W333Ter) is ∼36.8 kDa). C, we studied the monomer-dimer equilibrium of sNUCB1 with MALS of fractions with increasing protein concentration collected from Superdex200 10/30 HR size-exclusion chromatography in 50 mm Tris-HCl, pH 8.0, 150 mm NaCl. The weight average molecular weight of the complex at a given protein concentration was determined from a nonlinear least square fit of a collection of values determined for the apex fractions of each eluting peak. A monomer-dimer association model of the values as a function of sNUCB1 concentration gave an apparent dissociation constant (Kd) for dimerization of 0.26 ± 0.12 μm. The error bars indicate the extent of variation in molecular mass determination originating from the light scattering measurement. We next measured DLS chromatograms for sNUCB1 (D) and sNUCB1(W333Ter) (E). Each protein sample was injected onto a Superdex200 10/30 HR column, and the refractive index detector was used to analyze the protein peak. The change in refractive index as a function of protein concentration was used to compute the molecular mass as shown in green for sNUCB1 and red for sNUCB1(W333Ter). The molecular mass corresponds to a dimer for sNUCB1 and a monomer for sNUCB1(W333Ter). The insets show the correlation functions as each protein diffuses through the solution in the DLS experiment. The diffusion coefficient from the correlation function gives the hydrodynamic radius (Rh) using Stokes equation. The computed Rh values were 6.2 and 3.03 nm for sNUCB1 and sNUCB1(W333Ter), respectively.
The thermodynamics of the dimerization of sNUCB1 was further investigated using MALS. In a series of SEC experiments, Ca2+-free sNUCB1 was analyzed by gel filtration using a Superdex200 10/30 HR column. The eluted protein peak was fractionated and subjected to MALS analysis at seven different angles to estimate the molecular mass of the eluting species. As shown in Fig. 2C, a gradual increase in the weight-average molecular weight of the eluting species was observed with increasing concentrations of the protein. At concentrations around 10 nm, sNUCB1 exists as a monomer. As the concentration is gradually increased, a monomer-dimer equilibrium was observed with dimeric sNUCB1 dominating at concentrations greater than 1 μm. The data collected at different angles for each run were fit to a monomer-dimer equilibrium model giving a dissociation constant, Kd of dimerization, for sNUCB1 of 0.26 ± 0.12 μm. sNUCB1(W333Ter) in MALS measurements continued to exist as a monomer even at high protein concentrations. Thus, the leucine zipper region is responsible for the dimeric state of sNUCB1 in solution under physiological conditions.
sNUCB1 Is Structurally Asymmetric with an Elongated C Terminus
Ca2+-free sNUCB1 in SEC experiments elutes at a much higher volume than a globular protein with a molecular mass similar to that of a sNUCB1 monomer or a dimer (supplemental Fig. S3A). A plot of the elution parameter, Kav, versus log(molecular mass) for a number of globular protein standards was utilized to estimate an apparent molecular mass of 300 kDa for sNUCB1 (supplemental Fig. S3B). This implies that either sNUCB1 exists as a trimer of dimers (3× 100 kDa) or is asymmetric in its shape. Because AUC and MALS analysis convincingly ruled out any association of sNUCB1 dimers into higher order structures, we proceeded to derive structural information on sNUCB1 through DLS. DLS analysis of Ca2+-free sNUCB1 yields a hydrodynamic radius (Rh) of 6.2 nm and a frictional coefficient (f/f0) of 2.03 (Fig. 2D). The frictional coefficient of globular proteins usually lies in the range of 1.1–1.25 (40). A value of 2.03 for sNUCB1 indicates an asymmetric shape for the dimer, deviating considerably from a sphere. Interestingly, Ca2+-free sNUCB1(W333Ter) has an Rh of 3.3 nm and a f/f0 of 1.59 showing that the deletion of the C-terminal domain reduces the radius of hydration by almost 50% (Fig. 2E). This suggests that although the N terminus of sNUCB1 is relatively more compact and globular, the C-terminal domain of the protein is elongated, contributing to the observed asymmetry in the shape of sNUCB1.
sNUCB1 Preferentially Interacts with GDP-bound WT Gαi1 in the Absence of Ca2+
NUCB1 is a Ca2+-binding protein that has been shown to interact with Gi and Gs classes of α subunits (16). Physiologically, Gα subunits can individually exist both in the “inactive” or GDP-bound state and the “active” or GTP-bound state. To investigate the interaction of sNUCB1 with WT Gαi1, sNUCB1 was incubated with WT Gαi1·GDP (Fig. 3A) and WT Gαi1·GTPγS (Fig. 3B), respectively, and the complex formation was analyzed by SEC using a Superdex200 10/30 HR column. As shown in the top panels of Fig. 3, the Coomassie-stained gels of the fractionated peaks show elution of the complex containing both sNUCB1 and Gαi1 with excess Gαi1 fractionating at higher elution volume. The amount of Gαi1 eluting with sNUCB1 was significantly higher when Gαi1 was present in the GDP-bound state rather than in the GTPγS-bound state. Thus, Ca2+-free sNUCB1 preferentially interacts with Gαi1 in the GDP-bound form. Interestingly, the interaction with Gαi1 in both the GDP- and GTPγS-bound states is absent when sNUCB1 is bound to Ca2+ as shown in the lower panels of Fig. 3. The two protein subunits elute out as individual components as the complex is disrupted in the presence of Ca2+. The inhibition of the complex formation between sNUCB1 and Gαi1 in the presence of Ca2+ can be explained by the structural rearrangement of EF hands upon Ca2+ binding. Yeast two-hybrid analysis for interaction of Gα with deletion mutants of NUCB1 as bait proposed the EF hand intervening acidic region of NUCB1 to be involved in G protein binding (16). The conformational change induced upon Ca2+-binding can possibly result in the masking of the acidic region sandwiched between the two EF hands, thereby making it inaccessible for G protein binding.
FIGURE 3.

sNUCB1 forms a stable complex with Gαi1·GDP only in the absence of Ca2+. We used size-exclusion chromatography to determine the nature of the interaction between sNUCB1 and Gαi1 in solution under several conditions. sNUCB1 and Gαi1·GDP were incubated together in the absence (upper left) or presence (lower left) of Ca2+, and the mixture was subjected to size-exclusion chromatography using a Superdex200 10/30 HR column. Peak fractions were analyzed by SDS-PAGE with Coomassie Brilliant Blue staining as shown in the insets. In the absence of Ca2+, sNUCB1 and Gαi1·GDP formed a complex as judged by the following: (i) the slight shift (from ∼11.2 to 10.6 ml) and enhancement of the peak containing only sNUCB1 in the presence of Ca2+ and both sNUCB1 and Gαi1 in the absence of Ca2+, and (ii) the depletion, in the absence of Ca2+, of the peak eluting at about 14.6 ml, which contained only Gαi1. The same experiment was repeated with sNUCB1 and Gαi1·GTPγS in the absence (upper right) or presence (lower right) of Ca2+. Although the same general trend and Ca2+ dependence were observed, the apparent ability of Gαi1·GTPγS to form a stable complex with sNUCB1 was markedly diminished compared with that of Gαi1·GDP.
The thermodynamics of Ca2+-free sNUCB1 complex formation with Gαi1·GDP was investigated using ITC. The exothermic nature of the reaction shows a thermodynamically favorable binding event between the two protein subunits with ΔH0 = −1.45 ± 0.17 kcal mol−1 and ΔS0 = 16.9 cal mol−1 K−1. The data were best fit to a “one-set binding sites” model with dimeric sNUCB1 and monomeric G protein as the interacting partners. The fit provides a Kd value of 18.3 ± 1.45 μm suggesting that sNUCB1 interacts relatively weakly with GDP-bound Gαi1 (Fig. 4A). The association between Ca2+-free sNUCB1 and Gαi1·GDP was also monitored using light scattering. Protein samples with increasing concentrations of sNUCB1-Gαi1·GDP complex were injected onto a Superdex200 10/30 HR column, and MALS was done on the complex elution peak. The analysis shows that binding of Gαi1·GDP to sNUCB1 initiates only after dimerization of Ca2+-free sNUCB1. At lower concentrations, dimerization of sNUCB1 from molecular mass of 50–00 kDa was traced. Once the dimer was formed, binding of one Gαi1·GDP subunit to sNUCB1 was evident from an increase in molecular mass to 140 kDa (Fig. 4B). The trace observed for sNUCB1-Gαi1·GDP complex formation can be understood from the experimental Kd values. The Kd value for sNUCB1 dimerization is much smaller than the Kd value for binding to Gαi1·GDP, resulting in sNUCB1 dimerization prior to Gαi1·GDP binding. However, sNUCB1-Gαi1·GDP association curve does not rule out the ability of sNUCB1 monomer to interact with Gαi1·GDP.
FIGURE 4.

Interaction of sNUCB1 and Gαi1·GDP. A, we used ITC to measure the binding of Ca2+-free sNUCB1 to Gαi1·GDP. The heat released per injection of aliquots of a solution of Gαi1·GDP (200 μm) into a buffered solution of Ca2+-free sNUCB1 (50 μm) was recorded, and the area under the curve was integrated. The heat of dilution for the addition of Gαi1·GDP to buffer alone was subtracted. A nonlinear least squares fit of the calculated values using the one-set of sites model for dimeric sNUCB1 resulted in a satisfactory fit with a dissociation constant of 18.3 ± 1.45 μm as shown in the inset. We used MALS to measure the formation of a complex between sNUCB1 and Gαi1·GDP (B) or the truncation mutant sNUCB1(W333Ter) and Gαi1·GDP (C). MALS data were collected on protein complex peaks eluting from Superdex200 10/30 HR size-exclusion chromatography connected to a high performance liquid chromatography system equipped with an autosampler. Samples with increasing protein concentrations were successively injected onto the column, and the eluate was monitored using a photo-diode UV-visible detector, a differential refractometer, and a static multiangle laser light scattering detector. The weight average molecular weight of the complex was determined as a function of increasing protein concentrations. The magenta curve in B, inset, and the red curve in C indicate nonlinear least square fits for a complex formation model of weight average molecular weight values determined for the apex of each eluting peak at various concentrations. The error bars indicate extent of variation in molecular mass determination originating from the light-scattering measurement. The data for sNUCB1-Gαi1·GDP complex formation shows that association between the protein subunits occurs only after dimerization of sNUCB1 (blue curve). The data suggest that one Gαi1 subunit binds to a dimer of sNUCB1 and a monomer of sNUCB1(W333Ter), respectively, and that the binding site for Gαi1 lies in the sNUCB1(W333Ter) sequence.
MALS experiments were also conducted with the monomeric truncation mutant sNUCB1(W333Ter). A plot of the weight average molecular weight obtained from light scattering plotted versus the protein concentration shows that monomeric Ca2+-free sNUCB1(W333Ter) forms a 1:1 complex with Gαi1·GDP. The fit yields a Kd value of 1.99 ± 0.26 μm showing that the interaction of sNUCB1 with WT Gαi1·GDP becomes stronger on deletion of the C-terminal region of sNUCB1 (Fig. 4C). This suggests that the C-terminal region of sNUCB1 possibly acts as a regulatory domain modulating the interaction of sNUCB1 with the G protein α subunit. Alternatively, the structural rearrangement associated with dimerization can also result in the reduced affinity of sNUCB1 for Gαi1 in comparison with sNUCB1(W333Ter).
sNUCB1 Is a GDI
GPCRs activate heterotrimeric G proteins by catalyzing the exchange of GTP for GDP on Gα subunits. The receptor facilitates the nucleotide exchange by using the C-terminally linked α5 helix of Gα subunits. The α5 helix region is postulated to be a molecular microdomain on Gα that allosterically couples the activation signal from the receptor to the catalytic pocket of G proteins (29, 30, 41, 42). Farquhar and co-workers (28) have shown that NUCB1 possibly interacts with the C-terminal α5 helix region of Gαi3. We evaluated the effect of this interaction on G protein activation. We measured the nucleotide exchange on Gα in the presence of Ca2+-free sNUCB1. The concentrations of both sNUCB1 and Gαi1 were kept above the determined Kd value for sNUCB1-Gαi1 complex formation. Steady-state and time-based fluorescence experiments were performed by monitoring the enhancement in Trp fluorescence, which is coupled to activation of Gαi1. Any potential contribution from the Trp residues of sNUCB1 was eliminated by mutating both Trp-232 and Trp-333 to Ala, thereby generating sNUCB1(W232A/W333A) (supplemental Fig. S4A). The binding of sNUCB1(W232A/W333A) to Gαi1·GDP was confirmed using SEC and SDS-PAGE analysis. The interaction of sNUCB1(W232A/W333A) with Gαi1·GDP was present only in the absence of Ca2+ (supplemental Fig. S4B). Both AlF4− uptake (supplemental Fig. S4C) and GTPγS exchange (supplemental Fig. S4D) by Gαi1·GDP alone shows an increase in Trp fluorescence as Gαi1 molecules were activated with time. However, supplemental Fig. S4, C and D, shows that binding to sNUCB1(W232A/W333A) reduces the relative enhancement of Trp fluorescence coupled to Gαi1 activation. Furthermore, to simultaneously monitor the activation-associated conformational change in Gαi1 and the uptake of nucleotide, a mGTPγS-based FRET assay was performed. The binding of mGTPγS in the catalytic pocket of Gαi1 brings it in close vicinity of Trp-211 on the switch II region of Gαi1. Hence, FRET can be observed between the Trp and mant fluorophores upon Gαi1 activation (43). As shown in supplemental Fig. S4E, the FRET intensity for Gαi1 alone at each time point is significantly higher in comparison with the FRET intensity for Gαi1 associated with Ca2+-free sNUCB1(W232A/W333A). These experiments suggest that sNUCB1(W232A/W333A) might inhibit nucleotide exchange by Gαi1.
G protein activation was also monitored using a fluorescent nucleotide analogue BODIPY FL-GTPγS, which absorbs at 504 nm on binding to Gαi1. We isolated the BODIPY FL-GTPγS-bound fraction of Gαi1 in the absence and presence of sNUCB1 and calculated the amount of bound BODIPY FL-GTPγS in each case (Fig. 5). The supplemental Fig. S5 shows that binding of sNUCB1 to Gαi1 does not affect the λmax of bound BODIPY FL-GTPγS. However, binding of sNUCB1 to Gαi1 reduces the amount of nucleotide exchange by Gαi1 as seen by a decrease in the BODIPY absorbance. Thus, measuring absorbance of bound BODIPY FL-GTPγS provides a direct monitor of the number of bound nucleotides on Gαi1 in the presence or absence of sNUCB1. Fig. 5A shows BODIPY FL-GTPγS nucleotide exchange by Gαi1 alone (blue) and by Gαi1 in complex with sNUCB1 (orange). The results show a significant reduction in the number of bound BODIPY FL-GTPγS nucleotides to Gαi1 when complexed with Ca2+-free sNUCB1. This shows that binding of sNUCB1 to Gαi1 causes inhibition of nucleotide exchange.
FIGURE 5.

Evidence that sNUCB1 acts as a GDI of Gαi1. A, we estimated the number of available nucleotide-binding sites on Gαi1 in the absence or presence of Ca2+-free sNUCB1 by monitoring the absorbance of Gαi1-bound BODIPY FL-GTPγS at 504 nm. Gαi1·GDP alone (20 μm, black) or in complex with Ca2+-free sNUCB1 (100 μm, red) was allowed to undergo nucleotide exchange with 100 μm BODIPY FL-GTPγS in the reaction mixture. Samples were withdrawn at different time points, and unbound BODIPY FL-GTPγS was removed using a micro-spin desalting column. The amount of bound BODIPY FL-GTPγS was measured using the UV-visible absorbance spectroscopy at each time point. A relative decrease in the number of nucleotide-binding sites on Gαi1 was observed on association with sNUCB1. This shows that Ca2+-free sNUCB1 binds to Gαi1 in its GDP-bound form and inhibits nucleotide exchange. We further investigated the guanine nucleotide inhibitory activity of Ca2+-free sNUCB1 using the radioligand-based binding assay (B). WT Gαi1·GDP (20 μm) was incubated alone (blue) or with 100 μm Ca2+-free sNUCB1 (green)/sNUCB1(W333Ter) (red) in the presence of [35S]GTPγS, and samples were withdrawn at various time points to estimate the amount of radioligand bound upon nucleotide exchange. As a positive control, 100 μm of a C-terminal GoLoco motif peptide from RGS14, namely RGS14(496–531), with established GDI activity was used as a positive control (magenta). The data show that both Ca2+-free sNUCB1 or Ca2+-free sNUCB1(W333Ter) considerably inhibits nucleotide exchange with similar potency. The GDI peptide RGS14(496–531) shows complete inhibition of nucleotide exchange. Each experiment was repeated at least three times, and the data were plotted as mean ± S.E. among the independent measurements.
To further substantiate the effect of sNUCB1 on nucleotide exchange by WT Gαi1, we monitored the exchange of radioligand [35S]GTPγS by Gαi1 in the presence and absence of sNUCB1. Fig. 5B shows the kinetics of exchange of GDP for [35S]GTPγS by Gαi1 alone (blue). However, in the presence of sNUCB1 (Fig. 5B, green), the rate and extent of [35S]GTPγS exchange are significantly reduced. Thus, sNUCB1 on binding to Gαi1 influences the nucleotide exchange rate by substantially inhibiting GDP release and subsequently the GTP uptake.
GDI Activity of sNUCB1 Does Not Arise from a Canonical GoLoco Motif
Earlier work by Siderovski and co-workers (9, 14, 44–46) has unraveled several interacting partners of Gαi1·GDP, namely RGS12, RGS14, AGS3, GPSM2, PCP-2, and G18, all of which possess GDI activity. The GDI activity was attributed to the presence of a single or tandem repeats of a 19-amino acid-long conserved GoLoco motif present in each of these proteins. The crystal structure of Gαi1·GDP complexed to RGS14-derived GoLoco peptide showed that the conserved C terminus of the 19-amino acid GoLoco motif makes contacts with both the switch I region and the bound nucleotide. Each GoLoco motif ends in a conserved triad of (Asp/Glu)-Gln-Arg. On binding to Gα, the Arg side chain extends into the catalytic pocket to interact directly with the bound GDP and stabilize it. In addition, the highly conserved Asp/Glu and Gln residues are absolutely essential for binding to G protein α subunit as they are involved in both intra- and intermolecular H-bonding interactions (47). A sequence alignment of C-terminal residues of NUCB1(381–399) with GoLoco motifs of several GDIs of Gαi subunits reveals that NUCB1 also possesses the highly conserved triad of Glu-Gln-Arg and the Gln conserved at the 11th position of the GoLoco motifs (supplemental Fig. S6, A and B). The other residues in the aligned sequence of NUCB1 show relatively poor conservation across the GoLoco motif.
We synthesized the NUCB1 peptide corresponding to residue 381–419, namely NUCB1(381–419) (supplemental Fig. S6A). We first performed an ITC experiment to evaluate the interaction of the NUCB1(381–419) with Gαi1. The supplemental Fig. S6C shows that NUCB1(381–419) does not interact with Gαi1, suggesting that this C-terminal region of NUCB1 alone is not responsible for the observed GDI activity of sNUCB1. To identify further the region of sNUCB1 responsible for its GDI activity, we tested the effect of C-terminal truncation mutant sNUCB1(W333Ter) on the nucleotide exchange by Gαi1 using [35S]GTPγS exchange assay. Fig. 5B illustrates the ability of sNUCB1(W333Ter) to inhibit nucleotide exchange on Gαi1 in a manner similar to sNUCB1. This suggests that the GDI activity of sNUCB1 lies in the N-terminal part of the protein containing the two EF hands. Thus, the GDI activity of sNUCB1 may not depend on a peptide sequence that is similar to a canonical GoLoco sequence.
As a positive control, we also monitored the [35S]GTPγS uptake by Gαi1 in the presence of RGS14 GoLoco peptide RGS14(496–531). RGS14(496–531) completely inhibits the nucleotide exchange by Gαi1 (Fig. 5B) as reported by Siderovski et al. (54). Furthermore, using ITC, we tested the binding of RGS14(496–531) to Gαi1 both in the absence and presence of sNUCB1. As shown in supplemental Fig. S7A, RGS14(496–531) (200 μm) binds to Gαi1 (20 μm) alone with an affinity of 268.8 nm. However, when Gαi1 was complexed to sNUCB1, RGS14(496–531) did not show any binding to Gαi1 (supplemental Fig. S7B) even at a concentration of 500 μm. Similar results were obtained when Gαi1 was complexed to sNUCB1(W333Ter) (supplemental Fig. S7C). RGS14(496–531) has been postulated to compete off Gβγ for Gαi binding (47). In our experiments, RGS14(496–531) was not able to bind to Gαi1 in the presence of sNUCB1 suggesting that both RGS14(496–531) and sNUCB1 may bind to the same site on Gαi1. These results demonstrate that the GDI activity of sNUCB1 does not originate from the conventional GoLoco motif but that it competes for the same general binding site on Gαi1.
Physiological Relevance of GDI Activity of sNUCB1
To test the physiological relevance of the observed GDI activity of sNUCB1, we expressed the human WT Gαi1 in the HEK293 cells in the absence and presence of sNUCB1. We first measured forskolin-stimulated intracellular cAMP levels for each transfection. Forskolin directly activates adenylyl cyclase (AC), which converts adenosine monophosphate to cAMP. Thus the amount of cAMP production can evaluate the ability of Gαs or Gαi to stimulate or inhibit cellular AC activity. We used the cAMP assay to measure the effect of GDI activity of sNUCB1 on Gαi signaling (Fig. 6, inset). Intriguingly, the results show that sNUCB1 decreases Gαi-mediated AC inhibition after forskolin stimulation.
FIGURE 6.

sNUCB1 overexpression decreases apparent Gαi1-dependent inhibition of AC. The physiological role of interaction of sNUCB1 with Gαi1 was investigated by monitoring its effect on cellular cAMP production. We transfected HEK293 cells with CXCR4 alone (blue) or with sNUCB1 and CXCR4 (red) cDNA and monitored cAMP production upon ligand (SDF1α)-mediated receptor activation. To increase basal cAMP levels through AC activation at each ligand concentration, cells were stimulated with 10 μm forskolin. For CXCR4 alone (blue), we observe significant inhibition of cAMP production upon stimulation with increasing concentrations of receptor agonist ligand. However, upon co-expression of sNUCB1 with CXCR4, ligand-dependent receptor-mediated inhibition of cAMP production is markedly reduced (red). In the inset, we transfected HEK293 cells with mock (yellow), Gαi1 alone (orange), and with sNUCB1 and Gαi1 (green) cDNA, respectively, and then measured cAMP production. The bar in yellow shows the concentration of cAMP produced with mock transfection, and the bar in orange shows cAMP production with overexpression of Gαi1 alone, and the bar in green shows cAMP production when sNUCB1 was transfected with Gαi1. The expression of sNUCB1 results in significant enhancement of cAMP production. The results suggest that sNUCB1 affects the ability of Gαi1 to promote forskolin-stimulated cAMP production by AC; *, p < 0.05.
We further evaluated the effect of sNUCB1 on GPCR-mediated signaling. We transfected HEK293 cells with CXCR4 and monitored cAMP production as a function of SDF-1α concentration. SDF-1α is an agonist ligand of CXCR4, which activates the receptor to facilitate nucleotide exchange on Gαi. CXCR4-transfected cells show a dose-response curve for cAMP production upon SDF-1α stimulation with an EC50 value of 1 nm. However, when sNUCB1 was overexpressed along with CXCR4 and cells were stimulated with SDF-1α, no dose-dependent inhibition was observed. The amount of cAMP produced does not diminish upon stimulation with increasing concentrations of SDF-1α (Fig. 6.).
DISCUSSION
NUCB1 is a ubiquitously expressed multidomain Ca2+-binding protein whose physiological role is not well understood. Our results provide the first direct evidence that sNUCB1 is a Ca2+-regulated GDI for Gαi1. SEC experiments showed that Ca2+-free sNUCB1 interacts primarily with the GDP-bound state of Gαi1 and that no interaction occurs when sNUCB1 is bound to Ca2+ (Fig. 3). The role of Ca2+ binding as a regulatory switch for the association of sNUCB1 with Gαi1 can be explained structurally through the involvement of the acidic region of sNUCB1. The acidic region has been previously shown to be a G protein interaction site (28). Thus binding of Ca2+ may cause structural rearrangement that can occlude the G protein interaction site on sNUCB1. In addition, yeast two-hybrid experiments have suggested that the C-terminally linked α5 helix of G protein interacts with NUCB1 (28). Interestingly, the α5 helix is conformationally identical in both the Gαi1·GDP and Gαi1·GTPγS structures. However, as Gα undergoes transition from the inactive to the active state, the structurally dynamic switch regions (switch I to IV) of Gα subunits change conformation (48). Thus, the preferential binding of Ca2+-free sNUCB1 to Gαi1·GDP, which can modulate G protein activation, points toward a possible participation of switch regions in this interaction.
Analysis of the dimer of sNUCB1 using SEC (supplemental Fig. S3) and dynamic light scattering (Fig. 2D) revealed that it is considerably asymmetric in its shape with a long axis of 12.4 nm. This elongated shape may facilitate simultaneous interaction of sNUCB1 with both the α5 helix and the switch regions of Gαi1·GDP. Interestingly, deletion of the leucine zipper domain along with the C-terminal region of sNUCB1 increased the binding affinity of the monomeric truncation mutant sNUCB1(W333Ter) for Gαi1·GDP and decreased the Rh to 3.3 nm. This suggests that the deleted C terminus of sNUCB1 contributes significantly to the shape asymmetry of sNUCB1 and may play a role in regulating sNUCB1-Gαi1·GDP interaction.
NUCB1 is a major Ca2+-binding protein of Golgi and assists in the uptake, storage, and release of Ca2+ from the Golgi apparatus (49). Optimal Golgi Ca2+ levels are required for anterograde (endoplasmic reticulum to Golgi), retrograde (Golgi to endoplasmic reticulum), and intra-Golgi transport (50). Thus, NUCB1 along with Cab45 (51), the only other known Ca2+-binding Golgi resident protein, may be involved in maintaining the essential Ca2+ levels and gradient from cis- to trans-Golgi. The Ca2+ concentration in cis-Golgi is around 0.3 mm under unstimulated conditions (50). Thus, the NUCB1 pool in cis-Golgi should largely be Ca2+-bound given the Kd values for Ca2+ binding. However, at cytosolic Ca2+ concentrations of 100–125 nm (52), NUCB1 should essentially be in a Ca2+-free state. Similar to Gαi1, NUCB1 also exists both in the cytosolic and membrane fractions (17), and this creates an opportunity for these two protein subunits to interact and perform physiologically relevant functions. Our ITC experiments along with results from MALS analysis establish the interaction of the Gαi1·GDP subunit with a dimer of Ca2+-free sNUCB1 (Fig. 4), which is a variant of NUCB1 that lacks the N-terminal signal sequence and represents the cytosolic pool of NUCB1.
Our nucleotide-exchange assays with GTPγS and its fluorescent analogues (supplemental Fig. S4 and supplemental Fig. 5) together show that Ca2+-free sNUCB1 significantly inhibits nucleotide exchange by Gαi1·GDP. The GDI activity of sNUCB1 was established by monitoring exchange of radioligand [35S]GTPγS. Our results clearly show that sNUCB1 significantly inhibits Gαi1 activation. Interestingly, this ability was unaffected upon deletion of the C-terminal domain of sNUCB1, showing that the GDI ability of sNUCB1 does not depend on a region of the proteins that resembles a GoLoco motif. Thus, the mechanism of GDI activity of sNUCB1 is likely to be different from the GoLoco motif peptides derived from RGS proteins. The RGS peptides generally have nanomolar binding affinity for Gαi·GDP in comparison with micromolar binding affinities observed for GPSM2-, PCP-2-, or G18-derived GoLoco motif peptides (44–46). Unlike these peptide-based studies, our in vitro assays report the binding affinity of 18.3 μm for a 99-kDa sNUCB1 dimer with Gαi1. Yeast two-hybrid analysis by Lin et al. (16) showed that NUCB1 preferentially interacts with Gαi1 and Gαi3 with higher affinity than with Gαi2 and Gαs. Further structural studies now underway should reveal the mechanism of GDI activity of sNUCB1 and its relevance to other classes of G protein subunits.
G proteins are widely expressed in different tissue types and are involved in various signal transduction pathways interacting with a number of binding partners. The interaction of NUCB1 with Gαi1 has been previous established, but no guanine nucleotide exchange factor or GDI activity was observed because the in vitro experimental concentrations used were below the Kd values of association. Our results show that sNUCB1 inhibits G protein activation and that Ca2+ binding regulates interaction of Gαi1 with sNUCB1. Furthermore, to investigate the physiological importance of this interaction, we performed tissue culture-based experiments. Our results show that upon expression in HEK293 cells, sNUCB1 significantly decreases Gαi-mediated AC inhibition. In addition, even receptor-mediated inhibition of AC was prevented by overexpression of sNUCB1. This can be explained through the possible sequestration of Gαi subunits by sNUCB1, thereby alleviating inhibition of AC activity. Together with the biophysical studies, this result suggests that sNUCB1 and possibly NUCB1 can affect and regulate downstream signaling events involving heterotrimeric G proteins. In 2009, Lin et al. (17) showed that NUCB1 affects the dynamic distribution of the Gα subunits to the plasma membrane. It is possible that the association of the two proteins due to colocalization on the secretory granules may facilitate inhibition of G protein activation, which would tend to promote delivery of the Gα protein subunit primarily in the GDP-bound state to the plasma membrane for heterotrimer formation with Gβγ. Thus the cytosolic pool of NUCB1 may exclusively regulate G protein activation, whereas the membrane-bound pool would regulate both the activation and trafficking to facilitate receptor association upon heterotrimer formation.
In addition, NUCB1 also has a nuclear localization signal sequence within the DNA binding region. Recently, its role in affecting the biogenesis of amyloid precursor protein was reported (27). Such a potential for diverse functionality makes the comprehensive understanding of the physiological role of NUCB1 difficult. Based on our current knowledge, NUCB1 seems to be neither an RGS nor an AGS protein. Future experiments, including structural studies, will unravel the functional relevance of the interaction of NUCB1 with other Gα subunits. Understanding the role of NUCB1 in the context of different Gα subunits will highlight the signaling pathways modulated by NUCB1.
In summary, our results show that sNUCB1 is a physiological dimer that interacts with the GDP-bound form of Gαi1. The binding of sNUCB1 to Gαi1 inhibits GDP release and suggests that NUCB1 is a GDI. Ca2+ binding to sNUCB1 regulates sNUCB1-Gαi1·GDP complex formation and possibly modulates this interaction in different cellular compartments. We conclude that NUCB1 is a novel Ca2+-binding GDI. The elucidation of the Ca2+-regulated GDI activity of NUCB1 adds a new level of understanding to the role of NUCB1 and to the complexity of G protein-mediated signal regulation.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant GM078114. This work was also supported by Rockefeller/Sloan-Kettering/Weill Cornell Tri-Institutional Training Program in Chemical Biology.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7.
- GPCR
- G protein-coupled receptor
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- GDI
- guanine nucleotide dissociation inhibitor
- AC
- adenylyl cyclase
- GAP
- GTPase-accelerating protein
- DLS
- dynamic light scattering
- MALS
- multiangle light scattering
- SEC
- size-exclusion chromatography
- AUC
- analytical ultracentrifugation
- mGTPγS
- mant(2′,3′-O-(N-methyl-anthraniloyl)-GTPγS
- ITC
- isothermal titration calorimetry
- RI
- refractive index
- LS
- light scattering.
REFERENCES
- 1.Oldham W. M., Hamm H. E. (2008) Nat. Rev. Mol. Cell Biol. 9, 60–71 [DOI] [PubMed] [Google Scholar]
- 2.Sakmar T. P., Menon S. T., Marin E. P., Awad E. S. (2002) Annu. Rev. Biophys. Biomol. Struct. 31, 443–484 [DOI] [PubMed] [Google Scholar]
- 3.Gilman A. G. (1987) Annu. Rev. Biochem. 56, 615–649 [DOI] [PubMed] [Google Scholar]
- 4.Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 5.McCudden C. R., Hains M. D., Kimple R. J., Siderovski D. P., Willard F. S. (2005) Cell. Mol. Life Sci. 62, 551–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siderovski D. P., Willard F. S. (2005) Int. J. Biol. Sci. 1, 51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Vries L., Zheng B., Fischer T., Elenko E., Farquhar M. G. (2000) Annu. Rev. Pharmacol. Toxicol. 40, 235–271 [DOI] [PubMed] [Google Scholar]
- 8.Neubig R. R., Siderovski D. P. (2002) Nat. Rev. Drug Discov. 1, 187–197 [DOI] [PubMed] [Google Scholar]
- 9.Kimple R. J., De Vries L., Tronchère H., Behe C. I., Morris R. A., Gist Farquhar M., Siderovski D. P. (2001) J. Biol. Chem. 276, 29275–29281 [DOI] [PubMed] [Google Scholar]
- 10.Willard F. S., Kimple R. J., Siderovski D. P. (2004) Annu. Rev. Biochem. 73, 925–951 [DOI] [PubMed] [Google Scholar]
- 11.Takesono A., Cismowski M. J., Ribas C., Bernard M., Chung P., Hazard S., 3rd, Duzic E., Lanier S. M. (1999) J. Biol. Chem. 274, 33202–33205 [DOI] [PubMed] [Google Scholar]
- 12.Blumer J. B., Cismowski M. J., Sato M., Lanier S. M. (2005) Trends Pharmacol. Sci. 26, 470–476 [DOI] [PubMed] [Google Scholar]
- 13.Kimple R. J., Willard F. S., Siderovski D. P. (2002) Mol. Interv. 2, 88–100 [DOI] [PubMed] [Google Scholar]
- 14.De Vries L., Fischer T., Tronchère H., Brothers G. M., Strockbine B., Siderovski D. P., Farquhar M. G. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 14364–14369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mochizuki N., Hibi M., Kanai Y., Insel P. A. (1995) FEBS Lett. 373, 155–158 [DOI] [PubMed] [Google Scholar]
- 16.Lin P., Le-Niculescu H., Hofmeister R., McCaffery J. M., Jin M., Hennemann H., McQuistan T., De Vries L., Farquhar M. G. (1998) J. Cell Biol. 141, 1515–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin P., Fischer T., Lavoie C., Huang H., Farquhar M. G. (2009) Mol. Neurodegener. 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanai Y., Tanuma S. (1992) Immunol. Lett. 32, 43–48 [DOI] [PubMed] [Google Scholar]
- 19.Miura K., Titani K., Kurosawa Y., Kanai Y. (1992) Biochem. Biophys. Res. Commun. 187, 375–380 [DOI] [PubMed] [Google Scholar]
- 20.Miura K., Kurosawa Y., Kanai Y. (1994) Biochem. Biophys. Res. Commun. 199, 1388–1393 [DOI] [PubMed] [Google Scholar]
- 21.Kanuru M., Samuel J. J., Balivada L. M., Aradhyam G. K. (2009) FEBS J. 276, 2529–2546 [DOI] [PubMed] [Google Scholar]
- 22.Lavoie C., Meerloo T., Lin P., Farquhar M. G. (2002) Mol. Endocrinol. 16, 2462–2474 [DOI] [PubMed] [Google Scholar]
- 23.Wang S. N., Miyauchi M., Koshikawa N., Maruyama K., Kubota T., Miura K., Kurosawa Y., Awaya A., Kanai Y. (1994) Pathol. Int. 44, 844–849 [DOI] [PubMed] [Google Scholar]
- 24.Tsukumo Y., Tsukahara S., Saito S., Tsuruo T., Tomida A. (2009) J. Biol. Chem. 284, 27500–27510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leclerc P., Biarc J., St-Onge M., Gilbert C., Dussault A. A., Laflamme C., Pouliot M. (2008) PLoS One 3, e2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodeur J., Larkin H., Boucher R., Thériault C., St-Louis S. C., Gagnon H., Lavoie C. (2009) Traffic 10, 1098–1114 [DOI] [PubMed] [Google Scholar]
- 27.Lin P., Li F., Zhang Y. W., Huang H., Tong G., Farquhar M. G., Xu H. (2007) J. Neurochem. 100, 1505–1514 [DOI] [PubMed] [Google Scholar]
- 28.Lin P., Fischer T., Weiss T., Farquhar M. G. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 674–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapoor N., Menon S. T., Chauhan R., Sachdev P., Sakmar T. P. (2009) J. Mol. Biol. 393, 882–897 [DOI] [PubMed] [Google Scholar]
- 30.Marin E. P., Krishna A. G., Sakmar T. P. (2001) J. Biol. Chem. 276, 27400–27405 [DOI] [PubMed] [Google Scholar]
- 31.Marin E. P., Krishna A. G., Sakmar T. P. (2002) Biochemistry 41, 6988–6994 [DOI] [PubMed] [Google Scholar]
- 32.Folta-Stogniew E., Williams K. R. (1999) J. Biomol. Tech. 10, 51–63 [PMC free article] [PubMed] [Google Scholar]
- 33.Koppel D. E. (1972) J. Chem. Phys. 57, 4814 [Google Scholar]
- 34.Cummins P. G., Staples E. J. (1981) J. Physics E-Scientific Instruments 14, 1171–1177 [Google Scholar]
- 35.Vanholde K. E., Baldwin R. L. (1958) J. Phys. Chem. 62, 734–743 [Google Scholar]
- 36.de Alba E., Tjandra N. (2004) Biochemistry 43, 10039–10049 [DOI] [PubMed] [Google Scholar]
- 37.Phillips S. R., Wilson L. J., Borkman R. F. (1986) Curr. Eye Res. 5, 611–619 [DOI] [PubMed] [Google Scholar]
- 38.Cates M. S., Berry M. B., Ho E. L., Li Q., Potter J. D., Phillips G. N., Jr. (1999) Structure 7, 1269–1278 [DOI] [PubMed] [Google Scholar]
- 39.Landschulz W. H., Johnson P. F., McKnight S. L. (1988) Science 240, 1759–1764 [DOI] [PubMed] [Google Scholar]
- 40.Siegel L. M., Monty K. J. (1966) Biochim. Biophys. Acta 112, 346–362 [DOI] [PubMed] [Google Scholar]
- 41.Natochin M., Moussaif M., Artemyev N. O. (2001) J. Neurochem. 77, 202–210 [DOI] [PubMed] [Google Scholar]
- 42.Oldham W. M., Van Eps N., Preininger A. M., Hubbell W. L., Hamm H. E. (2006) Nat. Struct. Mol. Biol. 13, 772–777 [DOI] [PubMed] [Google Scholar]
- 43.Willard F. S., Kimple R. J., Kimple A. J., Johnston C. A., Siderovski D. P. (2004) Methods Enzymol. 389, 56–71 [DOI] [PubMed] [Google Scholar]
- 44.Willard F. S., McCudden C. R., Siderovski D. P. (2006) Cell. Signal. 18, 1226–1234 [DOI] [PubMed] [Google Scholar]
- 45.McCudden C. R., Willard F. S., Kimple R. J., Johnston C. A., Hains M. D., Jones M. B., Siderovski D. P. (2005) Biochim. Biophys. Acta 1745, 254–264 [DOI] [PubMed] [Google Scholar]
- 46.Kimple R. J., Willard F. S., Hains M. D., Jones M. B., Nweke G. K., Siderovski D. P. (2004) Biochem. J. 378, 801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimple R. J., Kimple M. E., Betts L., Sondek J., Siderovski D. P. (2002) Nature 416, 878–881 [DOI] [PubMed] [Google Scholar]
- 48.Mixon M. B., Lee E., Coleman D. E., Berghuis A. M., Gilman A. G., Sprang S. R. (1995) Science 270, 954–960 [DOI] [PubMed] [Google Scholar]
- 49.Lin P., Yao Y., Hofmeister R., Tsien R. Y., Farquhar M. G. (1999) J. Cell Biol. 145, 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pezzati R., Bossi M., Podini P., Meldolesi J., Grohovaz F. (1997) Mol. Biol. Cell 8, 1501–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scherer P. E., Lederkremer G. Z., Williams S., Fogliano M., Baldini G., Lodish H. F. (1996) J. Cell Biol. 133, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Usachev Y. M., Marchenko S. M., Sage S. O. (1995) J. Physiol. 489, 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.