

Abstract
Delayed release of neurotransmitter, also called asynchronous release, is commonly observed at synapses, yet its influence on transmission of spike information is unknown. We examined this issue at endbulb of Held synapses, which are formed by auditory nerve fibers onto bushy cells in the cochlear nucleus. Endbulbs from CBA/CaJ mice aged P6–P49 showed prominent delayed release when driven at physiologically relevant rates. In bushy cells from mice before the onset of hearing (P6–P12), spikes were driven by delayed release up to 100 ms after presynaptic activity. However, no such spikes were observed in bushy cells from mice after the onset of hearing (>P14). Dynamic-clamp experiments indicated that delayed release can drive spikes in older bushy cells provided synchronous release is absent, suggesting that activity normally suppresses these spikes. Application of apamin or α-dendrotoxin revealed late spikes in older bushy cells, suggesting that postsynaptic activation of KV1.x and SK channels during spiking suppresses the subsequent effects of delayed release. The developmental upregulation of these potassium channels would be highly adaptive for temporally precise auditory processing. Furthermore, delayed release appeared to influence synchronous neurotransmitter release. Enhancement of delayed release using strontium was correlated with lower firing probability in current clamp and smaller synchronous EPSCs in voltage clamp. EGTA-AM had the opposite effects. These effects were consistent with delayed and synchronous release competing for a single vesicle pool. Thus delayed release apparently has negative presynaptic and postsynaptic consequences at the endbulb, which are partly mitigated by postsynaptic potassium channel expression.
Introduction
Delayed release is the stochastic fusion of synaptic vesicles that continues for 100s of ms after the normal, synchronous EPSC (Barrett and Stevens, 1972; Rahamimoff and Yaari, 1973; Goda and Stevens, 1994; Atluri and Regehr, 1998). It is a major question whether delayed release plays a computational role at synapses, or whether it is a byproduct of synchronous release. Delayed release has been suggested to play a role at inhibitory synapses by enhancing a tonic component of inhibition (Lu and Trussell, 2000; Best and Regehr, 2009). However, its role at excitatory synapses is unknown, and could be very different. Delayed release could directly trigger randomly timed spikes, or interact with synchronous synaptic activity and modulate the timing of spikes. This could affect processing in systems where the timing of spikes encodes information, such as in the auditory system.
We considered the function of delayed release by focusing on the auditory system. Activity in the auditory nerve (AN) would be expected to be ideal for generating delayed release. AN fibers are highly active for long periods (Kiang, 1965; Sachs and Abbas, 1974; Taberner and Liberman, 2005), which would lead to a build-up of residual calcium, which drives delayed release (Zengel and Magleby, 1981; Atluri and Regehr, 1998). AN fibers form synapses, called “endbulbs of Held,” onto bushy cells (BCs) in the cochlear nucleus (Lorente de Nó, 1981; Limb and Ryugo, 2000). BCs play a role in the analysis and localization of sound based on precise timing of spikes. The quantal size at the endbulb of Held is particularly large (Isaacson and Walmsley, 1995). These factors suggest that delayed release could have a major impact on spiking. The impact could be positive, such as by enhancing the likelihood of bushy cell firing, or negative, such as by reducing the precision of evoked spiking.
We measured delayed release and found that it is, indeed, considerable at the endbulb. Current-clamp experiments showed that delayed release could disrupt the timing of evoked spikes as well as drive spikes long after the synchronous train, in immature synapses. However, these spikes did not occur in slices taken from mice after hearing onset. This difference may result from the upregulation of potassium conductances, particularly KV1.x and SK channels. These appear to be activated during normal trains of activity, and suppress the effects of delayed release. Furthermore, manipulations using EGTA-AM and strontium suggested that delayed release contributes to depletion of the synchronous release pool during trains of activation. Current-clamp experiments indicated that this depletion had a measurable impact on firing.
Materials and Methods
Slices were cut from P6–P49 CBA/CaJ mice as described previously (Yang and Xu-Friedman, 2008). Briefly, brains were dissected into low-sodium, ice-cold cutting solution containing the following (in mm): 76 NaCl, 26 NaHCO3, 75 sucrose, 1.25 NaH2PO4, 2.5 KCl, 25 glucose, 7 MgCl2, 0.5 CaCl2, bubbled with 95% O2: 5% CO2 (pH 7.8, 305 mOsm). Near-sagittal slices were cut and then incubated at 30°C for 40 min in standard recording solution containing the following (in mm): 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 20 glucose, 1 MgCl2, 1.5 CaCl2, 4 Na l-lactate, 2 Na-pyruvate and 0.4 Na l-ascorbate bubbled with 95% O2: 5% CO2 (pH 7.4, 310 mOsm). Slices were then maintained at room temperature until recording.
Patch-clamp recordings were performed using an Axopatch 700B amplifier (Molecular Devices) at 34°C in the presence of 10 μm strychnine to block glycine receptors. CPP (5 μm) (Tocris Bioscience) was also present for current-clamp recordings to block NMDA receptors. Recordings were made with 1–2 MΩ pipettes containing (in mm), for voltage clamp, 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, and 1 QX-314, and for current clamp 130 KMeSO3, 10 NaCl, 2 MgCl2, 0.16 CaCl2, 0.5 EGTA, 10 HEPES, 4 Na2ATP, 0.4 NaGTP, and 14 Tris-CrPhos (pH 7.2, 302 mOsm). In voltage clamp, cells were held at −70 mV, with access resistance 3–12 MΩ compensated to 70%. In current clamp, resting potential was maintained at −60 mV between trials by passing up to ±300 pA. BCs were identified in voltage clamp by EPSCs showing paired-pulse depression and having rapid decay kinetics (τ < 0.2 ms), and in current clamp by their response to strong depolarizing current pulses with 1–2 undershooting spikes (Oertel, 1983). AN fibers were stimulated using a glass microelectrode placed 30–50 μm away. Single or paired pulses were applied every 10 s. Train stimuli were applied every 30 s. Stimulation and data collection were done using an ITC-18 (Instrutech Corp.) controlled using custom-written software (mafPC) running in Igor (Wavemetrics). Average results are reported throughout as mean ± SEM.
Delayed release events were detected by their rapid rise. Quantification of rates of delayed release during trains were corrected for the ∼1 ms following stimulation when quantal events could not be detected.
Most chemicals were purchased from Sigma, with the exception of (from Tocris Bioscience) CPP, (from Alomone Labs) α-dendrotoxin, (from Ascent Scientific) ZD7288, XE991, apamin, and charybdotoxin, and (from Invitrogen) EGTA-AM. Apamin and charybdotoxin were applied in the presence of 0.1% BSA. One set of current-clamp experiments used CsMeSO3 in place of KMeSO3. EGTA-AM was applied at 20 μm for 5 min (stock 100 mm in anhydrous DMSO).
For dynamic clamp, BCs were patched with two 3–5 MΩ pipettes containing the current-clamp solution. One electrode measured cell voltage, while the other was used to pass current. The ITC-18 system performs dynamic clamp at 50 kHz for linear conductances such as AMPAR EPSCs. To allow better comparison between cells, the amplitude of synaptic conductances was scaled by the conductance threshold for each cell (Yang and Xu-Friedman, 2009). The average conductance threshold of synapses after the onset of hearing (P14–P49) was 21.4 ± 1.5 nS (N = 21 cells). This compares with an initial EPSC amplitude of 10.3 ± 3.4 nA (N = 39 cells), equivalent to a conductance of 147 ± 49 nS. Thus the average EPSC is ∼7 times the average conductance threshold. To approximate this range, we set the starting conductance of each input in dynamic clamp to 5 times threshold (Yang and Xu-Friedman, 2009). In immature slices (P6–P12), the average endbulb EPSC was much smaller, 2.1 ± 0.3 nA (30.4 ± 4.3 nS, N = 16), but the conductance threshold was nearly the same (24.1 ± 2.7 nS; N = 8). To reflect this difference in relative amplitude in immature slices, we set the starting conductance of each input in dynamic clamp to 1.3 times threshold. To allow arbitrary times for input conductances, we used a model of synaptic depression, as set out in Yang and Xu-Friedman (2009). In these experiments, we modeled four AN fibers, each with identical amplitude and plasticity, and jitter in arrival times of SD = 0.5 ms.
For dynamic clamp of delayed release, we used a stochastic model. The average rate of delayed release was based on the model of Atluri and Regehr (1998), in which delayed release followed an equation of the form: DR(t) = A[Ca2+]i + B [Ca2+]i n ∑ exp(−(t − t i)/τ), where A, B, n, and τ are parameters, and t i are the times of action potentials. Residual calcium, [Ca2+]i, incremented at each stimulus and decayed with a single exponential of 50 ms. We fit this equation to the delayed release profiles from 28 cells, and used principal components analysis to select a representative cell in the middle of the population (see Fig. 2). The delayed release model for this cell (with parameters A = 8.9, B = 10, n = 0.7, τ = 80 ms) was used to set the average delayed release rate, from which the timing of individual delayed release events were generated randomly. To test delayed release double the normal level, A = 17.8 and B = 20. The timing of individual events was convolved with a fixed quantal amplitude based on voltage-clamp measurements of average mEPSC amplitude: 1.7 nS for older synapses (≥P14; mEPSC amplitude = 114 ± 4 pA; N = 37), and 0.5 nS for immature synapses (<P14; mEPSC amplitude = 33.0 ± 3.2 pA; N = 7). BC spikes were discriminated in dynamic-clamp recordings using the derivative of the membrane potential, after subtracting off the predicted passive response of a simple RC model cell. Spike timing was taken as the peak of the first derivative.
Figure 2.

Delayed release is prominent in endbulbs of all ages. A, Representative example of delayed release for a P49 endbulb. Ai, Sample traces in response to AN fiber stimulation in trains (20 pulses at 200 Hz). Insets are magnified views of the later part of the train. Aii, Raster plot of the timing of delayed release events (top) and poststimulus time histogram of the raster data (bottom). Delayed release rates during and after the train are indicated by the open and shaded parts, respectively. The histogram is corrected for the period of the stimulus artifact and synchronous EPSC when delayed release cannot be measured. B, Levels of delayed release are fairly constant over development (r = 0.04, N = 43 cells). Delayed release is quantified as the total number of events for the 400 ms following the train (shaded region in A). C, Levels of delayed release correlate with initial EPSC amplitude (r = 0.24). D, Delayed release rates for a representative endbulb plotted as a histogram. The line is a fit to the data using the model of Atluri and Regehr (1998) (see Materials and Methods). E, Model parameters for 28 cells analyzed similarly to the cell in D. The slices were from P14–P49 mice. The filled circles mark the parameters for the cell in D. F, Principal components analysis of the fit parameters. The first two principal components are plotted for all 28 cells in the dataset. These accounted for 89% of the variance. A cell near the middle of this dataset (filled circle, also shown in D) was selected as the average amount of delayed release.
Results
Effects of delayed release on firing
We examined the effects of delayed release on firing using current-clamp recordings from BCs of different ages. We activated AN fibers in trains of stimuli at different frequencies using an electrode placed in the neuropil of the AVCN. Figure 1 A shows responses of a BC taken from a mouse before the onset of hearing (P10). Most spikes occurred in response to fiber stimulation. After the stimulation ended, there appeared to be considerable delayed release, which was accompanied by occasional spikes (“late spikes”). We did similar experiments in slices taken from mice after the onset of hearing (P14–P19; example in Fig. 1 B). No late spikes were observed in these older BCs, even when delayed release was visible after the train. Average results from before (P9–P10; N = 5) and after (P14–P19; N = 9) the onset of hearing are shown in Figure 1 C. Late spikes were common in young synapses, on average 1–2 spikes/trial, with more at higher frequencies. However, late spikes were not seen in older cells.
Figure 1.

Spikes are triggered in BCs long after the end of a train of synaptic activation in immature synapses, but not in older synapses. A, Example responses of a BC from a P10 slice. Presynaptic activation was done at 100, 200 and 333 Hz (vertical marks). The lower raster plots show the timing of spikes from 5 trials. These cells show the typical BC characteristic of undershooting action potentials (Oertel, 1983). B, Similar experiments in a P16 slice, showing no late spikes. C–F, Spiking behavior was quantified in cells before (P9–P10, N = 5) and after (P14–P19, N = 9) the onset of hearing. Quantified were late spikes (C), as well as spike probability (D), latency (E) and jitter (F) for pulses 11–20 of 20 pulse trains. Asterisks mark features that were significantly different in the older synapses (p < 0.05).
We also evaluated characteristics of spiking during the second half of the train (pulses 11–20) in BCs before and after the onset of hearing. Firing probability was slightly higher in younger BCs than older BCs (Fig. 1 D), and latency was shorter in younger BCs (Fig. 1 E). However, the variability of evoked spikes (“jitter”) was larger in younger BCs (Fig. 1 F).
These differences in response between young and old BCs could arise from a number of sources. Late spikes could be triggered by delayed release or a slowly inactivating conductance, which is less effective at driving spikes in older synapses. Enhanced jitter in younger BCs could result from delayed release modulating spiking timing, or (more trivially) variability in the precision of triggering action potentials in auditory nerve fibers using extracellular stimulation. Another factor is that it is difficult to assess in current-clamp recordings in immature slices whether it is single or multiple endbulbs being stimulated, because AN inputs tend to be smaller. To address these questions, we aimed to do dynamic clamp recordings, so that we could more carefully control the number of activated inputs as well as levels of delayed release.
Delayed release is significant at the endbulb at all ages
To allow realistic dynamic-clamp recordings, we first characterized delayed release using trains of AN fiber stimulation in voltage clamp. For 20 pulse, 200 Hz trains delayed release was considerable (Fig. 2 A). We quantified rates of delayed release by detecting individual events. Delayed release grew during trains, and decayed back to baseline over the following >100 ms. Delayed release was a feature of endbulbs at all ages examined (P7–P49), with variability between endbulbs (Fig. 2 B). This variability correlated with the size of the rested EPSC (EPSC1, r = 0.24; Fig. 2 C). This likely results from variability in the number of release sites and anatomical size of the endbulb. Systematic experiments indicated that delayed release at the endbulb depended on the frequency and duration of stimulus trains (Fig. 2 D; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This is consistent with delayed release being driven by residual calcium, which is expected to build up with increased presynaptic activity. These results indicate that the lack of late spiking in older cells in Figure 1 is not a result of a downregulation in levels of delayed release. On the contrary, delayed release is present in mature synapses in response to physiologically relevant levels of AN activity.
Dynamic clamp of delayed release
To investigate whether delayed release affects spiking in BCs, we used the dynamic-clamp technique. Dynamic clamp is used to mimic synaptic conductances, which are based on conductances recorded in voltage clamp, but are otherwise set by the experimenter. This approach allows us to control delayed release levels independently of synchronous release, and also to examine multiple active inputs. To deliver the synchronous component, we used a model of EPSC conductances during trains that was described by Yang and Xu-Friedman (2009). We mimicked 4 inputs, because mouse BCs normally receive input from as many as 5 endbulbs (Oertel, 1985). Each input fired at an average frequency of 100–333 Hz, with some variability in the timing of the synchronous EPSC (i.e., jitter), that was based on a Gaussian distribution with SD = 0.5 ms. We scaled the initial amplitude of the synchronous EPSCs relative to the threshold conductance, and this scaling depended on the postnatal age (see Materials and Methods). We based the delayed release component on a population of P14–P49 endbulbs, that we quantified and fit to the model of Atluri and Regehr (1998) (Fig. 2 D,E; see Materials and Methods). We used principal components analysis to select an “average” endbulb from this population (Fig. 2 F). We refer to this level of delayed release as “1×” or “normal.” The mEPSC and starting EPSC amplitude were set to values reflective of the specific age of each preparation (see Materials and Methods), but levels of delayed release and depression were held constant to allow direct comparison.
We examined the effects of no delayed release (“0×”), normal, and double (“2×”), while keeping the synchronous component the same. In young synapses (P6–P11), delayed release triggered a large number of late spikes (Fig. 3 A, middle). The late spikes were clearly not triggered nonspecifically by activity during the train, because no spikes were observed in the absence of delayed release (Fig. 3 A, left). We quantified BC spiking more precisely in the second half of the train (i.e., pulses 11–20). Delayed release significantly increased the probability of spiking for the normal condition (1×), and decreased the latency of spikes during the train (Fig. 3 C,D, left). This is consistent with delayed release being generally excitatory. These effects were even stronger when the amount of delayed release was twice normal, and further caused the timing of evoked spikes to be more jittery.
Figure 3.
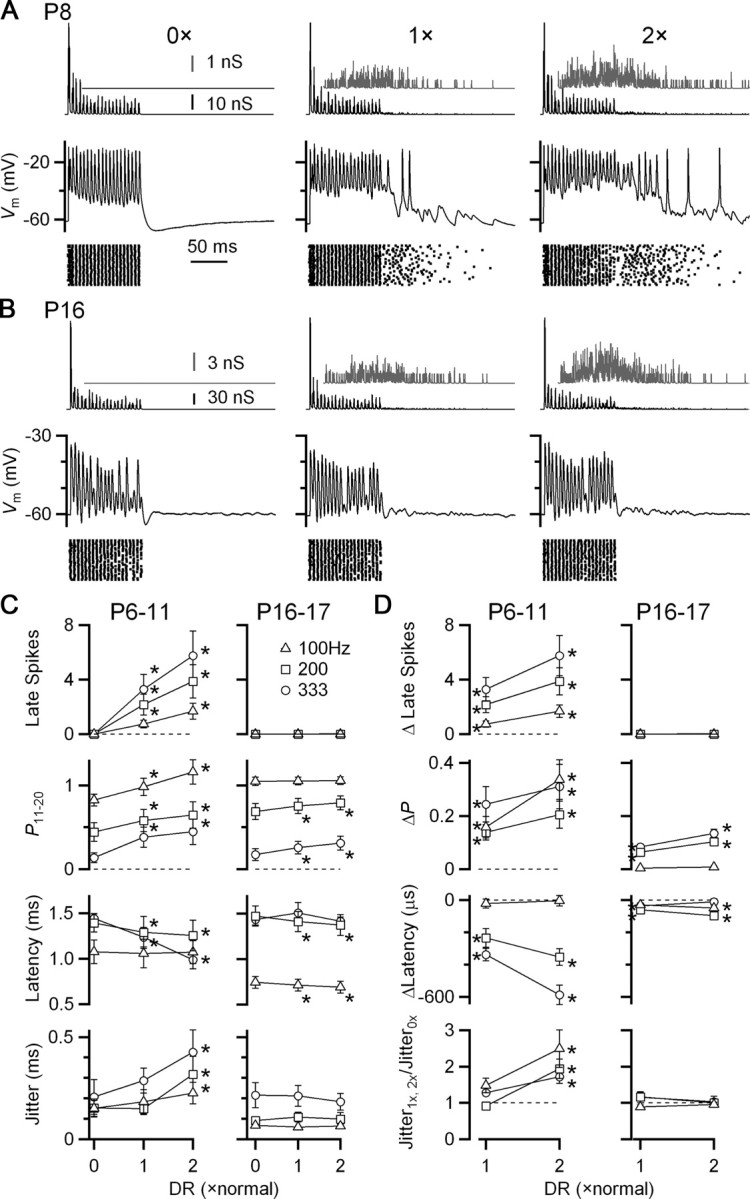
Delayed release drives significant late spikes in immature BCs, but not in older BCs controlled in dynamic clamp. A, Example dynamic-clamp experiment in an immature BC (P8). Dynamic clamp conductance waveforms are shown in the top traces, with the delayed release component isolated and magnified in the gray insets. Bottom traces show the BC response. The stimulus was 4 inputs, each active at an average rate of 200 Hz, with Gaussian-distributed jitter of SD = 0.5 ms, and EPSC amplitude based on a model of depression (see Materials and Methods). Delayed release levels ranged from none (0×) to twice normal (2×). The timing of BC spikes from many similar trials is shown in the rasters. B, Similar experiment in an older BC (P16). No spikes were observed following the synchronous train. C, D, Average effects of delayed release in immature (P6–P11; N = 15) and older (P16–P17; N = 7) BCs. C shows absolute data, while D shows data relative to 0× delayed release. Points significantly different from the 0× condition at α = 0.05 are indicated by asterisks.
Delayed release had weaker effects in older bushy cells (P16–P17). No late spikes were seen in any of the cells examined (Fig. 3 B–D). The probability of firing in the second half of the train increased significantly, and the latency decreased significantly, but the magnitude of the change was smaller than in younger synapses (Fig. 3 C,D). No significant effect on spike jitter was observed at all.
It is particularly striking that no late spikes were observed, even though delayed release is at quite high levels immediately following a train. It is possible that spike threshold has shifted in older synapses so delayed release is simply unable to trigger spikes directly. We tested that directly by conducting dynamic clamp experiments in which we presented only the delayed release component in the absence of any synchronous component. We applied the amount of delayed release that would have been contributed by 1–4 inputs resulting from 20 pulses at 333 Hz. An example response of a P20 BC is shown in Figure 4. As the amount of delayed release built up during the “train,” it was sufficient to cause significant depolarization, and there were occasional spikes elicited. The number of spikes was only appreciable when levels of delayed release equivalent to 4 inputs were applied. In that condition, delayed release contributed 0.5 spikes per trial, on average (Fig. 4 B). Thus, delayed release is by itself fully capable of driving spikes.
Figure 4.

Delayed release by itself is sufficient to drive BC spiking. A, Example dynamic-clamp experiment in a P20 BC showing the effects of delayed release on firing. Normal levels of delayed release were applied that would be observed for a 20 pulse, 333 Hz train in 0–4 inputs. The synchronous release component was excluded for this experiment. Top traces show conductance waveforms. Bottom traces show BC response. B, Average results of similar experiments (N = 5 cells, P18–P20).
The spikes elicited by delayed release alone occurred both during and after the train. While 60% of the spikes occurred during the train, 40% occurred after, which is equivalent to ∼0.2 spikes/trial. While that number is not large, it is striking that there were any spikes at all when none were observed in the presence of synchronous release in older synapses (Fig. 3), even with yet higher levels of delayed release (4 inputs at 2× normal, equivalent to 8 inputs in this experiment). Furthermore, no late spikes were observed in the current-clamp experiments of Figure 1.
Suppression of delayed spikes
The fact that in older synapses delayed release was capable of driving spiking, but no late spikes were observed after trains, suggested to us that the effects of delayed release were being suppressed. We considered that it was possible that some conductance were being activated by the strong depolarizations triggered by the synchronous release component during the train, which inhibited delayed release from triggering spiking. We tested this by doing dynamic clamp experiments similar to those in Figure 3, in the presence of various antagonists against channels that could conceivably be activated during trains. Using dynamic clamp allowed us to avoid potential confounding presynaptic effects of these drugs.
For example, to test the role of low-voltage-activated potassium channels (KV1.x), we applied 50 nm DTX (Fig. 5 A). When no delayed release was present (top trace and raster), there were no late spikes. When normal and twice normal levels of delayed release were included, there were late spikes observed. Delayed release also increased the probability and decreased the latency of firing. These effects were similar to those found in the absence of DTX (Fig. 5 B,C), so we do not attribute them to KV1.x channels. This suggests that KV1.x channels may have a minor effect on how delayed release influences firing during the train, but do play an important role in suppressing the effects of delayed release after presynaptic activity ends in older cells. This difference from younger cells is consistent with work showing that KV1.x channels are upregulated during development (Scott et al., 2005; Bortone et al., 2006).
Figure 5.
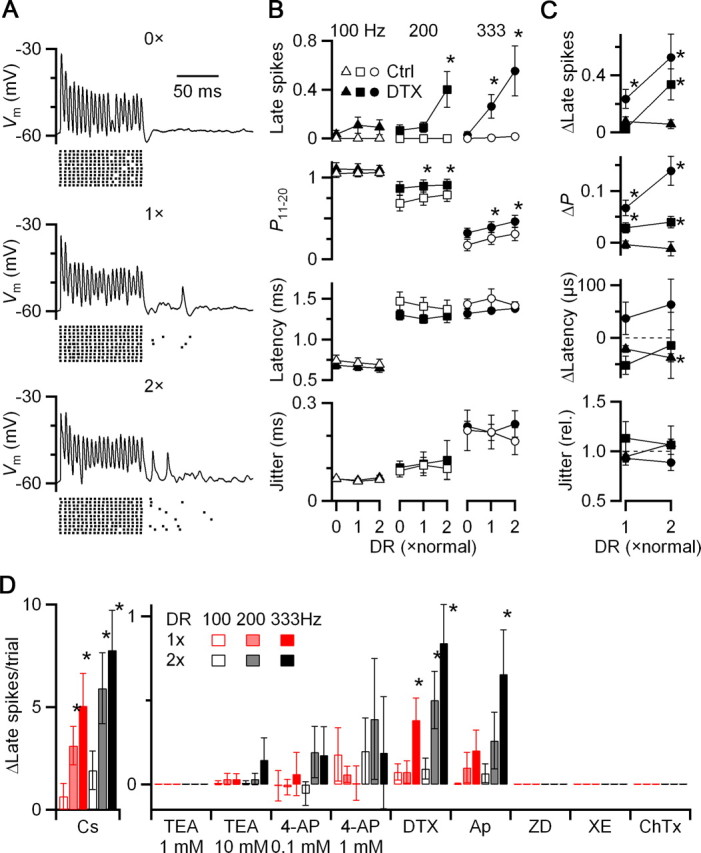
Delayed release can trigger late spikes in post-hearing-onset synapses when KV1.x and SK channels are blocked. A, Example dynamic-clamp experiment of a P16 BC in the presence of 50 nm DTX. Traces show individual responses to conductances similar to those in Figure 3 (i.e., four inputs firing at 200 Hz, each with 0.5 ms jitter). Rasters indicate spike times from additional trials. B, C, Average results from 7 experiments in P15–26 endbulbs. Absolute effects are shown in B, and changes from 0× for DTX are shown in C. DTX caused elevated response probability and shorter latency compared with control across all conditions. In addition, DTX led to the occurrence of late spikes, which were especially prominent at high activation rates. Asterisks mark significant differences from 0× at α = 0.05. D, Summary of effects on late spikes of CsMeSO4 internal (N = 6), 1 mm TEA (N = 5), 10 mm TEA (N = 5), 0.1 mm 4-AP (N = 6), 1 mm 4-AP (N = 6), DTX (50 nm, N = 5), apamin (100 nm, N = 5), ZD7288 (50 μm, N = 3), XE991 (20 μm, N = 3) and ChTX (10 nm, N = 3). CsMeSO4 internal, apamin, and DTX caused a significant increase in late spikes, implicating KV1.x and SK channels.
We further examined the effects of additional channel antagonists. Replacing potassium in the patch pipette with cesium triggered large numbers of late spikes (Fig. 5 D). This indicates that potassium channels are important in suppressing delayed release, although it is difficult to ascribe this to a particular subgroup of potassium channels, because Cs is such a wide-spectrum antagonist. Addition of TEA (1 or 10 mm) or 4-AP (0.1 or 1 mm), had no significant effect on late spikes, although they had clear effects on other aspects of firing (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This suggests that KV3.x channels play little role. No late spikes were seen with 50 μm ZD7288, 20 μm XE991, or 10 nm charybdotoxin, suggesting that KHCN (I h), KCNQ (I m), and BK channels are not involved. Late spikes were elevated in the presence of 100 nm apamin (blocker of SK calcium-sensitive potassium channels). This suggests that calcium influx during the train could activate these channels causing a drop in sensitivity to delayed release. These channels have not previously been suggested to influence BC firing, and mRNA levels for KCNN1–3 appear low in the AVCN (Lein et al., 2007). The conditions of SK-channel activation will require further investigation.
Manipulating delayed release from endbulbs
Our dynamic clamp results of Figure 3 suggested that delayed release could support increased BC firing during the period of auditory nerve activation even in cells after the onset of hearing. We wanted to confirm this prediction by making recordings in current clamp and stimulating real AN fibers. To evaluate the contribution of delayed release to firing, we reduced it using EGTA-AM (Van der Kloot and Molgó, 1993), and enhanced it using strontium (Zengel and Magleby, 1981; Goda and Stevens, 1994; Xu-Friedman and Regehr, 1999, 2000). Then we measured changes in probability, latency, and jitter of BC spikes. When 1 mm SrCl2 was added to the external solution, there was an evident increase in delayed release (Fig. 6 A), but a decrease in the probability of spiking in the latter half of the train in some conditions (Fig. 6 C, top; p < 0.05 for 200 Hz, open squares). By contrast, when the synapses were treated with 20 μm EGTA-AM for 5 min, there was a decrease in delayed release (Fig. 6 B), and a slight increase in the probability of spiking (Fig. 6 C, top). These changes were significant for higher train frequency, which is consistent with the higher levels of delayed release. There were also slight shifts in spike latency with Sr and EGTA-AM (Fig. 6 C, middle; p < 0.05 for 333 Hz, open circle; not significant for other cases). There were no significant changes in spike jitter (Fig. 6 C, bottom; p > 0.24 for all conditions).
Figure 6.

EGTA-AM and Sr influence BC firing. A, B, Example experiments showing effects of Sr (A, P19) and EGTA-AM (B, P19) in response to AN fiber stimulation. Changes in delayed release are clearly visible after the train. C, Effects of Sr (P17–21, N = 7 cells) and EGTA-AM (P17–P19, N = 8) on probability (top), latency (middle), and jitter (bottom) of BC spikes during the steady-state part of the train (pulses 11–20). Data are shown as changes with respect to control. Sr decreased the probability of firing, while EGTA-AM increased it for higher frequency trains. EGTA-AM also decreased the latency for 333 Hz trains. Effects on jitter are not significant. Points significant at the 0.05 level are indicated by asterisks. D, BC input resistance is unchanged by Sr (N = 4 cells) or EGTA-AM (N = 10). Inset, Example BC response to 50 ms current injection pulses. V m is averaged over the last 20 ms of the pulse. Action potential threshold is indicated by the arrow. E, Action potential threshold is unchanged by Sr or EGTA-AM. Spike threshold was identified using the first derivative of traces such as those in the inset in D.
The observed changes in firing were opposite to what we would have predicted based on delayed release alone, which we found to be excitatory in dynamic-clamp recordings (Fig. 3). It is unlikely that the changes in firing are caused by direct effects of Sr or EGTA-AM on postsynaptic intrinsic properties: the input resistance (Fig. 6 D) and spike threshold (Fig. 6 E) in response to current pulses appeared to be unchanged by Sr or EGTA-AM treatment (p > 0.12).
Presynaptic effects of delayed release
We suspected that the effects in current clamp were opposite to those in dynamic clamp because of a change in the synchronous EPSC. To determine whether that was the case, we examined the effects of EGTA-AM and Sr on the synchronous EPSC. Treatment with EGTA-AM produced a significant reduction of delayed release (36 ± 9%, p < 0.05, Fig. 7 A,G) as well as an increase in the synchronous EPSC amplitude late in trains (2.3 ± 0.1% for pulses 11–20; N = 11 cells; p < 0.05; Fig. 7 Ci,E). Addition of SrCl2 led to a significant increase in delayed release (22 ± 5%, p < 0.05, Fig. 7 B,G) as well as a decrease in EPSC amplitude (2.7 ± 0.1% for pulses 11–20; N = 8 cells; p < 0.05; Fig. 7 Cii,E). Neither of these treatments had a significant effect on the initial EPSC amplitude (Fig. 7 E; EGTA-AM: 0.1 ± 0.4%, N = 11, p > 0.47; SrCl2: 1.3 ± 0.5%, N = 8, p > 0.19).
Figure 7.
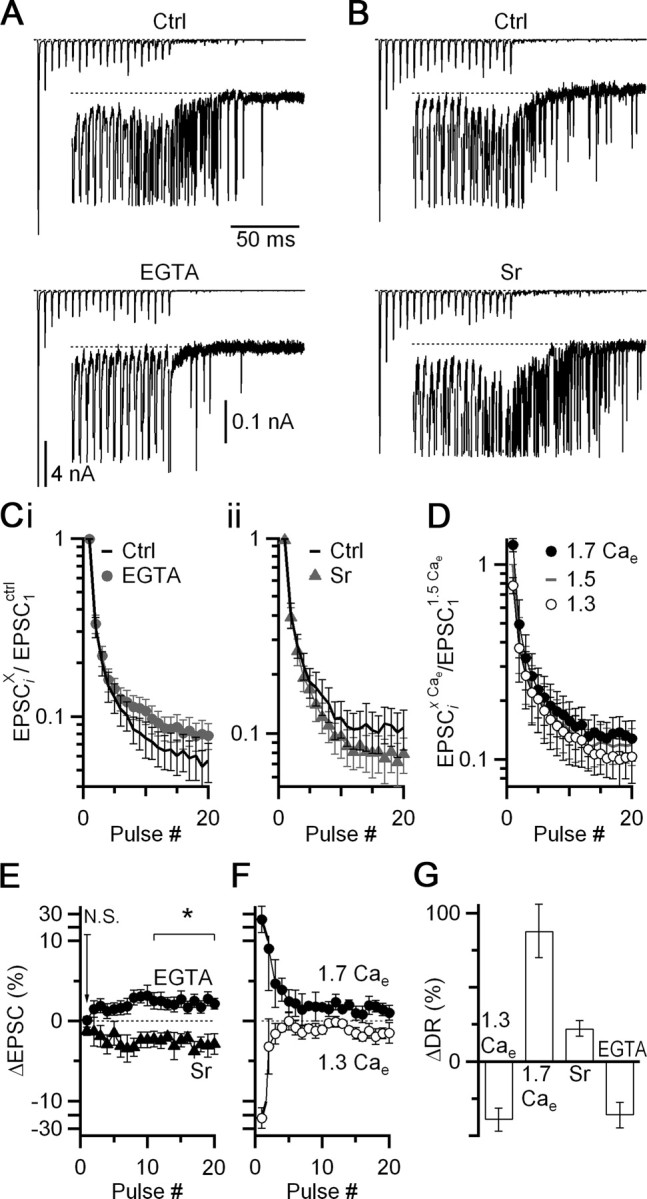
Strontium and EGTA-AM have opposite effects on synchronous and delayed release. A, B, Voltage-clamp recordings of synchronous and delayed release before and after incubating slices in 20 μm EGTA-AM for 5 min (A) or addition of 1 mm SrCl2 to the bath solution (B). Insets show delayed release on an expanded vertical scale, with synchronous EPSCs truncated. Stimuli are 20 pulses delivered at 200 Hz. C, Average EPSC amplitude before and after EGTA-AM (i, N = 8) or Sr (ii, N = 11) treatment. All EPSC amplitudes are normalized within-cell to EPSC1 in control, non-drug conditions. D Effects of changes in Cae on EPSCs during trains (N = 5 cells for all conditions). All EPSC amplitudes are normalized within-cell to EPSC1 in 1.5 Cae. E, F, Changes in the synchronous EPSC resulting from Sr or EGTA-AM treatment (E) or from changes in Cae (F). This is calculated for each treatment X as ΔEPSC = (EPSCi x − EPSCi ctrl)/EPSC1 ctrl. The effects of changing Cae do not match the effects of Sr and EGTA-AM, suggesting that these latter changes do not result from changes in P r. G, Effects of the manipulations in E and F on delayed release. The plot shows the change in total number of delayed release events over the 400 ms following the train, as a percentage of the initial levels. All points are significantly different from 0 (p < 0.05).
We wanted to determine the mechanism for these effects on the synchronous EPSC. We first tested whether these changes in EPSC amplitude resulted from changes in the probability of neurotransmitter release (P r). The reduction in depression by EGTA-AM could result from increased buffering of the peak calcium signal that drives release, and thereby a decrease in P r. The opposite effect of Sr on depression could result from increased divalent cation levels that drive release, and thereby an increase in P r.
We tested whether Sr and EGTA-AM acted through P r by changing external calcium (Cae). However these manipulations had very different effects. When P r was increased by changing from 1.5 mm to 1.7 mm Cae, EPSC1 increased considerably, and later EPSCs in the train showed smaller increases (Fig. 7 D,F). This contrasts with the effects of Sr, where EPSC1 did not increase at all, and subsequent EPSCs decreased (Fig. 7 Cii,E). When P r was decreased by changing from 1.5 mm to 1.3 mm Cae, EPSC1 decreased considerably and later EPSCs in the train showed smaller decreases (Fig. 7 D,F). This contrasts with the effects of EGTA-AM, where EPSC1 did not decrease at all, and subsequent EPSCs increased (Fig. 7 Ci,E). Thus, the effects of Sr and EGTA-AM are not consistent with changes in P r. The changes in Cae showed correlated effects on delayed release (Fig. 7 G).
An alternative possibility is that delayed release affects synchronous release through the number of releasable vesicles (N). It has been suggested that delayed and synchronous release draw on the same pool of releasable vesicles (Hagler and Goda, 2001; David and Barrett, 2003; Otsu et al., 2004). If this is the case, then high levels of delayed release could contribute to depletion of the synchronous EPSC, which could affect BC firing. When delayed release is reduced by EGTA-AM, that leaves more vesicles available for synchronous release, and EPSC amplitudes increase, but only later in trains when delayed release is significant in control. When delayed release is increased by Sr, that leaves fewer vesicles available for synchronous release, and EPSC amplitudes decrease, but only later in trains when delayed release builds up. Changing Cae changes P r as well as residual calcium, and therefore has quite different effects.
This hypothesis is supported by additional observations. First, higher levels of delayed release are correlated with greater depression in individual endbulbs (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Second, changes in delayed release resulting from EGTA-AM or Sr treatment are inversely correlated with changes in depression in individual endbulbs (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). Neither Sr nor EGTA-AM affected mEPSC frequency or amplitude (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), suggesting that other aspects of the release apparatus are not affected.
Discussion
We investigated the role of delayed release in driving spikes at the endbulb of Held synapse. Delayed release reaches significant levels at the endbulb, and the quantal conductance is large. In immature endbulbs, delayed release drove spiking long after presynaptic activation had ended, but such late spikes were never observed in endbulbs taken after the onset of hearing. Dynamic clamp experiments indicated that delayed release is capable of driving spikes even in these older cells. However, such spikes appear to be suppressed at least in part by the activation of KV1.x and SK channels triggered by spiking in response to synchronous release. Furthermore, we showed evidence that, by sharing releasable vesicle pools, delayed release effectively contributed to depression of the synchronous EPSC, and thereby reduced the probability of BC firing during the train.
Development of delayed release
Our voltage-clamp results indicate that delayed release is present in the youngest synapses and persists at roughly similar levels long after the onset of hearing. However, its effects on postsynaptic firing are developmentally regulated. In immature synapses, delayed release appears to drive BC spikes long after synchronous activity has ended, but these spikes are eliminated shortly after the onset of hearing. This is not because of a decrease in the strength of delayed release itself. We took particular advantage of the dynamic clamp technique to show that delayed release can cross threshold to trigger spikes, provided there is no synchronous release.
The elimination of spikes driven by delayed release appears to be highly adaptive. Before the onset of hearing, late spikes that are poorly timed to activity in the auditory nerve may have comparatively little impact. At this time, AN activity is driven by spontaneous activity in the cochlea, which is unrelated to sound stimuli (Tritsch et al., 2007; Tritsch and Bergles, 2010), and extra spikes would be unlikely to disrupt the developmental processes acting at this time. However, after the onset of hearing, the auditory nerve carries information about sounds encoded in the precise timing of spikes, which is further refined in BCs (Joris et al., 1994). This timing information is used in such processes as sound localization, which requires precision on the order of 10 μs (Carr, 1993; Oertel, 1999). The spikes triggered by delayed release are randomly timed, and therefore would be expected to interfere with these sorts of processes. Thus, the elimination of spikes triggered by delayed release appears to be highly adaptive for enhancing the precision of sound processing.
During maturation, a number of changes take place in the auditory pathway. One factor is the upregulation of potassium channels (Fitzakerley et al., 2000; Caminos et al., 2005; Bortone et al., 2006). We found that application of DTX and apamin both revealed late spikes. Both these antagonists target channels that would normally be activated during spiking activity, which probably explains their effects. While KV1.x channels have been implicated in driving the highly rectifying nature of BCs (Rothman and Manis, 2003; McGinley and Oertel, 2006), this is the first suggestion that SK channels also play a role in regulating their excitability. Further work will be needed to determine the Ca source that drives their opening. These could include NMDA receptors, which are present in mature BCs (Pliss et al., 2009), but these would not have been involved in these experiments because we conducted all experiments in the presence of CPP, to avoid any direct effect of NMDA receptors on spiking. Therefore, there must be additional calcium sources. Previous work in BCs has emphasized that activation of KV1.x channels reduces the input resistance and decreases the time constant of the cell. We have shown that these channels have additional effects that help to preserve timing information by eliminating late spikes triggered by delayed release.
Vesicle pools
The idea that synchronous and delayed release draw on the same pool of vesicles has been described at a number of synapses (Hagler and Goda, 2001; David and Barrett, 2003; Otsu et al., 2004; Hjelmstad, 2006). Our experiments at the endbulb also support this conclusion. Manipulations of delayed release using Sr and EGTA-AM had opposite effects on delayed and synchronous release. This was not merely due to changes in the probability of neurotransmitter release (P r), as changing external calcium had markedly different effects. In addition, the amount of depression was inversely correlated with levels of delayed release at individual endbulbs.
Our data indicate that this interaction between synchronous and delayed release has functional consequences. During periods of high activity, delayed release increases, which contributes to depression of the synchronous component. At the endbulb, this appears to result in the diversion of vesicles from providing precise timing information to randomly timed excitation that lasts at high levels for 10s of ms. BC firing was sensitive to small manipulations of delayed release during fiber stimulation, and these changes were opposite to those predicted by the delayed release itself, but consistent with the depletion of synchronous EPSC by delayed release (Fig. 6). The experiments described here may underestimate the effects of delayed release on synchronous release. The experimental approaches we used to manipulate delayed release were rather mild to avoid nonspecific effects on the synchronous EPSC and on activity-dependent forms of recovery that are present at the endbulb (Yang and Xu-Friedman, 2008). However, even these small changes appear to have measurable effects on firing, probably because BCs have a very steep input-output relationship (Chanda and Xu-Friedman, unpublished observations).
Why is delayed release present?
Our data provide support for the conclusion that delayed release does not serve a unique, positive function at the endbulb. On the contrary, it appears to have negative consequences both presynaptically (by diverting vesicles from synchronous release) and postsynaptically (in the triggering of poorly timed spikes). At the endbulb and BC, where spike timing and reliability seem to be of great functional importance, one might expect significant selective pressure to reduce delayed release presynaptically, and to suppress its effects postsynaptically.
Indeed, there are adaptations that appear suited to reduce delayed release. Postsynaptically, as discussed above, potassium channel upregulation as BCs develop contributes to the suppression of poorly timed spikes. Presynaptically, AN fibers in rodents express high levels of calretinin (Bazwinsky et al., 2008) (S. Chanda and M. A. Xu-Friedman, unpublished observations), which would be expected to reduce residual calcium, but evidently do not eliminate delayed release. Given the widespread prevalence of delayed release, it seems likely either that it serves some functional role we have not observed, or else that it is linked somehow to synchronous release.
A number of lines of evidence suggest that delayed release is partially separate from synchronous release. A process resembling delayed release is present even when synchronous release is eliminated in synaptotagmin knock-out mice (Geppert et al., 1994). Delayed release has different cooperativity compared with synchronous release at the calyx of Held (Sun et al., 2007). Synapses have been described that appear to naturally lack a major component of synchronous release, yet have delayed release (Best and Regehr, 2009). These observations suggest that it is possible to have delayed release without synchronous release.
Yet, to our knowledge, the converse has not been described at a normal or mutant synapse, i.e., synchronous release without delayed release. It is possible that such an effect has been overlooked, because most studies focus on synchronous release, especially using averaged traces, which tend to obscure delayed release. Alternatively, one might speculate that synchronous release may depend somehow on the mechanisms that drive delayed release.
Footnotes
This work was supported by National Institutes of Health Grant DC008125 to M.A.X.-F. We thank S. Chanda, G. Tao, and J. Trimper for technical help and useful comments. We also thank Dr. M. Slaughter for feedback on the manuscript.
References
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. J Physiol. 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazwinsky I, Härtig W, Rübsamen R. Characterization of cochlear nucleus principal cells of Meriones unguiculatus and Monodelphis domestica by use of calcium-binding protein immunolabeling. J Chem Neuroanat. 2008;35:158–174. doi: 10.1016/j.jchemneu.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Best AR, Regehr WG. Inhibitory regulation of electrically coupled neurons in the inferior olive is mediated by asynchronous release of GABA. Neuron. 2009;62:555–565. doi: 10.1016/j.neuron.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortone DS, Mitchell K, Manis PB. Developmental time course of potassium channel expression in the rat cochlear nucleus. Hear Res. 2006;211:114–125. doi: 10.1016/j.heares.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Caminos E, Vale C, Lujan R, Martinez-Galan JR, Juiz JM. Developmental regulation and adult maintenance of potassium channel proteins (Kv 1.1 and Kv 1.2) in the cochlear nucleus of the rat. Brain Res. 2005;1056:118–131. doi: 10.1016/j.brainres.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Carr CE. Processing of temporal information in the brain. Annu Rev Neurosci. 1993;16:223–243. doi: 10.1146/annurev.ne.16.030193.001255. [DOI] [PubMed] [Google Scholar]
- David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548:425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzakerley JL, Star KV, Rinn JL, Elmquist BJ. Expression of Shal potassium channel subunits in the adult and developing cochlear nucleus of the mouse. Hear Res. 2000;147:31–45. doi: 10.1016/s0378-5955(00)00118-0. [DOI] [PubMed] [Google Scholar]
- Geppert M, Ullrich B, Green DG, Takei K, Daniels L, De Camilli P, Südhof TC, Hammer RE. Synaptic targeting domains of synapsin I revealed by transgenic expression in photoreceptor cells. EMBO J. 1994;13:3720–3727. doi: 10.1002/j.1460-2075.1994.tb06681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagler DJ, Jr, Goda Y. Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol. 2001;85:2324–2334. doi: 10.1152/jn.2001.85.6.2324. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO. Interactions between asynchronous release and short-term plasticity in the nucleus accumbens slice. J Neurophysiol. 2006;95:2020–2023. doi: 10.1152/jn.01149.2005. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B. Counting quanta: direct measurements of transmitter release at a central synapse. Neuron. 1995;15:875–884. doi: 10.1016/0896-6273(95)90178-7. [DOI] [PubMed] [Google Scholar]
- Joris PX, Carney LH, Smith PH, Yin TC. Enhancement of neural synchronization in the anteroventral cochlear nucleus. I. Responses to tones at the characteristic frequency. J Neurophysiol. 1994;71:1022–1036. doi: 10.1152/jn.1994.71.3.1022. [DOI] [PubMed] [Google Scholar]
- Kiang NYS. Discharge patterns of single fibers in the cat's auditory nerve. Cambridge, MA: MIT; 1965. [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Limb CJ, Ryugo DK. Development of primary axosomatic endings in the anteroventral cochlear nucleus of mice. J Assoc Res Otolaryngol. 2000;1:103–119. doi: 10.1007/s101620010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente de Nó R. The primary acoustic nuclei. New York: Raven; 1981. [Google Scholar]
- Lu T, Trussell LO. Inhibitory transmission mediated by asynchronous transmitter release. Neuron. 2000;26:683–694. doi: 10.1016/s0896-6273(00)81204-0. [DOI] [PubMed] [Google Scholar]
- McGinley MJ, Oertel D. Rate thresholds determine the precision of temporal integration in principal cells of the ventral cochlear nucleus. Hear Res. 2006;216–217:52–63. doi: 10.1016/j.heares.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Oertel D. Synaptic responses and electrical properties of cells in brain slices of the mouse anteroventral cochlear nucleus. J Neurosci. 1983;3:2043–2053. doi: 10.1523/JNEUROSCI.03-10-02043.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel D. Use of brain slices in the study of the auditory system: spatial and temporal summation of synaptic inputs in cells in the anteroventral cochlear nucleus of the mouse. J Acoust Soc Am. 1985;78:328–333. doi: 10.1121/1.392494. [DOI] [PubMed] [Google Scholar]
- Oertel D. The role of timing in the brain stem auditory nuclei of vertebrates. Annu Rev Physiol. 1999;61:497–519. doi: 10.1146/annurev.physiol.61.1.497. [DOI] [PubMed] [Google Scholar]
- Otsu Y, Shahrezaei V, Li B, Raymond LA, Delaney KR, Murphy TH. Competition between phasic and asynchronous release for recovered synaptic vesicles at developing hippocampal autaptic synapses. J Neurosci. 2004;24:420–433. doi: 10.1523/JNEUROSCI.4452-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliss L, Yang H, Xu-Friedman MA. Context-dependent effects of NMDA receptors on precise timing information at the endbulb of Held in the cochlear nucleus. J Neurophysiol. 2009;102:2627–2637. doi: 10.1152/jn.00111.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahamimoff R, Yaari Y. Delayed release of transmitter at the frog neuromuscular junction. J Physiol. 1973;228:241–257. doi: 10.1113/jphysiol.1973.sp010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JS, Manis PB. Differential expression of three distinct potassium currents in the ventral cochlear nucleus. J Neurophysiol. 2003;89:3070–3082. doi: 10.1152/jn.00125.2002. [DOI] [PubMed] [Google Scholar]
- Sachs MB, Abbas PJ. Rate versus level functions for auditory-nerve fibers in cats: tone-burst stimuli. J Acoust Soc Am. 1974;56:1835–1847. doi: 10.1121/1.1903521. [DOI] [PubMed] [Google Scholar]
- Scott LL, Mathews PJ, Golding NL. Posthearing developmental refinement of temporal processing in principal neurons of the medial superior olive. J Neurosci. 2005;25:7887–7895. doi: 10.1523/JNEUROSCI.1016-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberner AM, Liberman MC. Response properties of single auditory nerve fibers in the mouse. J Neurophysiol. 2005;93:557–569. doi: 10.1152/jn.00574.2004. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Bergles DE. Developmental regulation of spontaneous activity in the mammalian cochlea. J Neurosci. 2010;30:1539–1550. doi: 10.1523/JNEUROSCI.3875-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–55. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- Van der Kloot W, Molgó J. Facilitation and delayed release at about 0°C at the frog neuromuscular junction: effects of calcium chelators, calcium transport inhibitors, and okadaic acid. J Neurophysiol. 1993;69:717–729. doi: 10.1152/jn.1993.69.3.717. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Presynaptic strontium dynamics and synaptic transmission. Biophys J. 1999;76:2029–2042. doi: 10.1016/S0006-3495(99)77360-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Probing fundamental aspects of synaptic transmission with strontium. J Neurosci. 2000;20:4414–4422. doi: 10.1523/JNEUROSCI.20-12-04414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA. Relative roles of different mechanisms of depression at the mouse endbulb of Held. J Neurophysiol. 2008;99:2510–2521. doi: 10.1152/jn.01293.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA. Impact of synaptic depression on spike timing at the endbulb of Held. J Neurophysiol. 2009;102:1699–1710. doi: 10.1152/jn.00072.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengel JE, Magleby KL. Changes in miniature endplate potential frequency during repetitive nerve stimulation in the presence of Ca2+, Ba2+, and Sr2+ at the frog neuromuscular junction. J Gen Physiol. 1981;77:503–529. doi: 10.1085/jgp.77.5.503. [DOI] [PMC free article] [PubMed] [Google Scholar]