

Abstract
Human transthyretin (TTR) is a homotetrameric protein involved in several amyloidoses. Zn2+ enhances TTR aggregation in vitro, and is a component of ex vivo TTR amyloid fibrils. We report the first crystal structure of human TTR in complex with Zn2+ at pH 4.6–7.5. All four structures reveal three tetra-coordinated Zn2+-binding sites (ZBS 1–3) per monomer, plus a fourth site (ZBS 4) involving amino acid residues from a symmetry-related tetramer that is not visible in solution by NMR. Zn2+ binding perturbs loop E-α-helix-loop F, the region involved in holo-retinol-binding protein (holo-RBP) recognition, mainly at acidic pH; TTR affinity for holo-RBP decreases ∼5-fold in the presence of Zn2+. Interestingly, this same region is disrupted in the crystal structure of the amyloidogenic intermediate of TTR formed at acidic pH in the absence of Zn2+. HNCO and HNCA experiments performed in solution at pH 7.5 revealed that upon Zn2+ binding, although the α-helix persists, there are perturbations in the resonances of the residues that flank this region, suggesting an increase in structural flexibility. While stability of the monomer of TTR decreases in the presence of Zn2+, which is consistent with the tertiary structural perturbation provoked by Zn2+ binding, tetramer stability is only marginally affected by Zn2+. These data highlight structural and functional roles of Zn2+ in TTR-related amyloidoses, as well as in holo-RBP recognition and vitamin A homeostasis.
Keywords: Amyloid, NMR, Vitamin A, X-ray Crystallography, Zinc, Retinol-binding Protein, Transthyretin
Introduction
Human transthyretin (TTR)5 is a 55 kDa, β-sheet-rich homotetramer found in serum and cerebrospinal fluid. It participates in thyroxine (T4) transport (1) and in the binding of holo retinol-binding protein (holo-RBP) and is believed to reduce the glomerular filtration of the relatively small (21 kDa) holo-RBP (2). In humans, TTR and its more than 100 variants are involved in amyloid diseases such as senile systemic amyloidosis (SSA) and familial amyloidotic polyneuropathy (FAP), among others (3). In SSA, deposits composed of the wild-type TTR (WT-TTR) are found in the heart. Autopsies show that at least 25% of all individuals over 80 years have such deposits, which prove fatal in 10% of these cases (4). FAP is caused by the accumulation of amyloid fibrils formed by TTR mutants, mainly around basal membranes of Schwann cells, inside the endoneurium of peripheral nerves (5, 6). Onset typically occurs in the third decade, with a life expectancy of ∼10 years (6). Several drugs are being tested against TTR amyloidosis (3), but so far, the only treatment for FAP is liver transplantation (7).
A widely accepted hypothesis for TTR amyloidogenesis presupposes tetramer dissociation into a misfolded monomer which aggregates, giving rise to amyloid fibrils. In vitro these events can be triggered by mild acidification (pH 5.0–4.0), although amyloid formation takes several days for completion (8). In vivo these events might occur inside lysosomes during TTR turnover (8). Recently, J. W. Kelly and co-workers (9) described an engineered monomer of TTR (M-TTR) in which the introduction of two bulky amino acid residues in the protein-protein interface (F87M/L110M) avoids oligomerization into dimers or tetramers. M-TTR aggregates in solution in the same pH range as WT-TTR, although for M-TTR this occurs on a much faster time scale because tetramer dissociation into monomers is the rate-limiting step in TTR aggregation (9).
Monomers (A–D) of TTR are composed of eight anti-parallel β-strands (A–H) arranged as a β-barrel (Greek key) with a short α-helix following strand E. The dimer (AB) is held together by hydrogen bonds involving the H and F strands located along the edge of each monomer. Two dimers (AB, CD) associate back-to-back via hydrophobic interactions between residues of loops connecting strands AB and GH (10) (Fig. 1, A and B).
FIGURE 1.

Crystallographic structure of TTR complexed with Zn2+. A, Cα alignment of Zn2+:M-TTR structures at pH 7.5 (red), 6.5 (magenta), 5.5 (blue), and 4.6 (orange) (PDB IDs 3GRG, 3GRB, 3GPS and 3DGD, respectively). Note the nearly perfect overlap among the four subunits (A–B) of the four structures. B, ribbon diagrams showing the overall crystal structure of Zn2+-free M-TTR (green, PDB ID 1GKO (9)) aligned with the Zn2+:M-TTR complex at pH 7.5 (red, PDB ID 3GRG). Zn2+ ions are shown as gray spheres. ZBS 1–3 are also highlighted. C, plots of the Cα r.m.s.d. for the four monomeric subunits (A–D) of M-TTR, comparing equivalent subunits of Zn2+-free M-TTR (PDB ID 1GKO (9)) with Zn2+-bound M-TTR at pH 7.5 (PDB ID 3GRG). Major Zn2+-dependent differences are seen in the region of the α-helix and F-loop (residues 74–90).
Numerous studies with WT and variants of TTR have attempted to unravel the structural rearrangements that precede amyloid formation (10–13). Only minor global and local changes are observed in most of these structures (10), providing no conclusive structural clues for the possible mechanisms behind TTR structural conversion and amyloidogenesis. Recently, the x-ray structures of WT-TTR and two variants, I84A and I84S, under acidic conditions (and without Zn2+) revealed structural disorder in the loop E-α-helix-loop F region in relation to the structure solved at pH 7.0, indicating a possible participation of this segment of the protein in fibril formation (14, 15).
Five decades of evidence support the interaction of TTR with Zn2+ in the biological milieu but nothing is known about the accompanying structural correlates (16, 17). The observation that concentrations of Zn2+ in the plasma of healthy individuals are ∼12–15 μm (18), while the Zn2+:TTR apparent dissociation constant (Kdapp) is 1 μm (19), suggests that TTR may circulate as a complex with Zn2+ in plasma. High concentrations of Zn2+ and Cu2+ can trigger TTR amyloid formation in vitro (19, 20), and chelating agents disrupt these amyloid structures (21). Zn2+ was found to be the main mineral in ex vivo ocular amyloid deposits from FAP patients bearing the V30M mutation (22). Altogether, these data suggest that binding of Zn2+ (or Cu2+) might induce structural changes that lead to TTR amyloidogenesis. The aims of this study are to dissect the structural modifications caused by Zn2+ binding and to explore their implications for TTR-mediated physiological recognition of holo-RBP, for TTR stability and for amyloidogenesis.
EXPERIMENTAL PROCEDURES
Proteins
Recombinant human WT-TTR and M-TTR were expressed and purified as previously described (23), using plasmids kindly provided by Dr. Jeffrey W. Kelly (9) (The Scripps Institute, San Diego, CA). Retinol-binding protein (RBP4) was obtained from Phoenix Pharmaceuticals (Burlingame, CA).
X-ray Crystallography
For crystallographic experiments, we used the double mutant F87M/L110M (M-TTR), a construct which displays a decreased self-association constant compared with WT-TTR (9). M-TTR crystals were obtained by the hanging-drop vapor-diffusion method at 20 °C. Crystals were grown in ∼5 days in 200 mm zinc acetate, 100 mm sodium citrate, 2.0 m ammonium sulfate, adjusted to the desired pH. X-ray diffraction images were collected in steps of 1° of oscillation per frame from single crystals cooled at 100 K. We used a wavelength of 1.458 Å (1.305 Å for the crystal obtained at pH 4.6, PDB ID 3DGD) at the MX2 beam line at LNLS (National Synchrotron Light Laboratory, LNLS, Campinas, Brazil (24)) and diffraction intensities were measured using a MarMosaic 225 detector (MAR Research GmbH, Norderstedt, Germany). Images were indexed, processed and integrated with Mosflm v. 7.0.3 (25). All M-TTR structures solved here belong to space group P21. We scaled experimental intensities, solved, and refined the structure with the CCP4 v. 6.0.2 suite (26). The complex was solved by molecular replacement, using monomer A with only Cα and Cβ atoms of PDB ID 1F41 (10) as search model, resulting in a solution for four monomers in the asymmetric unit. Model building and refinement were performed with Coot v. 0.5 (27) and the CCP4 v. 6.0.2 suite (26). Water molecules were added using Coot v. 0.5 (27).
Structural validation of the model performed with the CCP4 v. 6.0.2 suite (26) showed that almost all dihedral angles are within the favored region (for 3GRG (pH 7.5), 97.4% of residues are in favored regions, 2.0% are in allowed regions and 0.6% are in outlier regions; for 3GRB (pH 6.5), 98.5% of residues are in favored regions, 1.5% are in allowed regions and 0.0% are in outlier regions; for 3GPS (pH 5.5), 97.3% of residues are in favored regions, 2.0% are in allowed regions and 0.7% are in outlier regions; for 3DGD (pH 4.6), 99.1% of residues are in favored regions, 0.9% are in allowed regions and 0.0% are in outlier regions). Omit maps were generated with the CCP4 v. 6.0.2 suite (26). All crystallographic figures were generated with PyMol (28). Selected crystallographic parameters are found in supplemental Table S1.
Multiple Sequence Alignment and Conservation Analysis
A multiple sequence alignment (MSA) for the TTR family was obtained from the Pfam server (29). After the elimination of redundant and fragmented sequences, a total of 262 sequences remained. Overall site conservation and site-dependent conservation were calculated using C++ routines designed for statistical coupling analysis (SCA) (30).
NMR Spectroscopy
All NMR experiments were performed at 25 °C on an 800 MHz Bruker III Avance spectrometer equipped with a 1H[13C/15N] pulsed-field gradient probe (Bruker Corp., GmbH). Deposited sequential assignments of WT-TTR described elsewhere (BMRB 5507) (31) were used with adjustments due to differences between pH used in the deposited assignment and pH used in our experiments. To adjust assignment, we collected heteronuclear single-quantum coherence, transverse relaxation optimized spectroscopy (HSQC-TROSY) spectra using samples of WT-TTR 2H/15N (25 μm) at pH 5.7, 6.5 (buffer: MES 25 mm, KCl 50 mm, pH 5.7 or pH 6.5) and 7.5 (buffer: deuterated Tris-HCl 25 mm, KCl 50 mm, pH 7,5). We used the pulse sequence TROSYETF3GPSI, Avance version 12142007 (Bruker Corp., GmbH) (32), with 8 transients in 1H dimension with 1024 points of resolution and 16 transients in 15N dimension with 256 points of resolution.
For titration, the same HSQC-TROSY spectra were recorded using WT-TTR 2H/15N (100 μm) in deuterated Tris-HCl 25 mm, KCl 50 mm, pH 7.5. Titration experiments were performed by adding ZnCl2 to the sample to attain final concentrations of 100, 200, 300, 400, and 900 μm Zn2+. To the sample with 900 μm ZnCl2, a small volume of a concentrated stock of EDTA was added, reaching 1 mm EDTA. All spectra were processed using the TopSpin suite (Bruker Corp., GmbH) and were analyzed using Computer Aided Resonance Assignment (CARA) (33). CSP values were calculated using assignments of the zinc-free WT-TTR spectrum as reference, and using Equation 1 (34),
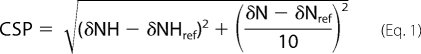 |
where δXref is the chemical shift of the reference spectrum and δX the chemical shift of the spectrum at each concentration of ZnCl2 for each amidic proton (NH) or amidic nitrogen (N). We chose 0.025 ppm as a threshold for considering a CSP to be significant.
For TROSY-HNCO we used the pulse sequence TRHNCOGP2H3D, Avance version 07042004 (Bruker Corp., GmbH) (35), with 8 transients in 1H dimension with 1024 points of resolution; 16 transients in 15N dimension with 40 points of resolution and 16 transients in 13C dimension with 50 points of resolution. For TROSY-HNCA we used the pulse sequence TRHNCAGP2H3D2, Avance version 07042004 (Bruker Corp., GmbH) (35), with 8 transients in 1H dimension with 1024 points of resolution; 16 transients in 15N dimension with 40 points of resolution and 16 transients in 13C dimension with 50 points of resolution. To perform these experiments, WT-TTR 2H/15N (100 μm) in deuterated Tris-HCl 25 mm, KCl 50 mm, pH 7.5 was used, and the spectra were collected in the absence or presence of 400 μm ZnCl2.
Holo-RBP Interaction with TTR
Titrimetric assays of WT-TTR interaction with RBP bound to retinol (holo-RBP) were performed by fluorescence anisotropy measurements in an ISS-PC1 spectrofluorometer (ISS Inc, Champaign, IL) equipped with Glan-Thompson polarizers in l-format as previously described (36). Excitation and emission wavelengths were set at 330 and 460 nm, respectively. Assays were performed in 10 mm Tris-HCl, 150 mm NaCl, pH 7.2, 400 nm holo-RBP, and at the indicated ZnCl2 concentrations. Dissociation constants were calculated as previously described (37), according to Equation 2,
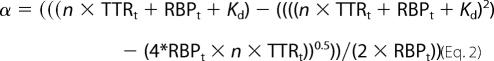 |
where α is the fraction of holo-RBP:WT-TTR formed, TTRt is the total WT-TTR concentration, Kd is the dissociation constant, n is the number of holo-RBP binding sites present on the WT-TTR molecule and RBPt is the total RBP concentration used in the assay. The fraction of holo-RBP:WT-TTR formed was calculated according to Equation 3,
where Aobs is the measured anisotropy at a given TTRt, and Ai and Af are the limiting initial and final anisotropies.
Thermodynamic Stability in the Presence of Zn2+
The influence of Zn2+ over M-TTR and WT-TTR stability was probed by both chemical (urea) and physical (pressure) variables. M-TTR or WT-TTR (3.5 μm) was incubated in 25 mm Tris-HCl, 50 mm KCl, pH 7.5 (25 °C) in the absence or presence of ZnCl2 (as stated in the legend of each figure). Pressure was raised to the desired value and equilibrated for 30 min before spectroscopic measurement. Tryptophan fluorescence emission was used as a sensor of the tertiary structure changes in the protein. The spectra were recorded by exciting samples at 280 nm and collecting emission from 300–400 nm. The center of spectral mass values (<ν> = ΣniFi/ΣFi, where Fi is the fluorescence emitted at wavelength νi) were calculated in nm and plotted against pressure applied. From these plots, the extent of reaction (α) was calculated and converted to a log scale to assess the free energy change of unfolding (ΔGunf) and the standard volume change of folding (ΔV) according to Equation 4,
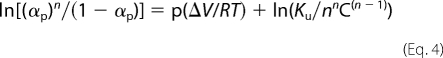 |
where αp is the extent of reaction at pressure p, n is the number of subunits (n = 1 for M-TTR; n = 4 for WT-TTR), p is pressure, R is the gas constant, C is the molar concentration of protein and T the temperature in K. The ΔV is calculated from the slope of the curve and Ku is the intercept on the ordinate (ΔGunf = −RTlnKu) (38).
M-TTR or WT-TTR (3.5 μm) was incubated in 25 mm Tris-HCl, 50 mm KCl, pH 7.5 (25 °C) for 72 h at varying concentrations of urea (0–8 m). ZnCl2 was added to desired concentration and tryptophan fluorescence emission (280 nm and collecting emission from 300–400 nm) was immediately measured. Then, the center of spectral mass of tryptophan emission of each spectrum was calculated and converted into the extent of reaction (α) as described under “Experimental Procedures” and plotted against [urea]. From these experiments, ΔGunf was calculated according to Equation 5,
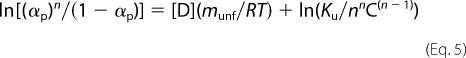 |
where α is the extent of reaction, n is the number of subunits, [D] is urea concentration, R is the gas constant, C is the concentration of protein in molar and T the temperature in K. The munf is calculated from the slope of the curve and Ku is the intercept on the ordinate (ΔGunf = −RTlnKu) (39).
Size-exclusion Chromatography (SEC)
SEC was performed in a TSK3000 (Tosoh Bioscience, Tokyo, Japan) column pre-equilibrated with 50 mm Tris-HCl, 100 mm KCl at pH 7.5 and with the indicated concentration of ZnCl2. Samples of 3.5 μm WT-TTR diluted in the absence or in the presence of ZnCl2 were injected into the column. TTR tetramers and monomers eluted at 7.5 and 9 min, respectively, as confirmed by previous column calibrations with RNase and bovine serum albumin as standards.
RESULTS
Crystallographic Determination of Zn2+-binding Sites in TTR
We solved the crystal structures of M-TTR in complex with Zn2+ at pH 7.5, 6.5, 5.5, and 4.6 at resolutions of 1.9, 1.7, 1.8, and 1.3 Å, respectively, with Cα r.m.s.d. among them less than 0.4 Å (Fig. 1A). Our attempts to solve the structure of WT-TTR in the presence of Zn2+ failed due to the fragility of the crystals. M-TTR, which at the concentrations used crystallizes as a tetramer (9), forms robust crystals in the presence of Zn2+.
The Zn2+:M-TTR complex presented the typical fold of TTR at all pH values (Fig. 1A), and in Fig. 1B the structure at pH 7.5 obtained in the presence of Zn2+ is superposed with that obtained at the same pH in the absence of Zn2+. However, despite their close resemblance, the structural alignment of Zn2+:M-TTR with Zn2+-free M-TTR shows large conformational differences in both secondary and tertiary structures, achieving 5 Å in some segments of the monomer (Fig. 1C). Fluctuating conformations in the loops connecting β-strands BC, DE, and FG were previously described (10). The more prominent structural modification, apart from local changes in residues involved in Zn2+ coordination, is a perturbation of the loop E-α-helix-loop F region and residues nearby (Fig. 1, B and C and Fig. 3A). These modifications occur in all four subunits of the tetramer (Fig. 1C).
FIGURE 3.

A close-up view of the loop E-loop F region flanked by ZBS 2 and ZBS 3 shows major structural modifications when Zn2+ is bound. A, structures of the four subunits of TTR (ABCD; orange, red, blue, and yellow) obtained in the presence of Zn2+ at different pH are overlapped with the Zn2+-free structure (green). Note the movement of the α-helix residues as the pH decreases from pH 7.5 to 4.6, causing the helix to unwind and form an extended loop. B, close-up view of the ZBS 2 and 3 region, in green, the apo form and in red the structure obtained in the presence of Zn2+ at pH 7.5, where the structural alterations in the α-helix region are better seen. Residues in pink are those that undergo major reorientation upon Zn2+ binding. Zn2+ atoms are shown as gray spheres. C, Zn2+ binding to WT-TTR decreases its affinity for holo-RBP. Isothermal holo-RBP:WT-TTR binding assays were performed by measuring anisotropy changes that take place upon TTR binding to holo-RBP in the absence (●) or in the presence of 15 μm ZnCl2 (▼) and 50 μm ZnCl2 (■). Data are normalized to fraction of holo-RBP:TTR complex formed. The Kd values (inset, error bars show ± S.D.) were calculated as described under “Experimental Procedures.” D, size-exclusion chromatography demonstrates that WT-TTR remains a tetramer upon Zn2+ binding. The elution profiles of WT-TTR without (black line) and in the presence of 20 μm (red line) and 100 μm (green line) ZnCl2 are shown. T, elution time of tetrameric TTR; M, elution time of monomeric TTR.
Zn2+ ions were coordinated at three Zn2+-binding sites (ZBS 1 to 3) in each monomer. All three sites displayed coordination spheres based on tight interactions of Zn2+ with a imidazole nitrogen from His, the thiol group from Cys, and carboxylic acid groups from Glu or Asp side chains (Fig. 2). These elements are typical of selective Zn2+-binding sites in proteins (40). In fact, the structures revealed the presence of a fourth ZBS (ZBS 4), which involves residues from the symmetry-related tetramer as explained bellow, and thus we assumed it was generated by crystallographic contacts (Fig. 2). NMR experiments confirmed the absence of ZBS 4 in solution (Figs. 4 and 5).
FIGURE 2.

Details of the three Zn2+-binding sites in TTR (ZBS 1–3), and a fourth site (ZBS 4) that is formed by amino acid residues from a symmetry-related tetramer. Close-up views of Zn2+-binding sites and schematic diagrams of Zn2+ coordination spheres. ZBS 1 is composed of Cys-10, His-56, and two water molecules (red spheres). ZBS 2 is composed of His-88, His-90, Glu-92, and one water molecule. ZBS 3 involves His31AU, Asp74AU and Glu72AU (at higher pH values) or Glu62symm (at pH4.6, ZBS 3′). ZBS 4 involves His31symm, Asp74symm, and Glu61AU in addition to water molecules (AU, asymmetric unit; symm, symmetry-related unit). Meshes are electron densities from 2 Fobs-Fcalc omit maps contoured at 1σ. The electron density displayed is limited to within 1.5 Å of the residues. Orange residues in ZBS 3 and ZBS 4 represent residues from neighboring tetramers of symmetrically related molecules in the crystal. The Zn2+-binding sites described here present the canonical constitution of other selective Zn2+-binding sites found in proteins (40).
FIGURE 4.

Mapping Zn2+ binding to TTR by TROSY-HSQC measurements at pH 7.5. The chemical-shift perturbations (CSP) in the NMR spectrum of fully 15N-2H labeled WT-TTR (100 μm) were measured at different concentrations of ZnCl2. CSP derived from differences between a reference spectrum of WT-TTR in the absence of Zn2+ and the spectra in the presence of Zn2+ were obtained for: (A) 100 μm ZnCl2; (B) 200 μm ZnCl2; (C) 300 μm ZnCl2; (D) 400 μm ZnCl2; (E) 900 μm ZnCl2, and (F) 900 μm ZnCl2 + 1 mm EDTA. Blue bars are residues whose signal vanished during the experiment; red bars are non-assigned residues; gray bars are prolines. Images at the right show where CSP occurred in WT-TTR at each Zn2+ concentration, marked in red in a ribbon representation of Zn2+:TTR pH 7.5 (PDB ID 3GRG). For clarity, only one of the four monomers is marked. Zn2+ atoms are represented as gray spheres. CSP was calculated for each assigned main-chain amide using Equation 1. Dotted lines show the threshold for considering a CSP significant (0.025 ppm).
FIGURE 5.

Mapping Zn2+ binding into TTR by TROSY-HNCO and TROSY-HNCA NMR measurements. Cα (upper plot) and carbonyl (lower plot) chemical-shift difference between the native WT-TTR and the random coil values (Δδ). Chemical shifts were derived from TROSY-HNCA and TROSY-HNCO spectra of WT-TTR in the absence and in the presence of 900 μm Zn2+. Δδ in the absence of Zn2+ are in black. Δδ in the presence of Zn2+ are colored red for Cα or in green for the carbonyl. Positive values of Δδ are typical of α-helix, while negative values are typical of the β-sheet. Note that Δδ for the residues comprising the α-helix remain unchanged in the presence of Zn2+, while those related to the nearby residues undergo perturbation (mainly the loop after the helix and the C-terminal of strand E). The resonance signal of these residues are missing probably because they are in conformational exchange, what can lead to the variation in the position of the α-helix observed in the crystal structure.
In ZBS 1, Zn2+ is coordinated by the thiol group of Cys-10 (Sγ atom), the imidazole nitrogen from His-56 (Nδ1 atom) and two water molecules (Fig. 2, upper left structure). In ZBS 2, Zn2+ is bound to the imidazole nitrogens of His-88 (Nϵ2 atom) and His-90 (Nδ1 atom), the carboxylic acid group of Glu-92 side chain (Oϵ1 atom) and one water molecule (Fig. 2, upper right structure). ZBS 1 and ZBS 2 presented this organization at all pH values.
ZBS 3 consists of one water molecule, the imidazole nitrogen of His-31 (Nδ1 or Nϵ2 atom), the carboxylic acid group of Asp-74 (Oδ1 or Oδ2 atom) and the carboxylic acid group of Glu-72 ([Oϵ1 or Oϵ2 atom], Fig. 2, lower left structure). However, in some subunits of structures obtained at pH 7.5, 6.5, 5.5 and in all subunits of the structure obtained at pH 4.6, Glu-62 replaces either Glu-72 or the water molecule (ZBS 3′, Fig. 2, lower middle structure). In ZBS 3, His-31, Glu-72, and Asp-74 are from the same asymmetric unit (H31AU, E72AU, D74AU, Fig. 2, lower middle structure). In ZBS 3′, His-31 and Asp-74 are both from the same monomer in the asymmetric unit (H31AU and D74AU, Fig. 2, lower middle structure), while Glu-62 is from a symmetry-related TTR molecule (E62symm, Fig. 2, lower middle structure). In solution, ZBS 3 might be composed of His31AU, Asp72AU, Glu74AU, and solvated accordingly.
ZBS 4 is composed of two residues from the symmetry-related tetramer (H31symm and D74symm, Fig. 2, lower right structure), while the carboxylic acid group of the Glu-61 side chain is from the asymmetric unit (E61AU, Fig. 2, lower right structure). Because it includes two residues from a symmetry-related tetramer, ZBS 4 is unlikely to exist as a functional Zn2+-binding site in TTR in solution, since stable Zn2+ coordination typically involves at least two amino acid interactions from the same molecule (40). Moreover, ZBS 4 is occupied in all four monomers exclusively at pH 4.6, being vacant in some of the monomers of the pH 5.5, 6.5, and 7.5 structures.
Analysis of multiple sequence alignments (MSA) (41, 42) for all non-redundant, non-fragmented TTR sequences obtained from Protein Families data base (Pfam (29)) shows that the amino acids that we identify within each ZBS are among the least conserved regions of TTR (e.g. Cys in position 10 is present in only 12.9% of sequences, while His occupies position 56 in only 14.8%) (supplemental Table S2).
An analysis of the amino acids occupying two distinct positions in a given protein family, if these residues vary and are not completely conserved, tells us if for some reason, normally related to the structure or function of the protein, these two positions co-evolved (42). Most of the residues that form ZBS 1, ZBS 2, and ZBS 3 tend to appear simultaneously for a subset of the sequences (e.g. all sequences with a Cys in position 10 have His in positions 56 and 88, and most of them have an Asp in position 74) (supplemental Table S2). Interestingly, the TTR sequences that simultaneously exhibit these correlated positions are predominantly from mammals (supplemental Fig. S1).
Structural Details of Zn2+:TTR Complex
In addition to the local changes in the amino acids that coordinate Zn2+, the binding of Zn2+ to the M-TTR structure causes striking structural modifications compared with the Zn2+-free state. The loops connecting β-strand E, the α-helix, and β-strand F, a region involved in the recognition of holo-RBP by TTR (2), are those that are most perturbed by Zn2+ binding (Fig. 3, A and B). ZBS 2 and ZBS 3 flank this region.
Interestingly, in going from neutral to acidic pH, the Zn2+:M-TTR structures show a complete disruption of the α-helix into a disordered loop comprising residues 74–90, but with a well defined electron-density map (supplemental Fig. S2). Even at pH 7.5, there is a substantial loss in the helix structure (Fig. 3, A and B) caused by Zn2+ binding, but this structural perturbation is very pronounced in the structure solved at pH 4.6, where the α-helix disappears in all four subunits (Fig. 3A). In the crystallographic structure of WT-TTR at pH 3.5 in the absence of Zn2+, the electron density of the amino acids from this region is completely disordered and not visible in the electron density map (15). Here, we could observe precisely the disruption of this specific region of the protein in the presence of Zn2+ upon acidification, a condition that triggers TTR aggregation.
At pH 7.5, the side chain of Asp-74 turns toward His-31, forming ZBS 3 (Fig. 3B). The His-90 side chain turns toward the consolidated ZBS 2, accompanied by an almost 180° flipping of the His-88 side chain (Fig. 3B) in all four subunits and at all pH values. Various main-chain hydrogen bonds of the α-helix are disrupted (Tyr-78 → Asp-74, Trp-79 → Thr-75, Lys-80 → Lys-76, Ala-81 → Ser-77, arrows point from donors to acceptors), leading to loss of the α-helix stability and structure.
Affinity of WT-TTR for holo-RBP in the Absence and Presence of Zn2+
The crystal structure of the TTR-holo-RBP complex indicates that surface of loops 63–67 and 92–98 of RBP penetrate into a crevice formed by the arrangement of three monomers in the tetramer of TTR (2). Point mutations in the α-helix of TTR lead to a decrease in its affinity for holo-RBP (36). Thus, we envisioned that the structural changes induced by Zn2+ binding in the region delimited by ZBS 2 and ZBS 3 could disrupt this topological complementarity between TTR and holo-RBP. Indeed, the extended segment of TTR resulting from the α-helix unwinding upon Zn2+ binding, which is very pronounced at acidic pH but considerable at pH 7.5, clashes with RBP (supplemental Fig. S3), a conformational change that might compromise the affinity of TTR for holo-RBP.
To address this issue, the affinities of WT-TTR for holo-RBP in the absence and presence of Zn2+ were measured using fluorescence anisotropy (Fig. 3C). In the absence of Zn2+, the dissociation constant of holo-RBP:WT-TTR complex was 227 ± 43 nm, in agreement with previous measurements (36, 43). In the presence of Zn2+, the binding curves shift to higher WT-TTR concentrations (Fig. 3C and inset), indicating a decrease in affinity of WT-TTR for holo-RBP. The dissociation constants obtained in the presence of 15 μm and 50 μm Zn2+ were 566 ± 105 nm and 1,000 ± 223 nm, respectively (± S.D.). Size-exclusion chromatography demonstrates that the oligomeric state of WT-TTR is not affected by the interaction with Zn2+, as indicated by the prevalence of tetramers at Zn2+ concentrations as high as 100 μm (Fig. 3D). This rules out the possibility that the decrease in the affinity of TTR for holo-RBP is due to TTR aggregation or changes in tetramer-monomer equilibrium.
Mapping the Zn2+-binding Sites by TROSY-HSQC NMR Measurements
To confirm that the Zn2+-binding sites and the structural changes involving the α-helix previously observed in the crystal structure of M-TTR are also present in solution and in the tetrameric WT-TTR, a stepwise titration of Zn2+ was performed using TROSY-HSQC experiments (2H15N-labeled WT-TTR, pH 7.5) (Fig. 4, supplemental Figs. S4 and S5). These experiments also allowed for the hierarchical identification of each ZBS in WT-TTR and the accompanying structural modifications evoked by Zn2+ binding to each one. Significant chemical-shift perturbations (CSP) were observed in several regions of WT-TTR upon stepwise additions of Zn2+.
As seen in Fig. 4A, incubating 100 μm WT-TTR (equivalent to 400 μm monomeric subunits) with 100 μm Zn2+ leads to significant changes in Cys-10 and in the adjacent amino acid residues of β-strand A, suggesting that ZBS 1 is likely the first Zn2+-binding site to be filled. Unfortunately, the resonance of His56, the other coordinating residue of ZBS 1, was not localized in the spectrum (red bar) at any concentration of Zn2+ added.
Increasing the concentration of Zn2+ to 200 μm (Fig. 4B) eliminates the resonance signal from Gly-57 (blue bar), adjacent to His-56. This might be related to Zn2+ binding to His-56. At this concentration of Zn2+, a prominent CSP occurs in the resonance of Glu-92, a residue that contributes to ZBS 2 (supplemental Fig. S5). Some of the residues of β-strands G and H are also perturbed, reflecting the fact that β-strands A, G, and H are anchored to each other by a network of backbone hydrogen bonds (44). With a further increase to 300 μm Zn2+ (Fig. 4C), the saturation of ZBS 2 is nearly complete (see residues His-88 and Glu-92 in Fig. 4C and supplemental Fig. S5). His-90 is only slightly perturbed at higher Zn2+ concentrations (Fig. 4, D and E). These data suggest that ZBS 2 is the second Zn2+-binding site to be occupied in solution.
At 400 μm Zn2+ (Fig. 4D), significant changes appear in ZBS 3 and in the residues in its vicinity, resulting in CSPs for Ile-73, Thr-75, Lys-76, and Trp-79, a segment that encompasses the end of β-strand E and the subsequent α-helix. The resonance signals from the other coordinating residues of ZBS 3, Asp-74 and Glu-72, vanish from the spectrum along with Val-71 (blue bars), indicating the induction of conformational change due to Zn2+ binding. Altogether, these modifications suggest a rearrangement in the vicinity of ZBS 2 and ZBS 3, to allow a proper spatial orientation for Zn2+ coordination. All these binding events lead to a reorganization of the secondary structure elements of TTR, which includes the partial unwinding of the α-helix at pH 7.5, resulting in the consolidation of the Zn2+:TTR complex, as observed in the crystallographic structure (Fig. 3). Interestingly, the resonance from Glu-62, a possible component of ZBS 3, does not undergo CSP even at the higher concentrations of Zn2+ (Fig. 4E). In the crystallographic structures, as mentioned, ZBS 3 was formed by Glu72AU at higher pH values or by Glu62symm at pH 4.6 (Fig. 2, ZBS 3 and ZBS 3′); the latter resonance is perturbed and vanishes from the spectrum. Thus, we postulate that ZBS 3 in solution comprises residues His-31, Glu-72, Asp-74 and one water molecule.
At 900 μm Zn2+ (Fig. 4E), CSP propagate throughout the whole WT-TTR structure. Nevertheless, the resonance of Glu-61, which contributes to ZBS 4, does not undergo perturbation, reinforcing the idea that ZBS 4, which involves residues from a symmetry-related subunit, cannot be formed stably in solution. The reversibility of all structural changes induced by Zn2+ binding was confirmed by the addition of 1 mm EDTA, a condition that was found to restore the original values of chemical shifts of the Zn2+-free protein (Fig. 4F).
It would be interesting to assess the dissociation constant (Kd) for Zn2+ binding to ZBS 1–3. The apparent Kd value already reported in the literature, based on fluorescence measurements, is ∼1 μm at pH 7.4 (19). This apparent Kd is not related to a specific Zn2+-binding site but to all of them. Unfortunately, the high protein concentrations necessary to obtain adequate signals in the NMR experiments make the precise calculation of the dissociation constants very difficult, because most of them are in the nm-μm range. Nevertheless, by plotting the CSP as a function of Zn2+ addition for residues located in ZBS 1–3 (supplemental Fig. S5) we were able to extract at least a C0.5 (Zn2+ concentration that promotes 50% change).
Regarding Cys-10, the addition of 100 μm Zn2+ was enough to promote the same CSP for Cys-10 as that observed with 200–900 μm. This suggests saturation of ZBS 1, which includes Cys-10. This behavior is typical of a binding equilibrium in slow-exchange regime, as expected for high-affinity binding sites (KD < 10−7 m). The slow-exchange regime is characterized as an on-off switch: the free and bound states of the protein are observed with their respective chemical shifts, but not the intermediate chemical shifts associated with partially bound species. Thus, we expect that ZBS 1 will have a Kd on the order of a few nm.
In contrast to what was observed with ZBS 1, the resonance signals associated with ZBS 2 and ZBS 3 change stepwise with Zn2+ addition (supplemental Fig. S5), showing intermediate values of CSP for intermediate Zn2+ concentrations. This behavior is typical of a fast-exchange regime with moderate- to low-affinity binding sites (Kd > 10−7 m).
Probing the Secondary Structural Changes Elicited by Zn2+ Binding through the Use of NMR
Because our crystallographic data suggest major perturbations in the loops connecting β-strand E, the α-helix, and β-strand F (Fig. 3, A and B), we triple-labeled WT-TTR (2H, 13C, 15N) and acquired triple resonance experiments (HNCA and HNCO) to assign Cα and carbonyl resonances. The chemical shifts of these resonances correlate well with secondary structural changes (45). The experiments were performed at pH 7.5. As seen in Fig. 5, addition of Zn2+ did not promote any significant CSP in the signals of the residues located in the α-helix (residues 74–82). These data unequivocally show that in solution at neutral pH, the α-helix persists when Zn2+ binds. The changes observed in the crystal probably arise from a dynamic equilibrium among different configurations of this α-helix. Note that there are pronounced chemical-shift changes in the Cα, carbonyl (Fig. 5), and HN (Fig. 4) for the residues comprising the nearby residues of this α-helix, mainly the loop before strand F and the C-terminal of strand E. These changes reflect fluctuations in the position of the α-helix. We also measured line broadening in HN resonances of the residues located in the loops close to the α-helix (supplemental Fig. S4). The broadening indicates conformational exchange on a time scale of milli- to microseconds (46). Fluctuations of these loops could ultimately lead to fluctuation of the helix that is perceived in the crystallographic structures.
Indeed, analyzing the Ramachandran plots of these residues (supplemental Fig. S6), we see that in the crystal structures obtained in the absence of Zn2+ (1GKO and 1F41), the torsion angles φ and ψ are located in a restricted, low-energy region of the Ramachandran plot. In the structures obtained in the presence of Zn2+ (3DGD, 3GPS, 3GRB, 3GRG), the angles adopted by these residues tend to be more dispersed within the allowed α-helix region, suggesting that in the presence of Zn2+, the α-helix shows conformational diversity, fluctuating between two positions.
Probing the Thermodynamic Stability of TTR in the Presence of Zn2+
As shown, Zn2+ binding induces broad tertiary structural changes in TTR, with a particular secondary structure change consisting in the perturbation of the α-helix and in other regions of the protein. To assess the stability cost of such structural effects, we challenged M-TTR and WT-TTR with urea and high hydrostatic pressure (HHP) denaturation (38, 47), using tryptophan fluorescence emission as a sensor of structural integrity.
Both urea and pressure isotherms show a typical denaturation curve for WT-TTR (Fig. 6, A and C, respectively). However, Zn2+ concentrations up to 600 μm promote only minor changes in the denaturation profile, indicating marginal effects of Zn2+ on stability of the WT-TTR tetramer. In contrast, with M-TTR there is a progressive change in the unfolding transition as a function of Zn2+ concentration, for both urea (Fig. 6B) and pressure (Fig. 6D).
FIGURE 6.

Zn2+ binding decreases monomer stability but is innocuous to the tetramers. WT-TTR (3.5 μm tetramers; panels A and C) or M-TTR (3.5 μm monomers; panels B and D) were subjected to denaturation by urea (panels A and B) or by high hydrostatic pressure (panels C and D) in Tris-HCl 25 mm, KCl 50 mm, pH 7.5 (25 °C) at varying ZnCl2 concentration (0–600 μM, as stated in the figure inset). The center of spectral mass of tryptophan emission of each spectrum was calculated and converted into the extent of reaction (α) as described under “Experimental Procedures” and plotted against the variables. From these plots and by using Equations 4 and 5, the ΔGunf (panel E) or the ΔV or munf parameters (panel F) for the unfolding of M-TTR were calculated. Circles represent urea denaturation and squares show HHP. Error bars in panels E and F are standard deviations obtained from non-linear regression analysis (most of them are smaller than the symbols). Details under “Experimental Procedures.”
A quantitative thermodynamic analysis of M-TTR denaturation reveals a steep decrease in ΔGunf up to 100 μm Zn2+ (Fig. 6E). From the urea denaturation experiments, the ΔGunf in the absence and in the presence of 100 μm Zn2+ decrease from 2.54 to 1.66 kcal/mol (ΔΔGunf = 0.88 kcal/mol) (filled circles in Fig. 6E). Additionally, the ΔGunf in the absence and in the presence of 100 μm Zn2+ calculated from the HHP experiments equal 2.97 and 2.41 kcal/mol, respectively, again showing a decrease in stability of M-TTR by ∼0.6 kcal/mol (squares in Fig. 6E), a change very similar to that obtained from the urea-induced unfolding. Fig. 6F shows the changes in the munf (circles) and ΔV (squares) parameters extracted respectively from the urea and HHP denaturation curves (Equations 4 and 5). Interestingly, in the presence of Zn2+ the volume change of folding (ΔΔV) decreases by ∼ 20 ml/mol (from 71 to 52 ml/mol), while the munf parameter decreases from 0.86 to 0.41 in the presence of 100 μm Zn2+. These data corroborate the idea that M-TTR in the presence of Zn2+ loses part of its tertiary structure, resulting in a lower cavity content and a decreased number of intra-chain contacts, changes that render the protein less stable.
DISCUSSION
The first human TTR crystallographic structure (PDB ID 2PAB (48)) revealed metal coordination involving Cys-10 and His-56, residues from ZBS 1, because the thiol group of Cys-10 underwent mercurization due to treatment with heavy metal for isomorphous derivatization. The crystals were then washed with chelating agent, thus the other ZBS were not detected (48). Occupation of ZBS 1 alone does not induce any global conformational changes, nor any modification in the α-helix or the EF loops, as presented here (Figs. 1 and 3). ZBS 1, the first site to be filled in low Zn2+ concentrations, might be permanently occupied in circulating TTR. This occupancy could prevent TTR aggregation by committing Cys-10 to the task of Zn2+ coordination and thereby avoiding SH-modifications (such as TTR-Cys, TTR-GSH, and TTR-CysGly), which have been implicated in TTR destabilization and amyloidogenesis in vitro (49).
In solution, Zn2+ binding to ZBS 2 and ZBS 3 results in a pronounced structural rearrangement in the EF loops and the α-helix (Figs. 3 and 5), a region that becomes more dynamic and flexible. Interestingly, a recently solved structure of TTR at acidic pH in the absence of metal ions, a condition that triggers TTR aggregation due to the formation of a misfolded amyloidogenic intermediate, displayed α-helix unwinding (15). Thus, we suggest that Zn2+ binding to TTR at neutral pH into ZBS 2 and 3 induces structural modifications that resemble those observed under acidic conditions, where TTR forms amyloid fibrils. Zn2+ binding might trigger TTR aggregation by perturbing the α-helix region and residues nearby. This structural perturbation is intensified at acidic pH, a condition in which the α-helix completely disappears, forming a long loop and exposing a segment of the protein rich in hydrophobic and aromatic residues (78YWKALGISF87, Tyr-78, Trp-79, and Phe-87), which may form hydrophobic and π-π stacking interactions with adjacent protein molecules, as well as hydrogen bonds, leading to aggregation. ZBS 4, which involves amino acids from adjacent subunits, might recruit more Zn2+, which could then bind and stabilize the quaternary structure of the amyloid fibril.
Interestingly, 2H-1H exchange experiments performed with WT-TTR at pH 4.5 (44) and with FAP variants at pH 7 (31) revealed that the CBEF sheet exhibits faster exchange rates than the ADGH sheet, which is the stable core of TTR. Thus, Zn2+ binding, mainly in ZBS 2 and 3, maps precisely to the labile region of the protein: the region around strands E and F.
These structural rearrangements involve the holo-RBP binding region in TTR, leading to a decrease in holo-RBP:WT-TTR affinity (36) (Fig. 3C and supplemental Fig. S3). Most holo-RBP circulates in serum bound to TTR (2), resulting in an interaction with mutual benefits such as the avoidance of RBP clearance, the stabilization of the retinol:RBP complex (2), and prevention of TTR dissociation and amyloid formation (43). Indeed, a 2-fold decrease in affinity observed in the presence of 15 μm Zn2+, the plasmatic concentration of Zn2+, is rather small. However, this small decrease is to be expected, since at this Zn2+ concentration, the structural perturbations caused by Zn2+ binding to TTR are not all complete and thus its affinity for holo-RBP would be only marginally affected. However, in the presence of 50 μm Zn2+, a concentration that is likely physiologically relevant at certain places, the affinity decreases 4–5-fold. In addition, one has to take into account the fact that TTR in the plasma is bound to other ligands, such as the hormone T4, and these other partners may modulate its affinity for holo-RBP when bound to Zn2+.
As shown here (Fig. 6, B, D, and E), Zn2+ binding decreases monomer stability, and this effect is probably related to all structural changes induced by Zn2+ binding. However, Zn2+ binding does not alter WT tetramer stability to a great extent (Fig. 6, A and C). This is expected since the tetrameric contacts are not severely affected by the formation of ZBS 1–3.
Aggregation of TTR presupposes the formation of a monomeric amyloidogenic intermediate, which is aggregation-prone (8). The existence of an altered tetramer with amyloidogenic properties has also been postulated (13, 50). It may be that a tetramer with ZBS 2 and 3 filled with Zn2+ serves as this tetrameric amyloidogenic intermediate, capable of giving rise to amyloid fibrils either directly or by populating a monomeric amyloidogenic species.
In FAP patients, the amyloid deposits of TTR variants are found mainly around basal membranes of Schwann cells, inside the endoneurium of peripheral nerves (5, 6). The myelin sheath accumulates a high concentration of Zn2+ (about 50 μm) (51). In addition, mutations predispose tetramer to dissociation and Zn2+ might trigger amyloidosis by occupying its binding sites in TTR, provoking the structural rearrangement described here. Thus, the high concentration of Zn2+ in the region in conjunction with the enhanced tetramer instability caused by the mutations, might explain the tropism of TTR amyloids to this specific site. The same scenario could be envisioned in the vitreous humor of the eye, where the concentration of Zn2+ is also high (2.4 ± 0.95 mg/liter; 36 ± 14 μm) (52), and is associated with Zn2+-rich TTR amyloid deposits (22) that cause vitreous opacity (53).
Fig. 7 summarizes the main findings of the present study. The concentration of Zn2+ in the plasma is sufficient to fill ZBS 1, which does not impede TTR-holo-RBP interaction and the efficient transport of retinol. However, when TTR encounters a place with an increased Zn2+ concentration, such as the endoneurium, ZBS 2 and 3 are occupied by Zn2+, leading to a structural reorganization in the region around the α-helix, which fluctuates-displaying structural diversity (Fig. 5). This rearrangement interferes with holo-RBP binding or favors protein-protein interactions, which are crucial for TTR aggregation.
FIGURE 7.

Scheme of the main findings of the present study. In the plasma (upper panel), ZBS 1 of TTR is occupied with Zn2+ (gray spheres), and it does not elicit any major structural alteration in TTR conformation, allowing its tight interaction with holo-RBP (orange structure). In the presence of an increased concentration of Zn2+, ZBS 2 and 3 are occupied by Zn2+; this triggers a conformational change mainly in the neighborhood of the α-helix (blown-up image). In the plasma, this change in conformation displaces holo-RBP (upper panel), while in the peripheral nerves it leads to TTR aggregation and fibril formation (lower panel). The intermediate species, which are present in the pathway from Zn2+-bound tetramers into amyloid fibrils, are unknown.
Evidence for the importance of metal ions in amyloidogenic diseases such as Alzheimer (54) and Parkinson disease (55) is growing (56–58); it may be that metal binding is a common feature of amyloids. Thus, a better characterization of the role and control of metal homeostasis in such pathologies could lead to a general therapeutic approach to ameliorate amyloid diseases (59).
Supplementary Material
Acknowledgments
We thank Emerson Gonçalves for technical assistance and Prof. Martha Sorenson for careful and critical reading of this manuscript.
This work was supported by CNPq (INCT-Bioimagem e Biologia Estrutural), CAPES Nanobiotec-Brasil 04-2008, and FAPERJ (to D. F. and L. M. T. R. L.).
The atomic coordinates and structure factors (codes 3GRG, 3GRB, 3GPS, and 3DGD) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1–S6.
- TTR
- transthyretin
- CSP
- chemical-shift perturbation
- FAP
- familial amyloidotic polyneuropathy
- holo-RBP
- holo-retinol-binding protein
- MSA
- multiple sequence alignment
- ZBS
- zinc-binding site
- SSA
- senile systemic amyloidosis.
REFERENCES
- 1.Palha J. A. (2002) Clin. Chem. Lab. Med. 40, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 2.Monaco H. L. (2000) Biochim. Biophys. Acta 1482, 65–72 [DOI] [PubMed] [Google Scholar]
- 3.Sekijima Y., Kelly J. W., Ikeda S. (2008) Curr. Pharm. Des. 14, 3219–3230 [DOI] [PubMed] [Google Scholar]
- 4.Kingsbury J. S., Théberge R., Karbassi J. A., Lim A., Costello C. E., Connors L. H. (2007) Anal. Chem. 79, 1990–1998 [DOI] [PubMed] [Google Scholar]
- 5.Inoue S., Kuroiwa M., Saraiva M. J., Guimarães A., Kisilevsky R. (1998) J. Struct. Biol. 124, 1–12 [DOI] [PubMed] [Google Scholar]
- 6.Sousa M. M., Saraiva M. J. (2003) Prog. Neurobiol. 71, 385–400 [DOI] [PubMed] [Google Scholar]
- 7.Ando Y. (2005) Med. Mol. Morphol. 38, 142–154 [DOI] [PubMed] [Google Scholar]
- 8.Lai Z., Colón W., Kelly J. W. (1996) Biochemistry 35, 6470–6482 [DOI] [PubMed] [Google Scholar]
- 9.Jiang X., Smith C. S., Petrassi H. M., Hammarström P., White J. T., Sacchettini J. C., Kelly J. W. (2001) Biochemistry 40, 11442–11452 [DOI] [PubMed] [Google Scholar]
- 10.Hörnberg A., Eneqvist T., Olofsson A., Lundgren E., Sauer-Eriksson A. E. (2000) J. Mol. Biol. 302, 649–669 [DOI] [PubMed] [Google Scholar]
- 11.Sebastião M. P., Saraiva M. J., Damas A. M. (1998) J. Biol. Chem. 273, 24715–24722 [DOI] [PubMed] [Google Scholar]
- 12.Yang M., Lei M., Bruschweiler R., Huo S. (2005) Biophys. J. 89, 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eneqvist T., Andersson K., Olofsson A., Lundgren E., Sauer-Eriksson A. E. (2000) Mol. Cell 6, 1207–1218 [DOI] [PubMed] [Google Scholar]
- 14.Pasquato N., Berni R., Folli C., Alfieri B., Cendron L., Zanotti G. (2007) J. Mol. Biol. 366, 711–719 [DOI] [PubMed] [Google Scholar]
- 15.Palaninathan S. K., Mohamedmohaideen N. N., Snee W. C., Kelly J. W., Sacchettini J. C. (2008) J. Mol. Biol. 382, 1157–1167 [DOI] [PubMed] [Google Scholar]
- 16.Scott B. J., Bradwell A. R. (1983) Clin. Chem. 29, 629–633 [PubMed] [Google Scholar]
- 17.Okunewick J. P., Schjeide O. A., Carlsen E. N., Hennessy T. G. (1963) Nature 198, 966–968 [DOI] [PubMed] [Google Scholar]
- 18.Gibson R. S., Hess S. Y., Hotz C., Brown K. H. (2008) Br. J. Nutr. 99, Suppl. 3, S14–S23 [DOI] [PubMed] [Google Scholar]
- 19.Wilkinson-White L. E., Easterbrook-Smith S. B. (2007) Biochemistry 46, 9123–9132 [DOI] [PubMed] [Google Scholar]
- 20.Martone R. L., Herbert J. (1993) Amyloid and Amyloidosis 1993: The Proceedings of the VIIth International Symposium on Amyloidosis, pp. 517–519, Parthenon Publishing, Kingston, Ontario, Canada [Google Scholar]
- 21.Herbert J., Martone R. (1993) Neurology 43, A175 [Google Scholar]
- 22.Susuki S., Ando Y., Sato T., Nishiyama M., Miyata M., Suico M. A., Shuto T., Kai H. (2008) Amyloid 15, 108–116 [DOI] [PubMed] [Google Scholar]
- 23.Lashuel H. A., Wurth C., Woo L., Kelly J. W. (1999) Biochemistry 38, 13560–13573 [DOI] [PubMed] [Google Scholar]
- 24.Guimarães B. G., Sanfelici L., Neuenschwander R. T., Rodrigues F., Grizolli W. C., Raulik M. A., Piton J. R., Meyer B. C., Nascimento A. S., Polikarpov I. (2009) J. Synchrotron Radiat. 16, 69–75 [DOI] [PubMed] [Google Scholar]
- 25.Leslie A. G. W. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography, Vol. 26 [Google Scholar]
- 26.Collaborative Computational Project, N. 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 27.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 28.DeLano W. (2008) DeLano Scientific LLC, San Carlos, CA, U.S.A. [Google Scholar]
- 29.Finn R. D., Tate J., Mistry J., Coggill P. C., Sammut S. J., Hotz H. R., Ceric G., Forslund K., Eddy S. R., Sonnhammer E. L., Bateman A. (2008) Nucleic Acids Res. 36, D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bachega J. F. R., Navarro M. V. A. S., Bleicher L., Bortoleto-Bugs R. K., Dive D., Hoffmann P., Viscogliosi E., Garratt R. C. (2009) Proteins 77, 26–37 [DOI] [PubMed] [Google Scholar]
- 31.Liu K., Cho H. S., Hoyt D. W., Nguyen T. N., Olds P., Kelly J. W., Wemmer D. E. (2000) J. Mol. Biol. 303, 555–565 [DOI] [PubMed] [Google Scholar]
- 32.Rance M., Loria J. P., Palmer A. G., 3rd (1999) J. Magn. Reson. 136, 92–101 [DOI] [PubMed] [Google Scholar]
- 33.Keller R. (2004) Optimizing the Process of Nuclear Magnetic Resonance Spectrum Analysis and Computer Aided Resonance Assignment. Ph.D. Thesis, ETH Zurich, Zurich, Switzerland [Google Scholar]
- 34.Ghosh M., Elsby L. M., Mal T. K., Gooding J. M., Roberts S. G., Ikura M. (2004) Biochem. J. 378, 317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salzmann M., Pervushin K., Wider G., Senn H., Wüthrich K. (1998) Proc. Natl. Acad. Sci. U. S. A. 95, 13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zanotti G., Folli C., Cendron L., Alfieri B., Nishida S. K., Gliubich F., Pasquato N., Negro A., Berni R. (2008) FEBS J. 275, 5841–5854 [DOI] [PubMed] [Google Scholar]
- 37.Lima L. M., Zingali R. B., Foguel D., Monteiro R. Q. (2004) Eur. J. Biochem. 271, 3580–3587 [DOI] [PubMed] [Google Scholar]
- 38.Silva J. L., Foguel D., Royer C. A. (2001) Trends Biochem. Sci. 26, 612–618 [DOI] [PubMed] [Google Scholar]
- 39.Bolen D. W., Santoro M. M. (1988) Biochemistry 27, 8069–8074 [DOI] [PubMed] [Google Scholar]
- 40.Auld D. S. (2001) BioMetals 14, 271–313 [DOI] [PubMed] [Google Scholar]
- 41.Kuser P., Cupri F., Bleicher L., Polikarpov I. (2008) Proteins 72, 731–740 [DOI] [PubMed] [Google Scholar]
- 42.Lockless S. W., Ranganathan R. (1999) Science 286, 295–299 [DOI] [PubMed] [Google Scholar]
- 43.White J. T., Kelly J. W. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 13019–13024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu K., Cho H. S., Lashuel H. A., Kelly J. W., Wemmer D. E. (2000) Nat. Struct. Mol. Biol. 7, 754–757 [DOI] [PubMed] [Google Scholar]
- 45.Wishart D. S., Sykes B. D. (1994) J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 46.Valente A. P., Miyamoto C. A., Almeida F. C. L. (2006) Curr. Med. Chem. 13, 3697–3703 [DOI] [PubMed] [Google Scholar]
- 47.Foguel D. (2005) Protein Pept. Lett 12, 245–249 [DOI] [PubMed] [Google Scholar]
- 48.Blake C. C., Geisow M. J., Swan I. D., Rerat C., Rerat B. (1974) J. Mol. Biol. 88, 1–12 [DOI] [PubMed] [Google Scholar]
- 49.Zhang Q., Kelly J. W. (2003) Biochemistry 42, 8756–8761 [DOI] [PubMed] [Google Scholar]
- 50.Ferrão-Gonzales A. D., Palmieri L., Valory M., Silva J. L., Lashuel H., Kelly J. W., Foguel D. (2003) J. Mol. Biol. 328, 963–974 [DOI] [PubMed] [Google Scholar]
- 51.Riccio P., Giovannelli S., Bobba A., Romito E., Fasano A., Bleve-Zacheo T., Favilla R., Quagliariello E., Cavatorta P. (1995) Neurochem. Res. 20, 1107–1113 [DOI] [PubMed] [Google Scholar]
- 52.Coutselinis A., Boukis D., Kalofoutis A. (1977) Clin. Chem. 23, 915–916 [PubMed] [Google Scholar]
- 53.Benson M. D., Kincaid J. C. (2007) Muscle Nerve 36, 411–423 [DOI] [PubMed] [Google Scholar]
- 54.Atwood C. S., Moir R. D., Huang X., Scarpa R. C., Bacarra N. M., Romano D. M., Hartshorn M. A., Tanzi R. E., Bush A. I. (1998) J. Biol. Chem. 273, 12817–12826 [DOI] [PubMed] [Google Scholar]
- 55.Uversky V. N., Li J., Fink A. L. (2001) J. Biol. Chem. 276, 44284–44296 [DOI] [PubMed] [Google Scholar]
- 56.Davis D. P., Gallo G., Vogen S. M., Dul J. L., Sciarretta K. L., Kumar A., Raffen R., Stevens F. J., Argon Y. (2001) J. Mol. Biol. 313, 1021–1034 [DOI] [PubMed] [Google Scholar]
- 57.Jobling M. F., Huang X., Stewart L. R., Barnham K. J., Curtain C., Volitakis I., Perugini M., White A. R., Cherny R. A., Masters C. L., Barrow C. J., Collins S. J., Bush A. I., Cappai R. (2001) Biochemistry 40, 8073–8084 [DOI] [PubMed] [Google Scholar]
- 58.Deng N. J., Yan L., Singh D., Cieplak P. (2006) Biophys. J. 90, 3865–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodríguez-Rodríguez C., Sánchez de Groot N., Rimola A., Alvarez-Larena A., Lloveras V., Vidal-Gancedo J., Ventura S., Vendrell J., Sodupe M., González-Duarte P. (2009) J. Am. Chem. Soc. 131, 1436–1451 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.