

Abstract
Expression of aquaporin-1 (AQP1) and -2 (AQP2) channels in the kidney are critical for the maintenance of water homeostasis and the operation of the urinary concentrating mechanism. Hypertonic stress induced in inner medullary (IMCD3) cells by addition of NaCl to the medium substantially up-regulated the mRNA and protein expression of AQP1, suggesting that its activation occurs at a transcriptional and a translational levels. In contrast, no up-regulation of AQP1 was observed when these cells were exposed to the same tonicity by addition of urea. To explore the transcriptional activation of aqp1 under hypertonic stress, we examined the role of the transcription factor associated with hypertonicity, TonEBP. Treatment of IMCD3 cells with the TonEBP inhibitor rottlerin or silencing its expression with specific shRNA technology led to a substantial reduction in AQP1 expression under hypertonic conditions. Moreover, we defined a conserved TonEBP binding site located 811 bp upstream of the aqp1 exon that is essential for its expression. Single site-directed mutation of this TonE site led to a 54 ± 5% (p < 0.01) decrease in AQP1 luciferase-driven activity under hypertonic stress. TonEBP mutant mice display marked decrement in the expression of AQP1 in the inner medulla. In conclusion, these data demonstrate that TonEBP is necessary for the regulation of AQP1 expression in the inner medulla of the kidney under hypertonic conditions.
Keywords: Gene Transcription, Kidney, NFAT Transcription Factor, Transcription Factors, Water Channel, AQP1, Hypertonicity, Osmotic Response, TonEBP
Introduction
The cells that inhabit the hypertonic environment of the inner medulla possess a number of adaptive mechanisms that allow them to survive this harsh environment. This survival is mediated initially by the activation of ion transport systems and thereafter by the cellular accumulation of a number of compatible organic osmolytes (1–4).
The classical osmotic stress response involves the prompt transcription of several target genes by the tonicity enhancer binding protein (TonEBP), also known as NFAT5 (5, 6). TonEBP enhances the transcription of genes that are important in the early osmotic stress response. These genes include aldose reductase (akr1b1), sodium-myoinositol transporter 1 (smit1), betaine/GABA transporter (bgt1), taurine transporter (taut), and the neuropathy target esterase (nte), which are implicated in the cellular accumulation of the organic osmolytes sorbitol, myo-inositol, betaine, taurine, and glycerophosphocholine, respectively (5, 7–11). In addition, four more genes have been identified to be transcriptionally regulated by TonEBP: heat shock protein 70 (Hsp70) (12), urea transporter A (13), tumor necrosis factor α (TNFα) (6), and the water channel, aquaporin-2 (AQP2) (14).
We have previously described a substantial up-regulation in the expression of aquaporin-1 (AQP1) in the inner medulla of the kidney under hypertonic stress conditions (15–17). In this region of the kidney AQP1 as well as AQP2 are important for water reabsorption from the lumen of the descending thin limb and in the collecting ducts, respectively (18–20). The water reabsorption in these two segments of the nephron is critical for the operation of the urinary concentrating mechanism (21–24). Therefore, knowledge of the upstream and downstream regulation of their expression is of importance to understand the processes that regulate renal water excretion, but these processes remain undefined. Therefore, the focus of this study was to determine the role of TonEBP in the osmotic up-regulation of AQP1.
MATERIALS AND METHODS
Materials
Cell culture medium, fetal calf serum, and antibiotics were from Invitrogen. Rabbit polyclonal antibody to AQP1 was purchased from BD/Clontech (Mount View, CA), whereas antibody to β-actin was from Cell Signaling (Danvers, MA). Monoclonal antibody to AQP1 employed in rat kidney immunofluorescence was purchased from Abcam (Cambridge, MA). Polyclonal antibody to TonEBP was a kind gift from Dr. H. Moo Kwon (University of Maryland) (8). Secondary antibodies conjugated with horseradish peroxidase were from Cell Signaling and those conjugated with Alexa dyes were purchased from Molecular Probes (Eugene, Oregon). All chemicals were purchased from Sigma.
Cell Culture
The established murine inner medullary collecting duct cell line (IMCD3) originally developed by Rauchman (25) was previously provided by Dr. Steve Gullans (Boston, MA). Cell stocks were maintained as described previously (26). IMCD3 clones silenced for TonEBP and empty vector control cells were developed in our lab as described previously for other proteins (27). In experiments involving hypertonic stress, the media in culture dishes were exchanged for that with added NaCl, NaAc, ChCl, mannitol, sucrose, or urea to the specified osmolality depending on the experiment. Osmolality was determined with an Advanced Instruments Micro-Osmometer (model 3300, Norwood, MA).
Gene Arrays
The mouse gene array 430-2.0 from Affymetrix (Affymetrix Inc., Santa Clara, CA) was employed, which contains 39,000 transcripts. RNA transcription to cDNA, biotinilylation to cRNA, fragmentation, hybridization to the chip, and chip analysis using a HP Gene Array scanner were as per the manufacturer's recommendations and performed by the University of Colorado Gene Array Core. Gene array data were analyzed using both the Affymetrix Microarray Suite (version 5.0) and GeneSpring (Silicon Genetics) software analysis programs.
TonEBP Stable Silencing in IMCD3 Cells
TonEBP silencing in IMCD3 cells was performed as described previously (28) employing a plasmid-based system generating fold-back stem-loop structures that are processed into the specific siRNA was used (pSM2-TonEBP, V2MM_2869, Open Biosystems). The 22-nucleotide sequence employed for silencing TonEBP (5′-ACCTGTATCAGTGGGAATATAT-3′) aligns 100% with the nucleotides 2026–2047 of mouse TonEBP transcript variant a (NM_133957) and the nucleotides 1964–1985 of transcript variant b (NM_018823). BLAST and alignment analysis were performed to validate the selectivity of the silencing sequence as compared with other genes, thus precluding off-target responses.
Mouse Kidney Tissues
C57/B6 mice were obtained from Jackson ImmunoResearch Laboratories (Bar Harbor, ME). Mice were subjected to food and ad libitum water or water was withheld for 36 h. Mice were harvested by cervical dislocation; urine samples were collected from the bladder for osmolality analysis; kidneys were removed, and papilla and cortex tissues were dissected and snap-frozen in liquid nitrogen. Tissues were homogenized using a glass tissue grinder on ice with lysis buffer and analyzed as described previously (27). Defective mutant TonEBP mice lacking exons 6 and 7, which encode critical amino acid residues of its DNA binding domain (residues 254–380), were developed by Go et al. (29) and were a kind gift from Dr. H. Moo Kwon. Due to the high embryo and perinatal lethality in different models of defective TonEBP mice (29, 30), pups from heterozygous (tonebp +/Δ) intercrosses were sacrificed, kidney was harvested at day 2 after birth, and AQP1 expression was determined by Western blot and immunohistochemistry. Genotyping was performed as described previously from collected kidneys (29).
Immunohistochemistry from 4-Day-old TonEBP Mutant Mice
Methyl Carnoy's fixed kidneys were processed and paraffin-embedded, and AQP1 expression was detected in 3-μm sections by indirect immunoperoxidase staining with an AQP1 polyclonal antibody (BD/Clontech). Negative controls consisted of omission of the primary antibody or substitution with an irrelevant antibody.
RNA Extraction, Analysis, and Message Quantification
Cytosolic RNA was isolated from confluent cultures using the RNeasy kit (Qiagen, Valencia, CA). RNA integrity was assessed by capillary electrophoresis using an Agilent Bioanalyzer (model 2100, Foster City, CA (using the 28 S to 18 S rRNA ratio)). RNA was converted to cDNA using the Omniscript Reverse Transcriptase kit (Qiagen) as described by the manufacturer. Quantitative PCR primers specific to AQP1 and β-actin were designed using Beacon Designer (version 5.0) software (Premier Biosoft International, Palo Alto, CA). Quantitative PCR for AQP1 was performed using 5′-CTGCTGGCGATTGACTACACTG-3′ (forward primer) and 5′-GGTTTGAGAAGTTGCGGGTGAG-3′ (reverse primer) (70 nm each) using a SYBR green master mix (JumpStart® Taq Readymix®, Sigma) on a Bio-Rad I-Cycler. QPCR3 runs were analyzed by agarose gel electrophoresis, and melt curve to verify the correct amplicon was produced. β-Actin RNA was used as an internal control, and the amount of RNA was calculated by the comparative CT method as recommended by the manufacturer.
Protein Extraction and Western Blotting
Cell protein lysates were prepared from confluent cell cultures as described previously (27). Sample protein content was determined by BCA protein assay (Pierce). 75 μg of total protein was loaded per lane for SDS-PAGE (12.5% (w/v)) analysis and then transferred to PVDF membranes. Membranes were incubated with primary antibodies and visualized using a horseradish peroxidase secondary antibody (Cell Signaling) and the HRP Immunstar® detection kit (Bio-Rad). Chemiluminescence was recorded with an Image Station 440CF, and results were analyzed with the one-dimensional Image Software (Kodak Digital Science, Rochester, NY).
Confocal Fluorescence microscopy
IMCD3 cells expressing endogenous AQP1 were grown to confluency on eight-well glass slides (catalog no. 177402, NUNC, Thermo Fisher Scientific), fixed for 2 min at −20 °C with methanol, and permeabilized at room temperature with PBS containing 0.3% Triton X-100 before staining. Immunohistochemistry was performed as described (31), using Wistar rat (Harlan Laboratories) kidneys perfused with 4% paraformaldehyde in PBS. After being blocked with 10% goat serum in PBS, kidney slices were incubated overnight with primary antibodies as indicated in the text, and the next day rinsed and incubated with Alexa Fluor-conjugated secondary antibody (Molecular Probes) against the specific IgG of the primary antibody. For nucleus identification, DAPI staining was used in combination with a fluorescence fading retardant (Vector Laboratories, Burmingdale, CA) before imaging by confocal microscopy. Immunostained preparations were imaged and analyzed using a laser-scanning confocal microscope (LSM510, Carl Zeiss, Thornwood, NY) with a 40× water immersion objective and the corresponding postacquisition software. Immunofluorescence analysis was performed from sections from three animals and by evaluation of >10 random fields each.
Chromatin Immunoprecipitation (ChIP) Assay
IMCD3 cells were resuspended in PBS to 5 × 105 cells/ml. DNA/protein interaction was cross-linked at room temperature for 10 min with paraformaldehyde at a final concentration of 1%. The cross-linking reaction was quenched by addition of glycine, and cells were pelleted and resuspended in lysis buffer containing: 5 mm PIPES, pH 8.0, 85 mm KCl supplemented with 0.5% Nonidet P-40, and mixture protease inhibitor (Roche Applied Science). The nuclear extract was pelleted by microcentrifugation at 2,000 rpm for 5 min, and genomic DNA was precleared by addition of protein A/G-agarose slurry and further centrifugation. TonEBP-DNA complexes were pulled down by overnight incubation at 4 °C with an anti-TonEBP antibody and further addition of protein A/G-agarose slurry. Agarose beads were washed twice with PBS supplemented with 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS, followed by four washes in buffer containing: 100 mm Tris, pH 8.0, 500 mm LiCl supplemented with 1% Nonidet P-40 and 1% deoxycholate. Agarose beads were collected by centrifugation, and cross-linking was reversed by overnight incubation at 67 °C. DNA was then isolated from proteins with phenol/chloroform/isoamyl alcohol, and PCR analysis for the mouse AQP1 promoter sequence was performed during 30 cycles employing the following primers: forward, 5′-AATCCAGTGCCAGTTGAAT-3′ and reverse, 5′-CAGTGATGGGATGATAGGCA-3′. These primers generate a 130-bp amplicon product corresponding to nucleotides −881 to −750 of the mouse aqp1 promoter. PCR products were separated using a 2% agarose gel and visualized by ethidium bromide staining.
Luciferase Reporter Assay and Site-directed Mutagenesis
Three different luciferase reporter constructs were engineered from the first 1 kb 5′upstream of the first exon of mouse aqp1, which were cloned before the luciferase cassette of the pGL3-Luc vector (Promega); constructs included complete (−1,014/+208, pGL3-AQP1), ΔTonEBP (−776/+208, pGL3-AQP1ΔTonEBP), and ΔTonEBP-like (−162 + 208, pGL3-AQP1ΔTonEBP-like). These constructs thus evaluate the importance of TonEBP or TonEBP-like consensus sites in the AQP1 promoter region. Site-directed mutagenesis (Stratagene, La Jolla, CA) was employed following the manufacturer's protocol to modify the TonE site within the promoter of aqp1. (Nucleotides GGAA were substituted for TTCG.) The primers employed were (substitution is shown underlined): forward, 5′-CTGGCCGCTGTCTCCTGCAAGGCTTTCGACCTCTTCCACTTTC-3′ and reverse, 5′-GAAAGTGGAAGAGGTCGAAAGCCTTGCAGGAGACAGCGGCCAG-3′. The luciferase activity was measured in IMCD3 cells using the Luciferase Assay System (Promega, Madison, WI) according to the manufacturer's instructions and as described previously (32). Briefly, IMCD3 cells in six-well plates grown to 50% confluence were transiently transfected with each construct and a β-galactosidase reporter (used for normalizing the transfection efficiency). Replicates were incubated at isotonic conditions (300 mosmol/kg H2O) or with acute exposure to hypertonicity (550 mosmol/kg H2O) for 24 h. Cells were then lysed directly in luciferase reporter lysis buffer (Promega). Supernatants from cell lysates were mixed with luciferase substrate and measured immediately with a Luminoskan Ascent DCReady luminometer (Labsystems). All transfection experiments were performed in triplicate. Luciferase activity was normalized to β-galactosidase expression as described previously (33).
Statistics and Data Analysis
All data are presented as the means ± S.E. Data graphics and statistical analysis were performed using Instat (version 3.0) and Prism 4 (both from GraphPad Software, San Diego, CA). Data were analyzed for normality tests using the Tukey-Kramer multiple comparison test. Multiple group corrections were performed using the method of Bartlett. In most cases, experiments were performed three times with independent replicates. Total data points (n) for each experiment are identified in the corresponding figure legend. p values < 0.05 were recognized as statistically significant.
RESULTS
Enhanced Transcriptional Activity of AQP1 Gene in IMCD3 Cells Exposed to Hypertonicity
Gene expression in IMCD3 cells maintained at isotonic conditions (300 mosmol/kg H2O) or chronically adapted to different levels of tonicity (600 and 900 mosmol/kg H2O) was analyzed by Affymetrix GeneChip Technology. As shown in Fig. 1A, expression of AQP1 mRNA was increased substantially under chronic hypertonic conditions (10.3×, p < 0.01 and 11.2×, p < 0.01, for 600 and 900 mosmol/kg H2O, respectively). This data were further validated by QPCR employing specific primers for murine AQP1 obtaining similar results (15.8x and 31.7x, p < 0.01 for 600 and 900 mosmol/kg H2O, respectively). To assess whether this difference in mRNA resulted in an increase in protein expression, we performed Western blot analysis from protein lysates obtained from the same cell conditions. As shown in Fig. 1B, little or no AQP1 protein expression is found in cells maintained at isotonic conditions. In contrast, a substantial AQP1 expression was found when cells were chronically exposed to hypertonicity (10.4×, p < 0.01 and 12.1×, p < 0.01, for 600 and 900 mosmol/kg H2O, respectively).
FIGURE 1.

Enhanced transcriptional activity of AQP1 under hypertonic stress. IMCD3 cells were exposed to isotonic conditions (300 mosmol/kg H2O) or chronically adapted to 600 or 900 mosmol/kg H2O and harvested for mRNA isolation. A, message levels were analyzed by gene array (left) and quantitative PCR (right). For both methods, data show up-regulation in AQP1 mRNA expression under hypertonic conditions. B, protein expression of AQP1 in cells under the same conditions as above was analyzed by Western blot. A representative Western blot is shown and includes β-actin protein levels as a loading control. Data for mRNA and protein experiments were collected from three identical experimental replicates and represent the mean ± S.E. for two independent experiments (n = 6).
To determine the expression of AQP1 under acute hypertonic stress and to assess whether the effects observed with sodium chloride stress were unique to this solute, we exposed IMCD3 cells to 550 mosmol/kg H2O for 48 h by addition of inorganic (sodium chloride, sodium acetate, and choline chloride) and organic (sucrose, mannitol, and urea) mediators. As depicted in Fig. 2A, in addition of NaCl, other osmotically active solutes, including sucrose and mannitol, also caused a marked increase in protein expression but not with an ineffective solute such as urea. Also, replacement of chloride with acetate inhibited the response, suggesting that chloride rather than sodium is the major inorganic driver in the hypertonic response. To better define whether the effect of urea on AQP1 expression was due to the absence in transcriptional activity or the translation of message, we performed QPCR to determine the level of expression of AQP1 mRNA. As shown in Fig. 2B, AQP1 mRNA is not overexpressed with urea, reflecting that its expression is mainly regulated at the transcriptional level. As a control, the acute hyperosmotic urea-dependent mRNA expression of other genes involved in the osmotic stress response, including aldose reductase, BGT1, and SMIT1, did not change significantly as compared with cells maintained at isotonic conditions.
FIGURE 2.

Effect of different osmotic mediators on AQP1 expression. A, AQP1 protein was analyzed by Western blot employing different osmotic mediators to obtain a tonicity of 550 mosmol/kg H2O for 48 h. Western blot analysis reveals that urea does not up-regulate AQP1 protein expression. B, no transcriptional activation with urea was observed for either AQP1 or other osmotic response genes including AR, BGT1, and SMIT1. a, p < 0.01; NS, no significant difference as compared with isotonically maintained IMCD3 cells. Data for mRNA and protein experiments was collected from three identical experimental replicates and represents the mean ± S.E. for two independent experiments (n = 6).
Role of TonEBP in AQP1 Expression under Acute Hypertonic Conditions; Rottlerin Inhibits AQP1 Expression in IMCD3 Cells under Acute Hypertonic Stress Conditions
We have previously described the activation of TonEBP in IMCD3 cells under hypertonic conditions (28). As shown in Fig. 3A, this activation is characterized by increased protein expression (top panel) and nuclear translocation of this transcription factor (bottom panel). To determine whether AQP1 mRNA expression in IMCD3 cells is dependent on TonEBP activity, we initially employed a pharmacological inhibitor, rottlerin (34), in conjunction with exposing cells to acute hypertonic stress (500 mosmol/kg H2O. In this experiment, we pretreated cells with rottlerin (at a concentration of 0.1 or 10 μm) for 24 h to inhibit TonEBP activity followed by 48 h of hypertonic shock plus rottlerin to first determine the expression of the TonEBP target gene, AR (7). As depicted in Fig. 3B, the expression of AR is markedly down-regulated by rottlerin under hypertonic conditions as compared with nontreated cells (94 ± 3%, p < 0.001). To determine the effect of rottlerin on AQP1 expression, we employed the same conditions as for the detection of AR. As shown in Fig. 3C, AQP1 mRNA expression is substantially up-regulated by hypertonic stress in cells treated with the vehicle alone (DMSO). In contrast, no mRNA up-regulation was observed when cells were incubated with 10 μm rottlerin in the setting of hypertonic stress. These data were validated by a loss in AQP1 protein expression in a dose-dependent manner (Fig. 3C). To further examine the effect of the inhibitor rottlerin on AQP1 expression, cells were analyzed by confocal immunofluorescence. Data depicted in Fig. 3D demonstrates that antibodies specific for AQP1 identify the protein at the apical surface of IMCD3 cell monolayers exposed to an acute hypertonic stress (upper panel, vehicle). In contrast, when cells were preincubated with the inhibitor rottlerin, AQP1 protein was essentially absent at the apical membrane under similar conditions (lower panel).
FIGURE 3.

Rottlerin inhibits AQP1 up-regulation under hypertonic stress conditions. A, TonEBP is activated by hypertonic stress in IMCD3 cells. This activation is reflected by an increased expression (top panel) and nuclear translocation (bottom panel). B, rottlerin (10 μm) inhibits AR expression under hypertonic stress. Representative Western blot. C, AQP1 mRNA expression was analyzed by QPCR (left), whereas protein expression was analyzed by Western blot (right). Rottlerin inhibited both AQP1 mRNA and protein expressions under hypertonicity. D, AQP1 immunofluorescence (green) demonstrates luminal localization in control cells (top panel). This localization is abolished by 10 μm rottlerin (bottom panel). Data for mRNA and protein experiments were collected from three identical experimental replicates and represent the mean ± S.E. for two independent experiments (n = 6).
Silencing the Expression of TonEBP in IMCD3 Cells Leads to Diminished Up-regulation of AQP1 under Hypertonic Stress
Because the TonEBP inhibitor rottlerin dramatically reduced AQP1 expression under hypertonic conditions, we performed a more direct approach to the study of the transcriptional up-regulation of AQP1 by TonEBP under hypertonic stress conditions. To this end, we stably silenced the expression of TonEBP in IMCD3 cells by shRNA technology as described under “Materials and Methods.” Expression of TonEBP protein by Western blot in silenced clones and empty vector control cells was assessed following incubation under acute osmotic stress for 24 h. From a total of 25 clones analyzed, we chose three clones for further study (clones 1, 2, and 3) considered as silenced (clone 1), partially silenced (clone 2), and nonsilenced (clone 3) for TonEBP expression, respectively (Fig. 4A).
FIGURE 4.

TonEBP silencing leads to inhibition of AQP1 expression on IMCD3 cells. A, TonEBP protein expression for control cells (pSM2-EV, empty vector-transfected cells) and three different stably clones transfected with the pSM2-TonEBP silencing construct demonstrate variable expression. Clone 1 was considered totally silenced, whereas clones 2 and 3 were considered partially or nonsilenced, respectively. B, QPCR analysis of AQP1 mRNA expression in TonEBP-silenced clones under hypertonic stress revealed an absence or major reduction in AQP1 mRNA expression in clones 1 and 2, respectively, whereas relatively normal expression of AQP1 is shown for clone 3. C, representative Western blot for TonEBP-silenced clones under hypertonic stress demonstrated an absence or slight AQP1 protein expression in clones 1 and 2, respectively, and substantial expression in clone 3. Data for mRNA and protein experiments were collected from three identical experimental replicates and represent the mean ± S.E. for two independent experiments (n = 6).
As shown in Fig. 4B, the expression of AQP1 mRNA can be directly correlated with the expression of TonEBP. No mRNA expression of AQP1 was detected in the totally silenced clone 1, whereas little AQP1 expression was noted in the partially silenced clone 2, and substantial expression was found in the nonsilenced clone 3 under acute hypertonic stress (500 mosmol/kg H2O) for 48 h. These results were further validated by Western blot for AQP1 protein expression (Fig. 4C).
Identification of a TonE Consensus Site within the AQP1 Promoter Sequence
To further verify that AQP1 is actually a target gene for TonEBP transcriptional activity under hypertonic stress, we investigated whether TonEBP may bind directly to the promoter region of aqp1. To this end, we first identified potential TonEBP binding sites within the promoter region of aqp1 (consensus site: TGGAAANNYNY, where N means any nucleotide and Y means any pyrimidine) (11). Sequence analysis revealed the existence of a TonEBP consensus site at −811 bp upstream of the first exon (TGGAAACCTCT) as well as a TonEBP-like site at −113 bp upstream the first exon (TGGAAGAATTT, mismatch in the sequence in boldface type). We also could identify TonEBP consensus sites in the promoter sequences of different species including human (TGGAAAATCAT, −1438 bp) (data not shown). To determine whether TonEBP binds to the promoter region of AQP1, we first performed a ChIP assay from IMCD3 cells acutely exposed to hypertonicity for 24 h as described under “Materials and Methods.” The resulting genomic DNA was analyzed by PCR employing primers that codified for a specific region in the aqp1 mouse promoter gene that included the potential TonEBP binding site. As shown in Fig. 5A, a single band of the expected size was found after PCR amplification, and sequencing analysis confirmed that this band contained the TonEBP binding site. As a control, we performed PCR amplification from pGL3 plasmids containing several parts of the promoter region of AQP1. As shown in the same Fig. 5A, the expected band was found only in the plasmid that included the first −1000 bp upstream of the first exon (pGL3-AQP1). In contrast, no signal was found from PCRs of plasmids containing less than the first −700 bp (pGL3-AQP1ΔTonE) or when the anti-TonEBP antibody used in the ChIP was substituted by IgG. Because the use of ChIP analysis for identification of TonEBP-DNA binding remains controversial due to its ability to encircle DNA and therefore potentially bind nonspecifically to DNA fragments (35), we also performed a luciferase-based assay in which the promoter region of aqp1 was cloned into the pGL3 vector (Promega) upstream of the luciferase cassette (pGL3-AQP1). Employing this system, we analyzed the transcriptional activation of this luciferase cassette in transfected IMCD3 cells under acute hypertonic stress as compared with isotonic conditions. As shown in Fig. 5B, the transcriptional activity of an AQP1 promoter containing the first −1000 bp upstream the first exon of aqp1, which included the TonEBP consensus site showed a 4.1 ± 0.2-fold increase (p < 0.01) under hypertonic stress conditions for 24 h. In contrast, there was no increase in luciferase activity when the promoter only included the TonEBP-like site (0.4 ± 0.1 for pGL3-AQP1ΔTonEBP) or when both sites were removed (0.5 ± 0.05 for pGL3-AQP1ΔTonEBP-like). To better assess the role of the TonEBP consensus site in AQP1 expression, we mutated the TonE site (TGGAAACCTCT to TTTCGACCTC; the mutation is underlined and confirmed by sequencing, pGL3-AQP1mut). As shown in Fig. 6, luciferase activity is blunted substantially in the pGL3-AQP1mut (54 ± 5%, p < 0.01) as compared with the wild type pGL3-AQP1 vector under hypertonic conditions indicating that this site is substantially responsible for the overexpression of AQP1 under hypertonic conditions.
FIGURE 5.

Identification of a hypertonically inducible TonEBP binding site within the AQP1 promoter. A, PCR product image from ChIP assay with specific primers codifying for a specific region in the aqp1 mouse promoter that included the potential TonEBP binding site (lane 2, ChIP). As a positive control, the pGL3 vector including the first 1000 bp in the promoter region upstream the first exon of mouse AQP1 is used (lane 3, pGL3-AQP1). As a negative control, the pGL3 vector including the first 700 bp in the promoter region is used (without the TonEBP consensus site) (lane 4, pGL3-AQP1ΔTonEBP). No product was obtained when the anti-TonEBP antibody was substituted by IgG in the ChIP assay (lane 5, IgG). B, luciferase (Luc) reporter assay employing different constructs of the aqp1 mouse promoter region. Full aqp1 promoter (pGL3-AQP1) including the first 1000 bp upstream of the first exon reveals a >4.1-fold increase in luciferase signal with acute hypertonic stress. In contrast, no significant increase in luciferase expression was observed when the TonEBP binding site was deleted alone (pGL3-AQP1ΔTonEBP) or with a TonEBP-like site closer to the first exon (pGL3-AQP1ΔTonEBP-like). a, p < 0.01; NS, no significant difference as compared with isotonically maintained IMCD3 cells. Data were collected from three identical experimental replicates and represent the mean ± S.E. for two independent experiments (n = 6).
FIGURE 6.
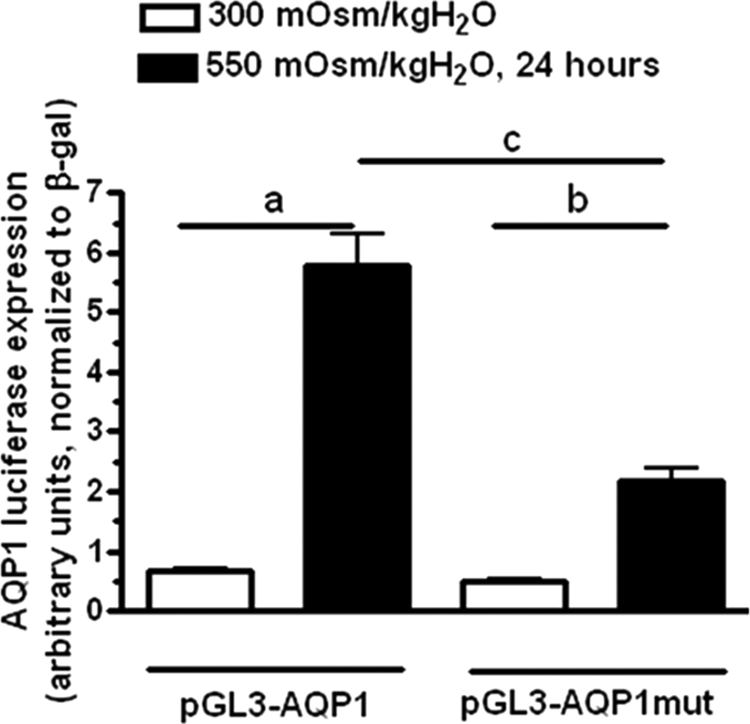
Mutation of TonEBP binding site inhibits AQP1 expression under hypertonic stress. AQP1 up-regulation under hypertonic stress was significantly blunted in the mutated plasmid (54 ± 5%, p < 0.01) as compared with the wild-type pGL3-AQP1 vector. No significant difference was found between both vectors in IMCD3 cells maintained at isotonic conditions. a, p < 0.01 and b, p < 0.05 as compared with luciferase expression at isotonic conditions; c, p < 0.01 between pGL3-AQP1 versus pGL3-AQP1mut at hypertonic conditions. Data were collected from three identical experimental replicates and represent the mean ± S.E. for two independent luciferase experiments (n = 6).
TonEBP and AQP1 Localize to Same Cells of Descending Thin Limb but Not to Proximal Tubules
To determine whether the results obtained in IMCD3 cells may be applicable in vivo, we analyzed the expression and localization of TonEBP and AQP1 in the kidney. We first analyzed the expression of AQP1 by Western blot analysis from kidney lysates. As previous studies reported (18) and as depicted in Fig. 7A, AQP1 was expressed in both the cortex and the medulla of the mouse kidney. However, the tonicity-dependent up-regulation of AQP1 induced in mice by withholding water from animals for 36 h (urine osmolality increased from 1424 ± 211 to 3105 ± 524 mosmol/kg H2O, n = 7) occurred substantially in the medulla of the kidney (85 ± 5% increase, p < 0.01) with no significant change in its expression in the cortex (8 ± 5% increase, p > 0.05) reflecting that the effect observed in IMCD3 cells may occur only at the medulla of the kidney but not in the cortex. The expression of TonEBP was studied in the same kidney tissues as for AQP1. As depicted in Fig. 7A, the expression of TonEBP is increased markedly in the medullary tissues of mice from which water was withheld as compared with ad libitum water mice (2.1 ± 1.1-fold increase, p < 0.01). Linear regression analysis between TonEBP and AQP1 expression in the medulla revealed a correlation value (r2) of 0.8059, indicating that the expression of TonEBP is associated with the expression of AQP1 (Fig. 7A, bottom). To better confirm this observation, we studied the in vivo localization of AQP1 and TonEBP in kidney tissues. As shown in Fig. 7B (left column), AQP1 (red) is expressed in cortical proximal tubules, mostly in the apical membrane domain. In contrast, we could not find any specific staining for TonEBP (green) indicating that AQP1 cortical expression is a TonEBP-independent process. The absence of TonEBP expression in the cortex of the kidney confirms previous results from a previous report (36). In the inner medulla, AQP1 has been identified previously as a marker of descending thin limb of Henle's loop tubules (18, 20), with no expression in either the ascending thick limb or in collecting ducts. As shown in Fig. 7B (right column), we identified that there is a substantial expression of TonEBP (green) in cells of the inner medulla expressing AQP1 (red). Furthermore, TonEBP expression seems to be mostly nuclear with little or no cytoplasmic staining as shown by its colocalization with the nuclear marker DAPI (colocalization shown in light blue). As a control, in vivo analysis of the localization of TonEBP in the inner medullary collecting ducts (as identified by AQP2 expression) showed no signal of AQP1 (data not shown), indicating that the transcriptional activity of AQP1 by TonEBP occurs only in the descending thin limb.
FIGURE 7.

In vivo localization of AQP1 and TonEBP in the kidney of hypertonically stressed mice. A, representative Western blot of cortex and medulla kidney tissues from mice with either ad libitum access to water or hypertonically stressed for 36 h (water was withheld) (n = 7 mice, representative Western blot depicts four animals per condition). AQP1 is expressed in both cortex and medulla of the kidney. However, the hypertonic up-regulation of AQP1 occurs predominantly in the medullary tissues. TonEBP is mostly present in the medullary tissues, medullary expression of TonEBP is significantly higher in animals from which water was withheld (2.1 ± 1.1-fold increase, p < 0.01). Correlation analysis between TonEBP and AQP1 expression in the medulla revealed an r2 linear regression value of 0.8059. B, left column, representative immunofluorescence image from cortex tissue showing AQP1 (red) but not TonEBP (green) expression. Right column, representative immunofluorescence image from medullary tissue showing AQP1 (red) in the same tubules that are expressing TonEBP (green). In these tubules, TonEBP is predominantly expressed in the nucleus where it colocalizes with DAPI (blue, merge is shown in light blue). Data were collected from seven mice per condition (ad libitum and from which water was withheld) and represent the mean ± S.E. for two independent experiments (n = 6).
As determination of descending limb tubules in the medulla is difficult in cryosections (very flattened epithelia; and it is often difficult to make out the borders of individual cells, identify nuclei, and distinguish them from closely adjacent IMCD and ascending thin limbs), we analyzed the functional relationship between TonEBP and AQP1 employing genetically created TonEBP-deficient mice (see “Materials and Methods”). As a lack of TonEBP induces high embryo and perinatal lethality (29, 30), we harvested kidneys from 4-day-old pups. At this age, in wild-type rodents, TonEBP is already shifted to the nucleus of Henle's loop and medullary collecting duct cells (37), so AQP1 expression should be present. Genotyping analysis confirmed only two homozygous mutant mice of 52 littermates analyzed (tonebp Δ/Δ, see Fig. 8A for genotyping analysis). Fig. 8B depicts AQP1 mRNA expression in the kidney of these mice. As shown, heterozygous mice express significantly less AQP1 than wild-type mice. This decrease in expression is significantly greater in the homozygous mice demonstrating direct correlation between TonEBP expression and AQP1 mRNA levels in the kidney. As shown in Fig. 8C. The protein expression of AQP1 and of another TonEBP target gene, AR, is substantially down-regulated in the surviving tonebp Δ/Δ mice. In contrast, we did not observe any significant change in expression between wild-type and TonEBP heterozygous mice, thus suggesting that there may be AQP1, and possibly other TonEBP target genes, post-transcriptional modifications in heterozygous mice to compensate for the lower mRNA expression. In this regard, we have not seen significant impairment in the ability to concentrate the urine between adult wild-type and heterozygous mice (data not shown). Immunohistochemistry analysis depicted in Fig. 8C demonstrates an almost complete absence of AQP1 in tonebp Δ/Δ mice, primarily in medullary tubules thus confirming the importance of this transcription factor in AQP1 medullary expression.
FIGURE 8.

AQP1 expression is nearly absent in kidneys from TonEBP-defective mice. A, representative genotyping analysis from wild type (+/+), heterozygous (+/Δ), and homozygous (Δ/Δ) TonEBP-deficient pups (4-day-old). The wild-type band corresponds to 284 bp, whereas the deficient band corresponds to 384 bp. B, AQP1 mRNA expression is nearly absent in tonebp Δ/Δ mice as compared with wild-type mice. Heterozygous TonEBP mice demonstrate an intermediate AQP1 expression between wild-type and homozygous. C, representative Western blot from kidneys of these pups demonstrating modest reduction in AR and marked reduction in AQP1 expression in the two surviving tonebp Δ/Δ mice. D, immunohistochemistry analysis demonstrating marked decrement AQP1 expression in medullary tubules of tonebp Δ/Δ mice. a, p < 0.01; b, p < 0.05 as compared with wild-type mice. Data were collected from four to six wild-type and heterozygous TonEBP mutant mice, n = two homozygous TonEBP mutant mice, and Western blot and immunohistochemistry results are representative of two independent experiments.
DISCUSSION
Changes in the cellular genome and proteome are required for cells of the renal inner medulla to survive and adapt to extreme changes in tonicity (2–4, 38). These processes ultimately allow for the ability of the kidney to concentrate and dilute the urine. In this regard, AQP1 and AQP2 play a key role in the urine concentrating mechanism by removing water from the lumen at the descending limb and collecting ducts, respectively. Knock-out mice lacking AQP1 are unable to create a hypertonic medullary interstitium by countercurrent mechanism, and humans that lack AQP1 cannot normally concentrate the urine (24). Also, hereditary non-X-linked nephrogenic diabetes insipidus is associated with a mutation in the AQP2 gene that leads to retention of this channel in the endoplasmic reticulum (21).
Previous studies have reported that the expression of AQP2 is up-regulated by hypertonicity (14, 39–41). This up-regulation is due to an increase in the activity of the essential transcription factor associated to hypertonicity, TonEBP (14, 41), which also is required for proper operation of the urine concentration mechanism (30). Under hypertonic stress conditions, TonEBP transcribes several genes involved mainly in the accumulation of compatible intracellular organic osmolytes that compensate the extracellular hypertonic gradient. Some of these genes include akr1b1, bgt1, smit1, and nte (5, 7–11), which increase the intracellular concentration of sorbitol, betaine, myo-inositol, and taurine, respectively.
Because AQP1 and AQP2 play the same role but in two different segments of the medullary part of the nephron, and in view of the previously described up-regulation of AQP1 by hypertonic stress (15, 17), we investigated whether the activation of AQP1 expression under hypertonic stress was mediated by TonEBP.To this end, we have employed both pharmacological and molecular approaches. One of the key features of a TonEBP target gene is that its expression is substantially up-regulated by hypertonicity induced by several different mediators but not by urea (42). This is because urea does not generate an osmotic gradient due to its high cellular permeability. In view of the impossibility of working with a well established immortalized mouse cell line from the descending thin limb of Henle's loop, we decided to employ mouse IMCD3 cells instead, which is a well characterized cell line that express both AQP1 (15, 16, 43) and TonEBP (28). When IMCD3 cells were subjected to either acute or chronic hypertonic stress, there was a substantial increase in the mRNA and protein expression of AQP1 confirming previous studies from ours and other groups (15, 16, 43). The expression and proper localization of AQP1 at the apical membrane domain of IMCD3 cells under hypertonic stress was confirmed by confocal immunofluorescence. In contrast, we did not observe any up-regulation of AQP1 mRNA or protein expression when IMCD3 cells were exposed to the same tonicity by addition of urea to the medium. To further study the role of TonEBP in the up-regulation of AQP1 under hypertonic stress, we employed methods to reduce or eliminate TonEBP activity under hypertonic stress by different approaches. Cells were first treated with rottlerin, a chemical inhibitor which reduces TonEBP expression through a PKCδ-independent mechanism as shown in a previous report (34). Our study confirmed that 10 μm rottlerin essentially eliminates mRNA expression of TonEBP and of the well-known TonEBP target gene aldose reductase, under hypertonic stress (data not shown). At this concentration, rottlerin also inhibited the up-regulation of AQP1 suggesting a role for TonEBP in the transcriptional regulation of AQP1. However, the inhibition of AQP1 expression by rottlerin may be secondary to the inhibition of other kinases and enzymes initially unrelated to TonEBP (44). Thus, to better determine whether TonEBP is involved in AQP1 transcriptional activation under hypertonic stress, we then employed silencing technology to specifically eliminate the expression of TonEBP in IMCD3 cells. In this regard, and after verifying the unique target sequence to reduce off-target effects due to the silencing, we studied AQP1 expression in three stable clones, which expressed different levels of TonEBP. In these clones, we showed that AQP1 expression correlates directly with TonEBP expression, with a complete failure of expression in the clone totally silenced for TonEBP. Further experiments employing ChIP revealed that TonEBP binds to a conserved TonE site positioned at −811 bp upstream of the first exon in the promoter region of AQP1. This site matches the sequence TGGAAANNYNY and represents an exact TonEBP consensus site (11). However, as ChIP data may be held suspect regarding certain transcription factors that may encircle DNA and thereby result in nonspecific binding (35), we also confirmed these results employing luciferase promoter analysis. Here, we analyzed TonEBP activity on a luciferase reporter cassette downstream the aqp1 promoter. In this way, we verified that the TonEBP consensus site on the aqp1 promoter is essential for its transcription under hypertonic stress. It is of note that the elimination of basis −776 to −1014 resulted in almost complete abolition of luciferase activity; however, the specific mutation at the TonEBP site resulted in the maintenance of some reporter activity, suggesting that there maybe quantitatively a less important regulatory site or more likely that the mutation did not render the site completely inactive.
We also have shown that TonEBP may be the driver of AQP1 expression under hypertonic stress conditions in vivo. The IMCD3 cells are immortalized cells originally derived from inner medullary collecting ducts (45). In contrast, the medullary expression of AQP1 occurs in the descending limb of Henle's loop with little or no expression in collecting ducts. Interestingly, some of the markers employed to characterize the collecting ducts including the expression of AQP2 are missing in IMCD3 cells (46). The loss of AQP2 in these cells may be compensated in this cell line by the overexpression of AQP1. In IMCD3 cells, AQP1 is mostly localized at the apical membrane domain (Fig. 3D), demonstrating a similar localization as AQP2 in the collecting duct (19, 47), thus suggesting a similar role under hypertonic conditions. To support the possibility that AQP1 expression in the descending limb of Henle's loop is mediated by TonEBP and considering the lack of a reliable well established cell line from this segment of the nephron, we have shown that TonEBP medullary expression under hypertonic stress correlates with AQP1 medullary expression, and TonEBP localizes to the same AQP1-expressing tubules of the inner medulla of hypertonically stressed mice but not in the cortex. The marked decrement in AQP1 expression in the medulla of TonEBP mutant mice further establishes the important role of this transcription factor in the expression of the water channel. Taken together, this data strongly suggests that the effect seen in IMCD3 cells may be translated in vivo to the inner medulla of the kidney. It is evident therefore that tonEBP can impact the renal water excretion by both osmolyte-dependent and osmolyte-independent pathways.
In summary, we have demonstrated for the first time that AQP1 is a TonEBP target gene in the inner medulla of the kidney under hypertonic stress. These data suggest that TonEBP is crucial for water reabsorption in the descending thin limb and collecting ducts by controlling the expression of both AQP1 and AQP2, respectively. Alternatively, as AQP1 is expressed ubiquitously in body tissues, we speculate that TonEBP may be important in its expression as well in these tissues and therefore in the overall homeostasis of the body water balance.
Footnotes
- QPCR
- quantitative PCR
- PIPES
- 1,4-piperazinediethanesulfonic acid
- AR
- aldose reductase.
REFERENCES
- 1.Handler J. S., Kwon H. M. (1996) Kidney Int. 49, 1682–1683 [DOI] [PubMed] [Google Scholar]
- 2.Burg M. B., Kwon E. D., Kültz D. (1997) Annu. Rev. Physiol. 59, 437–455 [DOI] [PubMed] [Google Scholar]
- 3.Ferraris J. D., Burg M. B. (2006) Contrib. Nephrol. 152, 125–141 [DOI] [PubMed] [Google Scholar]
- 4.Handler J. S., Kwon H. M. (2001) Nephron. 87, 106–110 [DOI] [PubMed] [Google Scholar]
- 5.Miyakawa H., Woo S. K., Chen C. P., Dahl S. C., Handler J. S., Kwon H. M. (1998) Am. J. Physiol. 274, F753–761 [DOI] [PubMed] [Google Scholar]
- 6.López-Rodríguez C., Aramburu J., Jin L., Rakeman A. S., Michino M., Rao A. (2001) Immunity 15, 47–58 [DOI] [PubMed] [Google Scholar]
- 7.Woo S. K., Dahl S. C., Handler J. S., Kwon H. M. (2000) Am. J. Physiol. Renal Physiol. 278, F1006–1012 [DOI] [PubMed] [Google Scholar]
- 8.Miyakawa H., Woo S. K., Dahl S. C., Handler J. S., Kwon H. M. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 2538–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito T., Fujio Y., Hirata M., Takatani T., Matsuda T., Muraoka S., Takahashi K., Azuma J. (2004) Biochem. J. 382, 177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallazzini M., Ferraris J. D., Kunin M., Morris R. G., Burg M. B. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 15260–15265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rim J. S., Atta M. G., Dahl S. C., Berry G. T., Handler J. S., Kwon H. M. (1998) J. Biol. Chem. 273, 20615–20621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woo S. K., Lee S. D., Na K. Y., Park W. K., Kwon H. M. (2002) Mol. Cell. Biol. 22, 5753–5760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakayama Y., Peng T., Sands J. M., Bagnasco S. M. (2000) J. Biol. Chem. 275, 38275–38280 [DOI] [PubMed] [Google Scholar]
- 14.Hasler U., Jeon U. S., Kim J. A., Mordasini D., Kwon H. M., Féraille E., Martin P. Y. (2006) J. Am. Soc. Nephrol. 17, 1521–1531 [DOI] [PubMed] [Google Scholar]
- 15.Umenishi F., Schrier R. W. (2002) Biochem. Biophys. Res. Commun. 292, 771–775 [DOI] [PubMed] [Google Scholar]
- 16.Umenishi F., Schrier R. W. (2003) J. Biol. Chem. 278, 15765–15770 [DOI] [PubMed] [Google Scholar]
- 17.Umenishi F., Yoshihara S., Narikiyo T., Schrier R. W. (2005) J. Am. Soc. Nephrol. 16, 600–607 [DOI] [PubMed] [Google Scholar]
- 18.Maunsbach A. B., Marples D., Chin E., Ning G., Bondy C., Agre P., Nielsen S. (1997) J. Am. Soc. Nephrol. 8, 1–14 [DOI] [PubMed] [Google Scholar]
- 19.Nielsen S., DiGiovanni S. R., Christensen E. I., Knepper M. A., Harris H. W. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 11663–11667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen S., Pallone T., Smith B. L., Christensen E. I., Agre P., Maunsbach A. B. (1995) Am. J. Physiol. 268, F1023–1037 [DOI] [PubMed] [Google Scholar]
- 21.van Lieburg A. F., Verdijk M. A., Knoers V. V., van Essen A. J., Proesmans W., Mallmann R., Monnens L. A., van Oost B. A., van Os C. H., Deen P. M. (1994) Am. J. Hum. Genet. 55, 648–652 [PMC free article] [PubMed] [Google Scholar]
- 22.Ma T., Yang B., Gillespie A., Carlson E. J., Epstein C. J., Verkman A. S. (1998) J. Biol. Chem. 273, 4296–4299 [DOI] [PubMed] [Google Scholar]
- 23.Schnermann J., Chou C. L., Ma T., Traynor T., Knepper M. A., Verkman A. S. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 9660–9664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King L. S., Choi M., Fernandez P. C., Cartron J. P., Agre P. (2001) N. Engl. J. Med. 345, 175–179 [DOI] [PubMed] [Google Scholar]
- 25.Rauchman M. I., Nigam S. K., Delpire E., Gullans S. R. (1993) Am. J. Physiol. 265, F416–424 [DOI] [PubMed] [Google Scholar]
- 26.Capasso J. M., Rivard C. J., Enomoto L. M., Berl T. (2003) Ann. N.Y. Acad. Sci. 986, 410–415 [DOI] [PubMed] [Google Scholar]
- 27.Lanaspa M. A., Almeida N. E., Andres-Hernando A., Rivard C. J., Capasso J. M., Berl T. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13672–13677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andres-Hernando A., Lanaspa M. A., Rivard C. J., Berl T. (2008) J. Biol. Chem. 283, 25082–25090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Go W. Y., Liu X., Roti M. A., Liu F., Ho S. N. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10673–10678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.López-Rodríguez C., Antos C. L., Shelton J. M., Richardson J. A., Lin F., Novobrantseva T. I., Bronson R. T., Igarashi P., Rao A., Olson E. N. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2392–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lanaspa M. A., Giral H., Breusegem S. Y., Halaihel N., Baile G., Catalán J., Carrodeguas J. A., Barry N. P., Levi M., Sorribas V. (2007) Am. J. Physiol. Renal Physiol. 292, F230–242 [DOI] [PubMed] [Google Scholar]
- 32.Wang X. Q., Li H., Van Putten V., Winn R. A., Heasley L. E., Nemenoff R. A. (2009) Mol. Biol. Cell 20, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Putten V., Refaat Z., Dessev C., Blaine S., Wick M., Butterfield L., Han S. Y., Heasley L. E., Nemenoff R. A. (2001) J. Biol. Chem. 276, 1226–1232 [DOI] [PubMed] [Google Scholar]
- 34.Zhao H., Tian W., Cohen D. M. (2002) Am. J. Physiol. Renal Physiol. 282, F710–717 [DOI] [PubMed] [Google Scholar]
- 35.Stroud J. C., Lopez-Rodriguez C., Rao A., Chen L. (2002) Nat. Struct. Biol. 9, 90–94 [DOI] [PubMed] [Google Scholar]
- 36.Cha J. H., Woo S. K., Han K. H., Kim Y. H., Handler J. S., Kim J., Kwon H. M. (2001) J. Am. Soc. Nephrol. 12, 2221–2230 [DOI] [PubMed] [Google Scholar]
- 37.Han K. H., Woo S. K., Kim W. Y., Park S. H., Cha J. H., Kim J., Kwon H. M. (2004) Am. J. Physiol. Renal. Physiol. 287, F878–885 [DOI] [PubMed] [Google Scholar]
- 38.Handler J. S., Kwon H. M. (2001) Kidney Int. 60, 408–411 [DOI] [PubMed] [Google Scholar]
- 39.Storm R., Klussmann E., Geelhaar A., Rosenthal W., Maric K. (2003) Am. J. Physiol. Renal Physiol. 284, F189–198 [DOI] [PubMed] [Google Scholar]
- 40.Kasono K., Saito T., Saito T., Tamemoto H., Yanagidate C., Uchida S., Kawakami M., Sasaki S., Ishikawa S. E. (2005) Nephrol. Dial. Transplant. 20, 509–515 [DOI] [PubMed] [Google Scholar]
- 41.Gajghate S., Hiyama A., Shah M., Sakai D., Anderson D. G., Shapiro I. M., Risbud M. V. (2009) J. Bone Miner. Res. 24, 992–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim J. A., Jeon U. S., Kwon M. S., Lim S. W., Kwon H. M. (2007) Methods Enzymol. 428, 253–267 [DOI] [PubMed] [Google Scholar]
- 43.Jenq W., Mathieson I. M., Ihara W., Ramirez G. (1998) Biochem. Biophys. Res. Commun. 245, 804–809 [DOI] [PubMed] [Google Scholar]
- 44.Soltoff S. P. (2007) Trends Pharmacol Sci 28, 453–458 [DOI] [PubMed] [Google Scholar]
- 45.Rauchman M. I., Nigam S. K., Delpire E., Gullans S. R. (1993) Am. J. Physiol. 265, F416–424 [DOI] [PubMed] [Google Scholar]
- 46.Umenishi F., Narikiyo T., Schrier R. W. (2005) Biochem. Biophys. Res. Commun. 338, 1593–1599 [DOI] [PubMed] [Google Scholar]
- 47.Fushimi K., Uchida S., Hara Y., Hirata Y., Marumo F., Sasaki S. (1993) Nature 361, 549–552 [DOI] [PubMed] [Google Scholar]