

SUMMARY
A hallmark of infection with the gram-negative pathogen Porphyromonas gingivalis is the induction of a chronic inflammatory response. P. gingivalis induces a local chronic inflammatory response that results in oral inflammatory bone destruction, which manifests as periodontal disease. In addition to chronic inflammation at the initial site of infection, mounting evidence has accumulated supporting a role for P. gingivalis-mediated periodontal disease as a risk factor for several systemic diseases including, diabetes, preterm birth, stroke, and atherosclerotic cardiovascular disease. A growing number of in vitro studies have demonstrated that P. gingivalis infection stimulates cell activation commensurate with expected responses paralleling inflammatory atherosclerotic-type responses. Furthermore, various mouse models have been used to examine the ability of P. gingivalis to stimulate chronic inflammatory plaque accumulation and recent studies have pointed to a pivotal role for innate immune signaling via the Toll-like receptors in the chronic inflammation associated with P. gingivalis infection. In this review we discuss the pathogen and host cell specificity of these responses and discuss possible mechanisms by which this oral pathogen can induce and maintain a chronic state of inflammation at sites distant from oral infection.
Keywords: atherosclerosis, bacterial persistence, immune evasion, innate immunity, Porphyromonas gingivalis, Toll-like receptor
INTRODUCTION
Porphyromonas gingivalis is a primary etiological agent of human periodontal disease, an inflammatory disease that affects approximately 100 million people in the USA. P. gingivalis induces a local chronic host inflammatory response that results in inflammatory bone destruction, which is manifested as periodontal disease (Oliver et al., 1998). In humans, the local chronic inflammatory lesions associated with P. gingivalis infection are initiated at the vascular endothelium in the oral cavity (Pihlstrom et al., 2005). During acute or active stages of periodontal disease, there is an initial neutrophilic cellular infiltrate that switches to a predominating monocytic and lymphocytic cellular infiltrate in chronic lesions. Macrophages in inflamed tissues differentiate to osteoclasts and also accelerate bone resorption through the production of proinflammatory cytokines (Adamopoulos et al., 2006). A number of proinflammatory mediators are expressed in periodontal inflammatory lesions and many of these are also expressed systemically (D'Aiuto et al., 2004). It has been postulated that the ability to control the expression of these proinflammatory mediators is crucial to controlling the bone loss and subsequent pathology associated with P. gingivalis infection.
SYSTEMIC COMPLICATIONS OF P. GINGIVALIS-INDUCED CHRONIC INFLAMMATION
Mounting evidence has accumulated supporting a role for P. gingivalis-mediated periodontal disease as a risk factor for several systemic diseases including atherosclerotic cardiovascular disease (Genco, 1996; Genco et al., 2002; Morrison et al., 1999). Although not all reports conclude that there is an association between P. gingivalis-mediated periodontal disease and atherosclerosis, recent results from the Oral Infections and Vascular Disease Epidemiology Study revealed that there was an association between periodontal pathogens and subclinical atherosclerosis (Desvarieux et al., 2005). Clinical identifiers of periodontal disease including increased probing depth, bleeding on probing, clinical attachment loss, tooth number, and radiographic assessments have been linked with endothelial dysfunction (Amar et al., 2003), aortic valve stenosis (Volzke et al., 2005), and early carotid atherosclerosis (Engebretson et al., 2005; Soder et al., 2005). Furthermore, periodontal treatment studies have been associated with improved endothelial dysfunction (Seinost et al., 2005). In addition to clinical studies, further evidence of linkage comes from reports that P. gingivalis-specific DNA is present in inflammatory atherosclerotic plaques (Padilla et al., 2006).
ANIMAL MODELS OF P. GINGIVALIS-INDUCED ATHEROSCLEROSIS
Inflammation plays a key role in the development/progression of atherosclerosis (Gibson et al., 2008; Libby, 2000) and various animal models have been used to examine the ability of P. gingivalis to stimulate an inflammatory response associated with atherosclerosis. Studies in rabbit and pig models have demonstrated that P. gingivalis challenge accelerates vascular intimal thickening and accelerates atherosclerosis (Brodala et al., 2005; Jain et al., 2003). In a murine apolipoprotein E-deficient (ApoE−/−) hyperlipidemic mouse model, repeated injection of P. gingivalis into the tail vein was demonstrated to accelerate atherosclerosis (Li et al., 2002). In a more biologically relevant ApoE−/− mouse model, we reported that P. gingivalis oral infection accelerates inflammatory atherosclerosis (Gibson et al., 2004). We also demonstrated that the P. gingivalis-accelerated atherosclerosis occurred early after infection and was prevented by immunization before challenge (Gibson et al., 2004; Miyamoto et al., 2006).
Adherence to and invasion of vascular tissue by P. gingivalis likely plays a key role in the processes that lead to stimulation of accelerated atheroma development, as oral challenge of ApoE−/− mice with a P. gingivalis fimA mutant failed to accelerate atherosclerosis (Gibson et al., 2004; Miyamoto et al., 2006). Mice infected with invasive P. gingivalis expressed increased levels of macrophages in the inflammatory lesions and increased expression of cell-associated intercellular adhesion molecule-1, vascular cell adhesion molecule-1, Toll-like receptor 2 (TLR2), and TLR4 early following infection (Gibson et al., 2004, Miyamoto et al., 2006). Although our data support the hypothesis that both the wild-type and the fimA mutant can gain access into the vasculature and to the aorta, the fimA mutant failed to elicit the induction of inflammatory molecules by endothelium, and resembled unchallenged controls (Gibson et al., 2004).
TLR-MEDIATED INFLAMMATORY PATHWAYS AND P. GINGIVALIS INFLAMMATION IN VIVO
Activation of proinflammatory cytokines occurs in part via the TLRs, a family of innate immune recognition receptors that detect conserved microbial patterns and endogenous ligands and play a key role in innate immune signaling and initiating inflammatory responses (Akira et al., 2006). Ligation of these receptors initiates the activation of several transcription factors including nuclear factor-κB, resulting in the expression of a wide array of inflammatory genes. Recent studies have established that in humans, polymorphisms in genes encoding TLRs and genes involved in subsequent signaling cascades are associated with atherosclerosis (Bjorkbacka, 2006; Hansson et al., 2006; Kiechl et al., 2002; Pryshchep et al., 2008). Animal studies employing hyperlipidemic mice have shown that TLR2, TLR4, and the downstream signaling molecules myeloid differentiation factor (MyD88) and interleukin-1 receptor-associated kinase 4 play an important role in the progression of atherosclerosis (Bjorkbacka et al., 2004; Liu et al., 2008; Michelsen et al., 2004; Mullick et al., 2005; Rekhter et al., 2008). Using an ApoE−/− mouse model we recently demonstrated that TLR2 contributes to the induction of proinflammatory responses in atherosclerotic lesions in response to infection with P. gingivalis (Hayashi et al., 2010). P. gingivalis infection in mice possessing functional TLR2 induced the accumulation of macrophages, and inflammatory mediators including CD40, interferon-γ and the proinflammatory cytokines interleukin-1β (IL-1β), IL-6 and tumor necrosis factor-α (TNF-α) in atherosclerotic lesions. The expression of these inflammatory mediators was reduced in atherosclerotic lesions from P. gingivalis-infected TLR2-deficient (TLR2−/−) mice. Interestingly we also observed that P. gingivalis infection resulted in greater changes in the levels of inflammatory mediators present in atherosclerotic plaque samples compared with the levels of inflammatory mediators observed in serum at the time of sacrifice. These results suggest that while P. gingivalis-induced systemic inflammation may contribute to atherosclerosis, the ability to modulate the levels of inflammatory mediators present in atherosclerotic plaques may ultimately be responsible for the induction of atherosclerosis. We also observed that while the absence of TLR2 diminished atherosclerosis in response to P. gingivalis infection, a TLR2- independent mechanism may also play a role in P. gingivalis-mediated inflammatory responses in atherosclerosis.
PATHOGEN SPECIFICITY IN INDUCTION OF CHRONIC INFLAMMATION AND ATHEROSCLEROSIS
We recently performed studies to determine if chronic bacterial infection and systemic inflammation are sufficient to drive atherosclerosis. In these studies we orally challenged groups of ApoE−/− mice with Helicobacter pylori, an organism that causes gastritis and peptic ulcers (Kusters et al., 2006) or P. gingivalis. Gastritis and oral bone loss were observed only in the groups of mice challenged with H. pylori or P. gingivalis, respectively (Fig. 1A–C and data not shown). Mice challenged with P. gingivalis exhibited increases in the distance between the alveolar bone crest to the cementum–enamel junction, relative to uninfected mice (P < 0.05), whereas in mice orally challenged with H. pylori the oral bone measurements resembled those of the control group (data not shown). Histological evaluation of stomach tissue cryosections revealed that ApoE−/− mice challenged with H. pylori presented with a characteristic submucosal cellular infiltrate (Fig. 1B). Viable H. pylori were isolated from stomach tissues (data not shown) confirming bacterial colonization. As expected, we did not observe a cellular infiltrate in gastric tissue samples obtained from P. gingivalis-infected ApoE−/− mice, and these tissues resembled the unchallenged, age-matched controls (Fig. 1C). Serum analysis revealed elevated levels of H. pylori-specific and P. gingivalis-specific immunoglobulin G only in the groups of mice challenged with the respective pathogen (data not shown). Morphometric analysis of digital micrographs following Sudan IV staining revealed that ApoE−/− mice challenged with P. gingivalis possessed significantly more atherosclerotic plaque in the aortic arch than either the H. pylori-infected or unchallenged groups of animals (P < 0.05 and P < 0.05, respectively; Fig. 1D–G). In contrast, Sudan IV staining of the aorta of H. pylori-infected ApoE−/− mice revealed similarity with the unchallenged control group. These results indicate that there is bacterial specificity in pathogen-induced chronic infection and acceleration of atherosclerosis. Analysis of sera from mice challenged with H. pylori and P. gingivalis revealed that only the ApoE−/− mice challenged with P. gingivalis expressed elevated serum levels of the proinflammatory cytokine IL-1β compared with unchallenged or H. pylori-challenged mice (Fig. 1H). Other studies have reported that the IL-1 pathway plays an important role in pathogen-accelerated atherosclerosis. Using mice that were heterozygous at the apoE allele (ApoE+/−) placed on high-fat diet and deficient in IL-1 receptor it was reported that P. gingivalis challenge did not accelerate atherosclerotic plaque (Chi et al., 2004). Despite several differences in assay conditions between our present study and the study of Chi et al., including diet, ApoE, and IL-1 receptor alleles, collectively these studies support a role for the IL-1 pathway in P. gingivalis- accelerated atherosclerosis.
Figure 1.
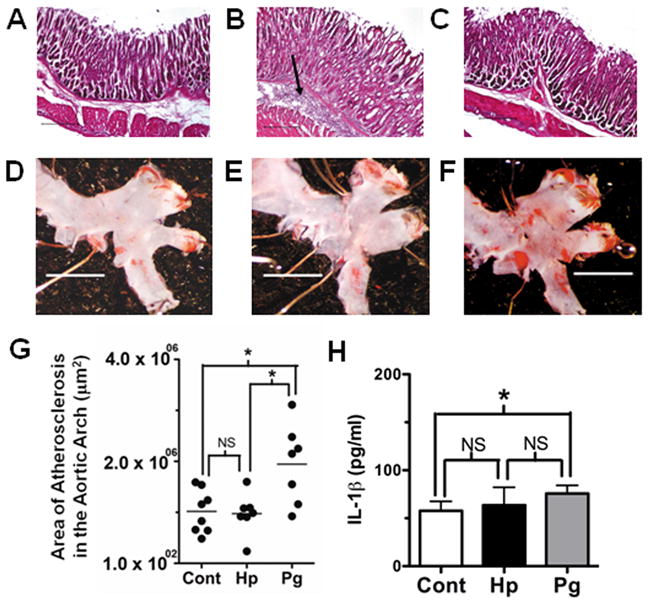
Chronic infection is established in apolipoprotein A-deficient (ApoE−/−) mice challenged with Porphyromonas gingivalis and Helicobacter pylori, yet only P. gingivalis-infected mice present with acceleration of atherosclerosis. Stomach tissue from representative uninfected (A), H. pylori-infected (B), and P. gingivalis-infected (C) mice reveal that H. pylori-infected mice (n = 8/group) present with submucosal cellular infiltrate (arrow); representative micrographs of Sudan IV staining for lipids (red) on the intimal surface of the aortic arch region from uninfected (D), H. pylori-infected (E), and P. gingivalis-infected (F) mice show increased staining only in P. gingivalis-challenged mice, which was confirmed by morphometric analysis (G). Horizontal line denotes the group median and black circle represents an individual mouse. Serum levels of the proinflammatory cytokine interleukin-1β (IL-1β) as measured by enzyme-linked immunosorbent assay (H); *P < 0.05 by analysis of variance with Dunn’s multiple comparisons; NS = not significantly different; scale bars in A–C = 25 μm, and D–F = 1 mm.
P. GINGIVALIS INDUCED INFLAMMATORY SIGNALING CASCADES
Both P. gingivalis and defined antigens of this organism induce a potent inflammatory response in various host cells and use both TLR2 and TLR4 for host cell signaling. However, the innate immune signaling pathways used by P. gingivalis are both host cell type and bacterial ligand specific. P. gingivalis lipopolysaccharide (LPS) has been demonstrated to activate host cells through both TLR2 and TLR4 and this may be dependent on the expression of various forms of P. gingivalis lipid A (Darveau et al., 2004). The major and minor fimbriae proteins of P. gingivalis induce the expression of proinflammatory cytokines and use both TLR2 and TLR4 for this response (Gibson et al., 2008; Hajishengallis et al., 2004, 2006). The major fimbriae use TLR1 or TLR6 for cooperative TLR2-dependent activation of transfected cell lines. We have demonstrated that fimbriae bind to TLR2 and signal through TLR2. Although fimbriae did not bind to TLR4 they were capable of signaling through TLR4 in the presence of MD2 (Davey et al., 2008).
Cell-specific innate immune signaling is the result of multiple factors including the pattern recognition receptors expressed by these cells, the location of these receptors, the adaptors expressed by these cells, whether or not they are phagocytic or non-phagocytic cells, and how they process microbes or their antigens (Harari et al., 2006). There also is evidence that tissue specificity can influence host cell signaling to microbial stimulation. We postulate that bacterial infection mediates inflammatory responses that involve specific innate immune pathways in defined host cells and that this can impact inflammatory outcomes in chronic inflammation. In addition to promoting inflammatory responses to P. gingivalis, recent studies point to cell-specific innate immune pathways, which function to control persistence of the organism. We predict that activation of signaling cascades leading to inflammatory cytokine expression will proceed independently of pathways leading to bacterial uptake and the generation of bactericidal molecules. A number of investigators have examined possible immune evasion strategies of P. gingivalis, which could potentially contribute to bacterial persistence and chronic inflammation. These are discussed below in the context of the specific cell type in which this has been examined.
P. GINGIVALIS PERSISTENCE AND INFLAMMATION IN DENDRITIC CELLS
Long-lived inflammatory cells such as dendritic cells, strategically poised along portals of entry, sample the local microenvironment and interact initially with P. gingivalis in the oral mucosa. Dendritic cells have a critical role in determining the nature of the immune reactions and in fine-tuning the balance between tolerance and the induction of inflammation. A number of studies have established that P. gingivalis can survive and replicate in dendritic cells. P. gingivalis expressing the major fimbriae are more efficient than fimbriae-deficient P. gingivalis in entering dendritic cells and in inducing a T helper type 1 inflammatory response (Jotwani et al., 2004). The P. gingivalis binds to dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) via the minor fimbriae protein, is internalized and routed in large numbers to intracellular vesicles of dendritic cells (Zeituni et al., 2009). These investigators also established that major fimbriae are immunostimulatory and the minor fimbriae are immunosuppressive and they proposed that the ability of P. gingivalis to uncouple dendritic cell maturation from the cytokine response could contribute to bacterial persistence. In addition, it is intriguing to postulate that P. gingivalis is trafficked to the atherosclerotic lesion via these immune cells including monocytes/macrophages or dendritic cells and that this may contribute to bacterial persistence in vivo (Fig. 3) (Hajishengallis et al., 2008b; Wang et al., 2007).
Figure 3.

Model for Porphyromonas gingivalis systemic infection and acceleration of atherosclerosis. 1. Platelets, monocytes, and dendritic cells are depicted in circulation. 2. Monocytes and dendritic cells are depicted in the oral mucosal lesion. Vaccination prevents oral colonization whereas antibiotics decrease the bacterial burden. Platelets, monocytes, dendritic cells, and foam cells are depicted at the atheroma. CAMs, cell adhesion molecules; IFN, interferon; IL, interleukin; Mφ, macrophages; LDL, low-density lipoprotein; PD, periodontal diseases; SE, sulcular epithelium; SRs, scavenger receptors; TNF, tumor necrosis factor; VE, vascular endothelium.
P. GINGIVALIS-INDUCED PLATELET ACTIVATION
Platelet aggregation is important in the formation of atherosclerotic plaque (Ross, 1993). The expression of functional TLRs in platelets provides a possible mechanism by which platelets interact with bacteria and further highlights the importance of platelets in the immune and inflammatory responses. We recently demonstrated that in mice in vivo challenge with live P. gingivalis induced a platelet-dependent proinflammatory response and this response was significantly reduced in TLR2-deficient mice (Blair et al., 2009). A role for fimbriae in P. gingivalis-mediated platelet aggregation was also confirmed using human platelets. As shown in Fig. 2, enhanced platelet aggregation was observed when platelets were incubated with wild-type P. gingivalis (381), but not with a major fimbriae mutant (DPG3). We also observed that wild-type P. gingivalis, but not DPG3, induced human platelet neutrophil aggregation in vitro (Fig. 2B). The ability of P. gingivalis to induce platelet aggregation via fimbriae-mediated stimulation may play an important role in P. gingivalis-mediated inflammatory responses leading to atherosclerosis. It has also been suggested that platelets may function to transport P. gingivalis to the atheroma, although bacterial persistence in platelets has not been examined in detail.
Figure 2.

Porphyromonas gingivalis induces human platelet aggregation. (A) P. gingivalis 381 (P.g) or DPG3 (DPG3) at 108 colony-forming units (CFU)/ml, was added to a suspension of human platelets (2 × 108/ml) and stirred continuously for 10 min in a PAP-4 aggregometer. The % aggregation relative to a collagen control is indicated. *P < 0.001 vs. collagen control. (B) P. gingivalis 381 (P.g) or DPG3 (DPG3) (108 CFU/ml) was added to suspensions of human whole blood (final platelet concentration, 2 × 108/ml) and stirred continuously in a whole blood aggregometer and formation of platelet neutrophil aggregates was determined by flow cytometry. Black = no P. gingivalis addition; gray = + P. gingivalis. *P < 0.001 vs. unchallenged.
P. GINGIVALIS PERSISTENCE AND INFLAMMATION IN ENDOTHELIAL CELLS
Local and distant chronic inflammatory lesions associated with P. gingivalis infection are initiated at the vascular endothelium (Moughal et al., 1992). P. gingivalis invades endothelial cells, where it survives, replicates, and induces a proinflammatory response within these cells (Gibson et al., 2006, 2008). In primary human arterial endothelial cells (HAEC) of the coronary artery P. gingivalis can be found within autophagosomes and may use components of the autophagocytic pathway as a means to survive within these cells (Progulske-Fox et al., 1999). P. gingivalis also appears to regulate host genes differently during invasion of HAEC (Chou et al., 2005). Invasive P. gingivalis has also been reported to increases the adhesion of mononuclear leukocytes to aortic HAEC (Roth et al., 2007). We demonstrated in HAEC that P. gingivalis induces an IL-1β response that is dependent on P. gingivalis invasion and precedes the monocyte chemoattractant protein-1 (MCP-1) and IL-8 response (Takahashi et al., 2006). Stimulation of HAEC with purified P. gingivalis fimbriae did not stimulate IL-1β but stimulated IL-8 and MCP-1. In contrast to TNF-α and IL-6, activation and release of IL-1β requires two distinct signals (Mariathasan et al., 2007). The first signal can be triggered by TLR activation via nuclear factor-κB activation, which leads to the synthesis of pro-IL-1β. The second signal involves caspase-1 activation, which is controlled via the inflammasome, a cytosolic signaling complex. Activation and release of IL-1β are both pathogen and host cell type-specific. Although P. gingivalis induces a potent IL-1β response in endothelial cells and macrophages in vitro, how IL-1β is induced in these cells, and how this modulates immune cell functions are not known.
We also demonstrated that P. gingivalis wild-type bacteria, but not the fimA mutant, stimulate TLR expression on the surface of the endothelium and that these primed cells respond to defined TLR-specific ligands (Yumoto et al., 2005). Increased cell surface expression of TLRs on HAEC was not observed after treatment with purified fimbriae. Our results suggest that P. gingivalis modifies the levels of TLRs expressed on the surface of HAEC and that P. gingivalis infection can convert HAEC from TLR ligand hyporesponsive cells to TLR ligand hyper-responsive cells. Chronic and episodic stimulation of the endothelium with P. gingivalis may therefore sensitize the endothelium to microbial ligands or other endogenous TLR ligands and in this way contribute to the progression of atherosclerosis (Fig. 3).
P. GINGIVALIS PERSISTENCE AND INFLAMMATION IN MONOCYTES/MACROPHAGES
Monocytes represent one of the primary cell populations present in chronic periodontal lesions and also play a role in foam cell formation in atherosclerosis. Gene expression profiling studies have demonstrated that macrophages respond differently to live P. gingivalis when compared with LPS or the FimA protein in the context of cytokine secretion and induction of intracellular molecules (Zhou et al., 2005). P. gingivalis stimulation of human or mouse monocytes leads to the expression of TNF-α, IL-1β, MCP-1, IL-6, IL-8, nitric oxide, and prostaglandin E2 and this is dependent on expression of fimbriae (Gibson et al., 2006). Expression of TNF-α and IL-6 is dependent on both TLR2 and TLR4. P. gingivalis fimbriae and LPS require lipid rafts for signaling and use similar yet distinct receptor complexes for interaction with host cells (Hajishengallis et al., 2006). Intracellular survival of P. gingivalis is diminished in TLR2-deficient or CD11b-deficient macrophages, indicating that this pathogen manipulates complement and TLRs for persistence in macrophages (Wang et al., 2007).
P. gingivalis and fimbriae or LPS have also been demonstrated to stimulate TLR2 surface expression on macrophages (Ukai et al., 2008). Neither P. gingivalis fimbriae nor LPS was observed to upregulate TLR4 on mouse macrophages. Increased cell surface expression of TLR2 on mouse macrophages following stimulation with P. gingivalis fimbriae protein was associated with an increase in TNF-α following subsequent challenge with live P. gingivalis. Hence, similar to what was observed in endothelial cells, P. gingivalis-mediated stimulation of TLR expression on monocytes may sensitize these host cells to microbial ligands, or other endogenous TLR ligands, and in this way contribute to atherosclerosis progression. There is also evidence of cross-talk between endothelial cells and monocytes/macrophages resulting in enhanced inflammatory responses via the secretion of soluble mediators that have pleiotropic functions in inflammation.
Inflammation in macrophages that is induced by P. gingivalis also contributes to the formation of foam cells (Giacona et al., 2004), one of the primary cells that make up the atherosclerotic lesion (Libby, 2000; Stary et al., 1994). The accumulation of macrophages within the arterial wall sets the stage for progression of atheroma to more complicated plaques such as vulnerable plaques, i.e. those that are prone to rupture, that are thought to be responsible for occlusive cardiovascular disease (Libby, 2000). Accumulation of lipids inside cells such as macrophages occurs by scavenger receptor (SR) -mediated uptake of modified LDL species (Moore et al., 2006). The SRs are a broad group of surface expressed trans-membrane, multi-domain proteins that function as pattern recognition receptors. They are regulated on the surface of macrophages in response to stimulation, and demonstrate broad ligand specificity for polyanionic molecules. SRs also bind gram-negative and gram-positive bacteria, bacterial structures such as LPS and lipoteichoic acid, as well as advanced glycation end products.
CELL SPECIFICITY OF P. GINGIVALIS-INDUCED INFLAMMATORY RESPONSES EXAMINED IN VIVO
Although the focus of this review is on P. gingivalis-mediated chronic inflammation and atherosclerosis, it is worth briefly discussing the cell specificity of P. gingivalis-induced inflammation in the context of other inflammatory outcomes of infection. A recent study examined the role of TLR2 in the control of P. gingivalis acute pulmonary infection (Hajishengallis et al., 2008a). These investigators observed that TLR2-deficient mice exhibited reduced proinflammatory responses in the lung and had impaired clearance of P. gingivalis compared with wild-type mice, but that the levels of macrophages and polymorphonuclear cells in the lungs were similar in both strains of mice. These authors concluded that TLR2 plays an important role in clearance of P. gingivalis in acute lung infection. However, we and others have demonstrated that TLR2 mediates destructive effects in oral infection and oral bone loss (Gibson et al., 2008) and in atherosclerosis (Hayashi et al., 2010; Madan et al., 2008). Hence, TLR2 may play diverse and contextual roles in P. gingivalis infections exacerbating certain chronic inflammatory conditions (oral bone loss and atherosclerosis) while providing protection against acute conditions (pulmonary infection). This may also explain recent data in a mouse subcutaneous infection model in which mice deficient in the TLR adaptor proteins MyD88 and Toll-IL-1 receptor domain-containing adaptor-inducing interferon-β were shown to exhibit similar proinflammatory cytokine responses following challenge with P. gingivalis (Burns et al., 2010). However, MyD88-deficient mice exhibited a deficiency in the ability to clear P. gingivalis systemic infection in this model in which the majority of cells encountering P. gingivalis following subcutaneous challenge are neutrophils. The role of TLR2 and MyD88 in promoting or clearing infection may therefore depend on the particular cell types present at the site of infection. These studies further highlight that activation of signaling cascades leading to inflammatory cytokine expression may proceed independently of pathways leading to bacterial uptake and the generation of bactericidal molecules, and may be related to the specificity of sites of bacterial colonization and inflammatory responses.
P. GINGIVALIS DISSEMINATION, PERSISTENCE AND INFLAMMATION LEADING TO ATHEROSCLEROSIS
The shift from periodontal health to periodontal disease involves a complex series of events that stem from colonization with P. gingivalis in the subgingival plaque and development of an inflammatory nidus that leads to periodontal disease. We propose two non-exclusive mechanisms by which P. gingivalis oral infection can induce and maintain inflammation at sites distant from oral infection (Fig. 3).
1
P. gingivalis disseminates in circulation (blood or lymphatics) where it interacts with platelets and vascular endothelial cells. The P. gingivalis binds to the vascular endothelium via major and minor fimbriae using a lipid raft-based cell receptor that is comprised of CAM/TLR/CD36. The infected vascular endothelium responds by the production of cytokines, chemokines, and surface molecules. P. gingivalis and antigens in the vascular intima serve as part of the stimulus that drives naive immune cell localization/activation. In support of this pathway we previously reported that P. gingivalis could be detected by 16S polymerase chain reaction in both blood and aortic tissue during the oral infection period, but not at later time points after bacterial challenge (Gibson et al., 2004). Polymerase chain reaction analysis also revealed detection of the P. gingivalis fimA mutant in both blood and aortic tissue, although the fimA mutant did not induce atherosclerosis. Although we have not detected P. gingivalis by culture techniques in aortic tissue, this does not exclude the possibility that viable organisms are present. Based on our earlier studies in other mouse model systems we propose that P. gingivalis can localize to atherosclerotic lesions. Using a subcutaneous chamber model we have previously determined that P. gingivalis can be cultured from chamber exudates, heart, liver, kidney, and spleen 3 days after infection (Genco et al., 1991, 2002; Mydel et al., 2006). Using the chamber model, we established that P. gingivalis infection results in significant bacteremia with detection of 2 × 103 to 3 × 103 colony-forming units of P. gingivalis in blood samples reported, as well as high levels of LPS (Mydel et al., 2006). Likewise, other investigators have detected viable P. gingivalis in the lung following intratracheal inoculation (Hajishengallis et al., 2008a). It therefore appears that in vivo P. gingivalis can survive in blood and host tissues. What remains to be determined is if P. gingivalis colonizes aortic tissue for extended periods of time leading to inflammatory cascades or if P. gingivalis transiently colonizes and sensitizes the endothelium via stimulation of innate immune receptors, which then respond to other endogenous ligands.
2
P. gingivalis enters immune cells such as monocytes/macrophages or dendritic cells in the diseased/inflamed oral mucosal lesion, these cells then leave the inflamed tissues of the oral cavity, enter the circulation, localize, and diapedese into the vascular intima at sites of activated vascular endothelium (pathogen-activated or activated by inflammation associated with atherosclerosis). Rupture-prone unstable plaques in particular, are infiltrated with high numbers of activated SIGN+ myeloid dendritic cells (Soilleux et al., 2002). Although the sources of these dendritic cells in atherosclerosis are not clear, lesions obtained from patients with chronic periodontitis contain an intense infiltrate of SIGN+ dendritic cells (Jotwani et al., 2003).
Immune cells activated by P. gingivalis drive modification of low-density lipoproteins to forms such as ox-LDL that are taken up by macrophages, leading to foam cell formation. These atherosclerotic lesions progress, develop a necrotic core, and when ruptured form an occlusive thrombus leading to a downstream ischemic event. It has been shown experimentally that vaccination to prevent oral bone loss also mitigates P. gingivalis acceleration of atherosclerosis. Use of antibiotics as part of aggressive periodontal clinical treatment effectively reduces the overall pathogen burden at the site of treatment and would result in lowering the risk for development of atherosclerosis.
FUTURE DIRECTIONS
Although epidemiological and experimental studies link infectious agents with the development of inflammatory atherosclerosis, the precise mechanisms contributing to this link have remained elusive. Recent seminal studies with the oral pathogen P. gingivalis have defined pathogen-specific persistence strategies and the modulation of cell-specific innate immune inflammatory pathways required for initiation and maintenance of P. gingivalis induced chronic inflammation at sites distant from oral infection. Despite these significant studies a number of questions remain (Table 1). We encourage future studies to examine both P. gingivalis and host-cell-specific pathways for induction of inflammatory cascades and bacterial persistence. These studies will advance our understanding of the roles of specific immune cells that participate in chronic inflammation and atherosclerosis in response to oral infection with P. gingivalis, and will provide a promising avenue for novel therapies for chronic inflammatory diseases induced by bacterial infection.
Table 1.
Questions that remain and areas that should be pursued in future studies
| Pathogen |
| Does P. gingivalis disseminate to the atherosclerotic lesion? |
| Does P. gingivalis colonize within the atherosclerotic lesion? |
| What immune evasion strategies allow P. gingivalis to survive? |
| Host |
| What host cells are used for innate immune signaling? |
| What proinflammatory mediators are required? |
| Are there different pathways for bacterial clearance versus inflammatory mediator production? |
| Overall |
| Are there differences in pathogen-mediated and host-mediated mechanisms required for oral colonization and inflammation versus colonization and inflammation at sites distant from infection? |
Acknowledgments
This study was supported by National Institutes of Health Public Health Service grant HL080387 (C.A.G.). We acknowledge Drs. Jane Freedman, Lea Beaulieu, and Price Blair for the human platelets studies.
References
- Adamopoulos IE, Sabokbar A, Wordsworth BP, Carr A, Ferguson DJ, Athanasou NA. Synovial fluid macrophages are capable of osteoclast formation and resorption. J Pathol. 2006;208:35–43. doi: 10.1002/path.1891. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Amar S, Gokce N, Morgan S, Loukideli M, Van Dyke TE, Vita JA. Periodontal disease is associated with brachial artery endothelial dysfunction and systemic inflammation. Arterioscler Thromb Vasc Biol. 2003;23:1245–1249. doi: 10.1161/01.ATV.0000078603.90302.4A. [DOI] [PubMed] [Google Scholar]
- Bjorkbacka H. Multiple roles of Toll-like receptor signaling in atherosclerosis. Curr Opin Lipidol. 2006;17:527–533. doi: 10.1097/01.mol.0000245258.25387.ec. [DOI] [PubMed] [Google Scholar]
- Bjorkbacka H, V, Kunjathoor V, Moore KJ, et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med. 2004;10:416–421. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- Blair P, Rex S, Vitseva OL, et al. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. 2009;104:346–354. doi: 10.1161/CIRCRESAHA.108.185785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodala N, Merricks EP, Bellinger DA, et al. Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. 2005;25:1446–1451. doi: 10.1161/01.ATV.0000167525.69400.9c. [DOI] [PubMed] [Google Scholar]
- Burns E, Eliyahu T, Uematsu S, Akira S, Nussbaum G. TLR2-dependent inflammatory response to Porphyromonas gingivalis is MyD88 independent, whereas MyD88 is required to clear infection. J Immunol. 2010;184:1455–1462. doi: 10.4049/jimmunol.0900378. [DOI] [PubMed] [Google Scholar]
- Chi H, Messas E, Levine RA, Graves DT, Amar S. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model: pharmacotherapeutic implications. Circulation. 2004;110:1678–1685. doi: 10.1161/01.CIR.0000142085.39015.31. [DOI] [PubMed] [Google Scholar]
- Chou HH, Yumoto H, Davey M, et al. Porphyromonas gingivalis fimbria-dependent activation of inflammatory genes in human aortic endothelial cells. Infect Immun. 2005;73:5367–78. doi: 10.1128/IAI.73.9.5367-5378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Aiuto F, Parkar M, Andreou G, et al. Periodontitis and systemic inflammation: control of the local infection is associated with a reduction in serum inflammatory markers. J Dent Res. 2004;83:156–160. doi: 10.1177/154405910408300214. [DOI] [PubMed] [Google Scholar]
- Darveau RP, Pham TT, Lemley K, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey M, Liu X, Ukai T, et al. Bacterial fimbriae stimulate proinflammatory activation in the endothelium through distinct TLRs. J Immunol. 2008;180:2187–2195. doi: 10.4049/jimmunol.180.4.2187. [DOI] [PubMed] [Google Scholar]
- Desvarieux M, Demmer RT, Rundek T, et al. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST) Circulation. 2005;111:576–582. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebretson SP, I, Lamster B, Elkind MS, et al. Radiographic measures of chronic periodontitis and carotid artery plaque. Stroke. 2005;36:561–566. doi: 10.1161/01.STR.0000155734.34652.6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genco CA, Cutler CW, Kapczynski D, Maloney K, Arnold RR. A novel mouse model to study the virulence of and host response to Porphyromonas (Bacteroides) gingivalis. Infect Immun. 1991;59:1255–1263. doi: 10.1128/iai.59.4.1255-1263.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genco R, Offenbacher S, Beck J. Periodontal disease and cardiovascular disease: epidemiology and possible mechanisms. J Am Dent Assoc. 2002;133(Suppl):14S–22S. doi: 10.14219/jada.archive.2002.0375. [DOI] [PubMed] [Google Scholar]
- Genco RJ. Current view of risk factors for periodontal diseases. J Periodontol. 1996;67:1041–1049. doi: 10.1902/jop.1996.67.10.1041. [DOI] [PubMed] [Google Scholar]
- Giacona MB, Papapanou PN, Lamster IB, et al. Porphyromonas gingivalis induces its uptake by human macrophages and promotes foam cell formation in vitro. FEMS Microbiol Lett. 2004;241:95–101. doi: 10.1016/j.femsle.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Hong C, Chou HH, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Ukai T, Genco CA. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Front Biosci. 2008;13:2041–2059. doi: 10.2741/2822. [DOI] [PubMed] [Google Scholar]
- Gibson FC, 3rd, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J Dent Res. 2006;85:106–121. doi: 10.1177/154405910608500202. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Sojar H, Genco RJ, DeNardin E. Intracellular signaling and cytokine induction upon interactions of Porphyromonas gingivalis fimbriae with pattern-recognition receptors. Immunol Invest. 2004;33:157–172. doi: 10.1081/imm-120030917. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Tapping RI, Harokopakis E, et al. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006;8:1557–1570. doi: 10.1111/j.1462-5822.2006.00730.x. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G, Wang M, Bagby GJ, Nelson S. Importance of TLR2 in early innate immune response to acute pulmonary infection with Porphyromonas gingivalis in mice. J Immunol. 2008a;181:4141–4149. doi: 10.4049/jimmunol.181.6.4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Wang M, Liang S, et al. Subversion of innate immunity by periodontopathic bacteria via exploitation of complement receptor-3. Adv Exp Med Biol. 2008b;632:203–219. doi: 10.1007/978-0-387-78952-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- Harari OA, Alcaide P, Ahl D, Luscinskas FW, Liao JK. Absence of TRAM restricts Toll-like receptor 4 signaling in vascular endothelial cells to the MyD88 pathway. Circ Res. 2006;98:1134–1140. doi: 10.1161/01.RES.0000220105.85182.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi C, Madrigal AG, Liu X, et al. Pathogen mediated inflammatory atherosclerosis is mediated in part via TLR2 induced inflammatory responses. J Innate Immun. 2010 doi: 10.1159/000314686. Published Online: May 10, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Batista EL, Jr, Serhan C, Stahl GL, Van Dyke TE. Role for periodontitis in the progression of lipid deposition in an animal model. Infect Immun. 2003;71:6012–6018. doi: 10.1128/IAI.71.10.6012-6018.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jotwani R, Cutler CW. Fimbriated Porphyromonas gingivalis is more efficient than fimbria-deficient P. gingivalis in entering human dendritic cells in vitro and induces an inflammatory Th1 effector response. Infect Immun. 2004;72:1725–1732. doi: 10.1128/IAI.72.3.1725-1732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jotwani R, Pulendran B, Agrawal S, Cutler CW. Human dendritic cells respond to Porphyromonas gingivalis LPS by promoting a Th2 effector response in vitro. Eur J Immunol. 2003;33:2980–2986. doi: 10.1002/eji.200324392. [DOI] [PubMed] [Google Scholar]
- Kiechl S, Lorenz E, Reindl M, et al. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Messas E, Batista EL, Jr, Levine RA, Amar S. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation. 2002;105:861–867. doi: 10.1161/hc0702.104178. [DOI] [PubMed] [Google Scholar]
- Libby P. Changing concepts of atherogenesis. J Intern Med. 2000;247:349–358. doi: 10.1046/j.1365-2796.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- Liu X, Ukai T, Yumoto H, et al. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis. 2008;196:146–154. doi: 10.1016/j.atherosclerosis.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan M, Amar S. Toll-like receptor-2 mediates diet and/or pathogen associated atherosclerosis: proteomic findings. PLoS ONE. 2008;3:e3204. doi: 10.1371/journal.pone.0003204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- Michelsen KS, Wong MH, Shah PK, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Yumoto H, Takahashi Y, Davey M, Gibson FC, 3rd, Genco CA. Pathogen-accelerated atherosclerosis occurs early after exposure and can be prevented via immunization. Infect Immun. 2006;74:1376–1380. doi: 10.1128/IAI.74.2.1376-1380.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43. [DOI] [PubMed] [Google Scholar]
- Morrison HI, Ellison LF, Taylor GW. Periodontal disease and risk of fatal coronary heart and cerebrovascular diseases. J Cardiovasc Risk. 1999;6:7–11. doi: 10.1177/204748739900600102. [DOI] [PubMed] [Google Scholar]
- Moughal NA, Adonogianaki E, Thornhill MH, Kinane DF. Endothelial cell leukocyte adhesion molecule-1 (ELAM-1) and intercellular adhesion molecule-1 (ICAM-1) expression in gingival tissue during health and experimentally-induced gingivitis. J Periodontal Res. 1992;27:623–630. doi: 10.1111/j.1600-0765.1992.tb01746.x. [DOI] [PubMed] [Google Scholar]
- Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mydel P, Takahashi Y, Yumoto H, et al. Roles of the host oxidative immune response and bacterial antioxidant rubrerythrin during Porphyromonas gingivalis infection. PLoS Pathog. 2006;2:e76. doi: 10.1371/journal.ppat.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver RC, Brown LJ, Loe H. Periodontal diseases in the United States population. J Periodontol. 1998;69:269–278. doi: 10.1902/jop.1998.69.2.269. [DOI] [PubMed] [Google Scholar]
- Padilla C, Lobos O, Hubert E, Gonzalez C, Matus S, Pereira M, Hasbun S, Descouvieres C. Periodontal pathogens in atheromatous plaques isolated from patients with chronic periodontitis. J Periodontal Res. 2006;41:350–353. doi: 10.1111/j.1600-0765.2006.00882.x. [DOI] [PubMed] [Google Scholar]
- Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- Progulske-Fox A, Kozarov E, Dorn B, Dunn W, Jr, Burks J, Wu Y. Porphyromonas gingivalis virulence factors and invasion of cells of the cardiovascular system. J Periodontal Res. 1999;34:393–399. doi: 10.1111/j.1600-0765.1999.tb02272.x. [DOI] [PubMed] [Google Scholar]
- Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118:1276–1284. doi: 10.1161/CIRCULATIONAHA.108.789172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekhter M, Staschke K, Estridge T, et al. Genetic ablation of IRAK4 kinase activity inhibits vascular lesion formation. Biochem Biophys Res Commun. 2008;367:642–648. doi: 10.1016/j.bbrc.2007.12.186. [DOI] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Roth GA, Ankersmit HJ, Brown VB, Papapanou PN, Schmidt AM, Lalla E. Porphyromonas gingivalis infection and cell death in human aortic endothelial cells. FEMS Microbiol Lett. 2007;272:106–113. doi: 10.1111/j.1574-6968.2007.00736.x. [DOI] [PubMed] [Google Scholar]
- Seinost G, Wimmer G, Skerget M, et al. Periodontal treatment improves endothelial dysfunction in patients with severe periodontitis. Am Heart J. 2005;149:1050–1054. doi: 10.1016/j.ahj.2004.09.059. [DOI] [PubMed] [Google Scholar]
- Soder PO, Soder B, Nowak J, Jogestrand T. Early carotid atherosclerosis in subjects with periodontal diseases. Stroke. 2005;36:1195–1200. doi: 10.1161/01.STR.0000165916.90593.cb. [DOI] [PubMed] [Google Scholar]
- Soilleux EJ, Morris LS, Trowsdale J, Coleman N, Boyle JJ. Human atherosclerotic plaques express DC-SIGN, a novel protein found on dendritic cells and macrophages. J Pathol. 2002;198:511–516. doi: 10.1002/path.1205. [DOI] [PubMed] [Google Scholar]
- Stary HC, Chandler AB, Glagov S, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89:2462–2478. doi: 10.1161/01.cir.89.5.2462. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Davey M, Yumoto H, Gibson FC, 3rd, Genco CA. Fimbria-dependent activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic endothelial cells. Cell Microbiol. 2006;8:738–757. doi: 10.1111/j.1462-5822.2005.00661.x. [DOI] [PubMed] [Google Scholar]
- Ukai T, Yumoto H, Gibson FC, 3rd, Genco CA. Macrophage-elicited osteoclastogenesis in response to bacterial stimulation requires Toll-like receptor 2-dependent tumor necrosis factor-alpha production. Infect Immun. 2008;76:812–819. doi: 10.1128/IAI.01241-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volzke H, Schwahn C, Hummel A, et al. Tooth loss is independently associated with the risk of acquired aortic valve sclerosis. Am Heart J. 2005;150:1198–1203. doi: 10.1016/j.ahj.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Wang M, Shakhatreh MA, James D, et al. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. 2007;179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- Yumoto H, Chou HH, Takahashi Y, Davey M, Gibson FC, 3rd, Genco CA. Sensitization of human aortic endothelial cells to lipopolysaccharide via regulation of Toll-like receptor 4 by bacterial fimbria-dependent invasion. Infect Immun. 2005;73:8050–8059. doi: 10.1128/IAI.73.12.8050-8059.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeituni AE, Jotwani R, Carrion J, Cutler CW. Targeting of DC-SIGN on human dendritic cells by minor fimbriated Porphyromonas gingivalis strains elicits a distinct effector T cell response. J Immunol. 2009;183:5694–5704. doi: 10.4049/jimmunol.0901030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Desta T, Fenton M, Graves DT, Amar S. Cytokine profiling of macrophages exposed to Porphyromonas gingivalis, its lipopolysaccharide, or its FimA protein. Infect Immun. 2005;73:935–943. doi: 10.1128/IAI.73.2.935-943.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]