

Abstract
STAT3 is a transcription factor that plays a critical role in heart development and protection. New developments in understanding its molecular chemistry have revealed the importance of STAT3 in controlling mitochondrial respiration, independent of its function as a transcription factor, and in modulating inflammatory signaling through interactions with other transcription factors and co-factors. The purpose of this article is twofold. First, we summarize some recent insights into the function of STAT3. Second, we seek to illustrate the complexity of targeting a particular cellular protein for therapeutic purposes and the need to consider context when attempting to decipher the role of a particular signaling pathway in the heart. In this case, inflammation, aging, hypertrophy, and heart failure provide new environments that certainly impact on the functioning of STAT3 and on the gene profile linked to its activation.
Introduction
Various genetic and nongenetic animal models have revealed that STAT3 has a role in protecting the heart from stress resulting from ischemia-reperfusion, pressure-overload hypertrophy, and aging, thereby preventing heart failure.1–4 This transcription factor has been implicated in both early and late ischemic preconditioning, as well as ischemic postconditioning. Clear evidence has been found that gene expression contributes to the protective actions of STAT3 in late ischemic preconditioning and aging. Genes shown to be upregulated are involved in extracellular matrix remodeling, angiogenesis, and protection from oxidants and apoptosis.2 But poorly understood nongenomic actions of STAT3 that likely impact on mitochondrial permeability transition pore opening and NF-κB transcriptional activity may be involved as well. That is assuredly the case in early (classical) preconditioning and postconditioning due to the short time frames involved. As discussed here, differential STAT3 phosphorylation and cellular distribution may contribute to the nongenomic actions of STAT3. Our recent work indicates that the former may impact as well on the particular gene profile linked to STAT3 activation by affecting the association of STAT3 with other transcription factors and cofactors.
Not your everyday transcription factor
STAT3 was initially described as a latent cytoplasmic transcription factor that dimerizes and translocates to the nucleus upon phosphorylation of a tyrosine residue (Y705) near the C-terminus by an activated JAK or Src family kinase.2 STAT3 homodimers or STAT3-STAT1 heterodimers induce the expression of target genes that have a GAS-like element in their promoter. The COOH-terminal region of STAT3 also has a transactivation domain (TAD) that contains a serine residue (S727) that modulates its transcriptional activity (Figure 1). Moving towards the NH2-terminus, other notable features of STAT3 are a linker domain, a DNA binding domain, a coiled–coiled domain that is important in protein–protein interactions, and a NH2-terminal domain. Phosphorylation of S727 within the TAD is generally thought to be essential for maximal STAT3 transcriptional activity by allowing for the recruitment of co-factors, such as the histone acetyltransferase p300. The extent of S727 phosphorylation thus introduces another level of regulation in STAT3-mediated gene expression. There is also evidence that prior STAT3 S727 phosphorylation precludes phosphorylation at Y705, at least in response to the IL-6-type cytokines.2 In any case, STAT3 has been shown to interact independent of DNA binding with several other transcription factors, including NF-κB p65 and p50 to induce or repress gene expression. In most cases, as with p300, these interactions have been mapped to the coiled-coil domain of STAT3, which has been postulated to interact with the TAD. Phosphorylation of S727 may disrupt this intramolecular interaction, thereby providing ready access to the coiled-coil domain. However, a mandatory role for STAT3 S727 phosphorylation in these interactions is not established. Anyway, these observations suggest the possibility that STAT3 may impact gene expression independently of tyrosine phosphorylation through association with other transcription factors or co-factors. Such a scenario was recently proposed as the mechanism responsible for interleukin-10 (IL-10)-dependent NF-κB recruitment to the interleukin 1 receptor antagonist (IL-1ra) promoter in LPS-stimulated monocytes and neutrophils.5 IL-1ra is a negative regulator of the inflammatory response. It was suggested that IL-10 activated-STAT3 enhances IL-1ra promoter hyperacetylation via its association with p300, thereby facilitating accessibility of the LPS-activated NF-κB components to the DNA elements.
Figure 1.
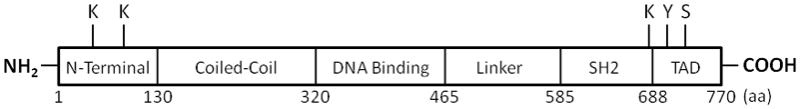
Domain structures of STAT3 showing the key sites for phosphorylation (Y705 and S727) and acetylation (K49, K87, and K685). SH2, src homology 2; TAD, transcription activation domain.
To complicate matters further, others have recently reported that contrary to an older report showing enhanced nuclear export of STAT3 phosphorylated on S727 alone in T lymphoma cells,6 STAT3 is constitutively phosphorylated on S727 in chronic lymphocytic leukemia cells, shuttles to the nucleus, binds DNA, and activates transcription of STAT3 responsive genes – all independently of Y705 phosphorylation.7 No molecular or cellular basis for these noncanonical findings comes to mind, nor is it known what role this process plays in other cell types if it does in fact occur in other cells. In any case, neither S727 nor Y705 phosphorylation would seem to be mandatory for the presence of STAT3 in the nucleus, since constitutive shuttling of STAT3 between the nucleus and cytoplasm has been observed and is attributable to a novel R214/215 sequence in the coiled-coil domain and a conserved sequence element in the DNA binding domain.8,9
Two other recent developments concerning STAT3 warrant mentioning. First, STAT3 itself is a substrate for p300, being acetylated on multiple lysine (K) residues including K685 in the TAD and K49 and K87 in the NH2-terminal domain. STAT3 K685 acetylation is required for its dimerization and DNA binding; K49 and K87 acetylation is important for p300 recruitment and enhanceosome assembly.10 Second, STAT3 phosphorylated on S727 was shown to be present in mitochondria of cultured cells and primary tissues, including the heart.11 Moreover, mitochondrial STAT3 is required for optimal activities of complexes I and II of the electron transport chain.
New insights in the role of STAT3 in the heart
Classically, STAT3 mediates the acute phase responses to cytokines such as IL-6. In the heart, STAT3 activation has been linked to cell protection and angiogenesis. Paradoxically, STAT3 activation has also been linked to left ventricular hypertrophy, the most powerful predictor for development of heart failure. Exactly how STAT3 activates cardiac hypertrophy is not known, but a recent study with transgenic mice overexpressing the angiotensin II type 1 receptor specifically in cardiac myocytes has revealed a possible contributory mechanism.12 Hearts of these transgenics exhibited an increase in STAT3 mRNA and protein, but not in STAT3 phosphorylated on either Y705 or S727 residues. Moreover, development of cardiac hypertrophy and dysfunction correlated with nuclear accumulation of unphosphorylated STAT3 (U-STAT3), which was further linked to abnormal expression of osteopontin. In a mouse model, osteopontin was shown to modulate compensatory cardiac hypertrophy in response to chronic pressure overload, and in humans, osteopontin is a marker of cardiac hypertrophy and severity of heart failure.13,14 As in certain cancers, therefore, U-STAT3 may serve as a “rogue transcription factor”.
Circulating levels of leptin, a hormone that regulates cellular metabolism and reduces appetite, are elevated in heart failure and paracrine production within the heart may be a contributing factor. A recent study has shown that the failing human heart increases expression of leptin and its receptor, and mechanical unloading attenuates these increases.15 Leptin is known to activate STAT3 and levels of Y705 phosphorylated STAT3 were elevated as well in failing human hearts. A ventricular assist device (VAD) reversed the increase in STAT3 phosphorylation. The pathological relevance of these changes will need to be explored.16
Vascular inflammation and STAT3
Cardiac vascular/microvascular inflammation occurs in various pathologies associated with aging, including coronary heart disease, myocardial infarction, and both systolic and diastolic heart failure. In fact, the normal aging process is associated with impaired microvascular endothelial function and age is a primary risk factor for heart disease.17 Vascular inflammation is initiated by stimuli acting on endothelial cells (ECs) to enhance leukocyte adherence and chemotaxis. In chronic inflammation, persistence of activated macrophages in the vessel wall creates a self-sustaining cycle of cytokines and ROS leading to vascular remodeling, a prothrombotic state, and endothelial dysfunction. A key mediator of EC inflammatory response is the transcription factor complex NF-κB, and a clinical feature of vascular inflammation is increased circulating IL-6, a pro-inflammatory cytokine that is induced by NFκB activation. However, the role of IL-6 in vascular inflammation is not well defined.18 Notably, IL-6 cytokines signal in ECs mainly through STAT3.
With an aging population the economic and social burden of heart-related diseases is predicted to increase dramatically.19–22 Therefore, the need for novel pharmacotherapeutic strategies to treat heart disease is great. IL-6 and STAT3 EC signaling has universally been studied under non-inflammatory conditions in vitro and in vivo. By studying the impact of inflammatory events on the so-called pro-inflammatory signaling of the IL-6 type cytokines in cardiac microvascular endothelial cells (ECs), our laboratory seeks to provide a new framework for designing such strategies. IL-6 type cytokine STAT3 signaling in other cells types has both protective and inflammatory actions.2 We contend that STAT3 activation in human cardiac microvascular ECs is likely to have both beneficial and detrimental effects depending upon the relative level of interaction this transcription factor has with other transcription factors, such as NF-κB p65 and Sp1 (Figure 2). Such interactions could be targeted pharmacologically for instance by blocking or enhancing STAT3 S727 phosphorylation. Of note, endothelial cell STAT3 was found to play a critical role in limiting cardiac myocyte apoptosis due to ischemia-reperfusion.23 Yet little research has been done examining IL6 type cytokine STAT3 signaling in cardiac endothelial cells.
Figure 2.
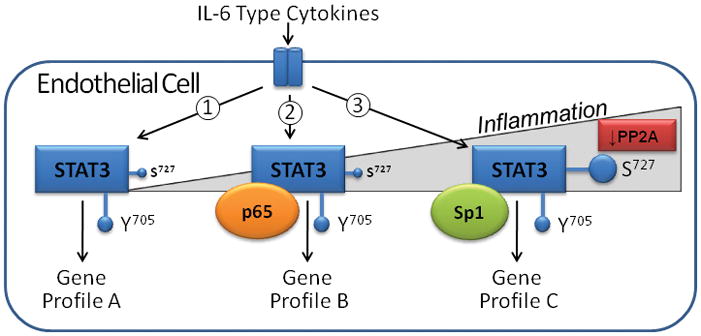
Hypothetical impact of increasing inflammatory stress on transcriptional response of endothelial cells to IL-6 type cytokine stimulation. The IL-6 cytokines activate STAT3 by increasing phosphorylation of Y705 and to a lesser extent S727: ➀No stress situation with minimal S727 phosphorylation; ➁STAT3 - NF-κB p65 association with mild inflammation; NF-κB would be activated by oxidative stress or some cytokine like TNFα; ➂Notable inflammation with markedly increased S727 phosphorylation and recruitment of the transcription factor Sp1. S727 phosphorylation might be increased by stress kinases or inhibition of the phosphatase PP2A due to oxidative stress. PP2A inhibition would affect Sp1 activity as well.
STAT3 and NF-κB
Several recent studies have found that NF-κB family members, p65 and p50 physically associate with STAT3 to regulate transcription of specific genes. ➊Non-Y705 phosphorylated STAT3 (U-STAT3) was shown to bind to nonactive p65 to form a U-STAT3/U-NF-κB complex that activates a subset of κB-dependent genes, such as RANTES, Il-6, and IL-8.24 This process is postulated to have importance in amplifying the pro-inflammatory IL-6 stimulus in certain cancers and possibly chronic inflammation, as STAT3 induces its own expression allowing for accumulation of U-STAT3. Evidence that this process contributes to TNFα-mediated RANTES production by vascular smooth muscle cells following arterial injury in vivo was recently reported.25 ➋Using a human hepatoma cell Hep3B, others reported that IL-1 and IL-6 induced a STAT3 (ostensibly nonphosphorylated Y705)/p65 complex that promoted expression of the IL-8 promoter off a GAS/κB element; the same complex inhibited classical STAT3 GAS promoter activity, while p50 co-operated with STAT3 bound to GAS sites.26 ➌Overexpressed p65 and/or p50 along with STAT3 and C/EBPβ were found to synergistically enhance expression of C-reactive protein in Hep3B.27 ➍Serum amyloid A expression in HepG2 cells in response to IL-1 and IL-6 cotreatment was found to result from a p65/STAT3/p300 complex that bound to a nonconsensus NF-κB promoter sequence.28 Evidence suggested that Y705 and S727 phosphorylation were both involved. ➎Enhanced interaction between STAT3 and p65 at the α-2-macroglobulin promoter in rat liver was reported during the acute-phase response, but STAT3 phosphorylation status was not assessed.29 Lastly, ➏STAT3 was found to inhibit induction of iNOS by both IL-1β and IFNγ + LPS by complexing with p65.30
STAT3 and Sp1
Sp1 is a ubiquitous transcription factor involved in diverse cellular processes, including apoptosis, immune response, and angiogenesis.31,32 Contrary to initial thinking, recent evidence shows that transcriptional activity of Sp1 is regulated by several posttranslational mechanisms, including phosphorylation. Of particular note in this regard is phosphorylation of S59, which can have either positive or negative effects on transcriptional activity of Sp1 depending upon the promoter.31–35 The endogenous phosphatase responsible for Sp1 S59 dephosphorylation is PP2A,31,32 the same phosphatase the targets STAT3 S727. In human microvascular ECs, Sp1 is activated by inflammatory stimuli, such as high-mobility group protein-1 (HMGP-1) and TNFα, and along with STAT3 and NF-κB constitutes a triad of transcription factors linked to an inflammatory response.36,37 Several studies with other cell types have shown that STAT3 and Sp1 can form a complex to enhance transcription,38–45 but the importance of this complex in vascular ECs is largely unexplored. In a recent study, STAT3 was reported to undergo S727 and Y705 phosphorylation, with the former particularly prominent, as a result of ischemia-reperfusion in the heart or after hypoxia–reoxygenation in vascular endothelial cells.38 Under these conditions STAT3 formed a complex with Sp1 and activated the promoter for intercellular adhesion molecule-1 (ICAM-1), which is upregulated rapidly on endothelial cells during ischemia-reperfusion and mediates tissue leukocyte accumulation.
Conclusions and future directions
Once seen as a paradigm of simplicity, STAT3 has revealed itself as an essential and complicated protein. STAT3 is now known to function as a transcription factor it its own right, as a partner with other transcription factors, as a co-factor for transcription or transcriptional enhancer, and as a modulator of mitochondrial respiration. Integration of these various functions is likely context-dependent with age, inflammatory events, and heart failure having importance in shaping the actions of STAT3. Critical factors that determine STAT3 function identified so far are relative levels of STAT3 S727 and Y705 phosphorylation, STAT3 acetylation levels, expression levels of total STAT3, and the activation status of other transcription factors. Could STAT3 be a potential therapeutic target in coronary artery disease or heart failure? Ideally, one would want to enhance the pro-survival and pro-angiogenic actions of STAT3, while diminishing its pro-inflammatory actions. A better understanding of the complex relationship between the function of STAT3 and its cellular environment may make that goal achievable.
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute to G. W. Booz (5R01HL088101-03 and 3R01HL088101-02S1) and grants from the Lebanese University and the Lebanese National Council for Scientific Research (CNRS) to M. Kurdi.
References
- 1.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008;120:172–185. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J Cardiovasc Pharmacol. 2007;50:126–141. doi: 10.1097/FJC.0b013e318068dd49. [DOI] [PubMed] [Google Scholar]
- 3.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–12934. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lacerda L, Somers S, Opie LH, Lecour S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc Res. 2009;84:201–208. doi: 10.1093/cvr/cvp274. [DOI] [PubMed] [Google Scholar]
- 5.Tamassia N, Castellucci M, Rossato M, Gasperini S, Bosisio D, Giacomelli M, Badolato R, Cassatella MA, Bazzoni F. Uncovering an IL-10-dependent NF-κB recruitment to the IL-1ra promoter that is impaired in STAT3 functionally defective patients. FASEB J. 2010;24:1365–1375. doi: 10.1096/fj.09-145573. [DOI] [PubMed] [Google Scholar]
- 6.Woetmann A, Nielsen M, Christensen ST, Brockdorff J, Kaltoft K, Engel AM, Skov S, Brender C, Geisler C, Svejgaard A, Rygaard J, Leick V, Odum N. Inhibition of protein phosphatase 2A induces serine/threonine phosphorylation, subcellular redistribution, and functional inhibition of STAT3. Proc Natl Acad Sci U S A. 1999;96:10620–10625. doi: 10.1073/pnas.96.19.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, Shanker S, Ferrajoli A, Keating MJ, Estrov Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115:2852–2863. doi: 10.1182/blood-2009-10-230060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma J, Cao X. Regulation of Stat3 nuclear import by importin α5 and importin α7 via two different functional sequence elements. Cell Signal. 2006;18:1117–1126. doi: 10.1016/j.cellsig.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proc Natl Acad Sci U S A. 2005;102:8150–8155. doi: 10.1073/pnas.0501643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou T, Ray S, Lee C, Brasier AR. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J Biol Chem. 2008;283:30725–30734. doi: 10.1074/jbc.M805941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yue H, Li W, Desnoyer R, Karnik SS. Role of nuclear unphosphorylated STAT3 in angiotensin II type 1 receptor-induced cardiac hypertrophy. Cardiovasc Res. 2010;85:90–9. doi: 10.1093/cvr/cvp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stawowy P, Blaschke F, Pfautsch P, Goetze S, Lippek F, Wollert-Wulf B, Fleck E, Graf K. Increased myocardial expression of osteopontin in patients with advanced heart failure. Eur J Heart Fail. 2002;4:139–146. doi: 10.1016/s1388-9842(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 14.Singh M, Foster CR, Dalal S, Singh K. Role of osteopontin in heart failure associated with aging. Heart Fail Rev. 2010 Feb 3; doi: 10.1007/s10741-010-9158-6. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15.McGaffin KR, Moravec CS, McTiernan CF. Leptin signaling in the failing and mechanically unloaded human heart. Circ Heart Fail. 2009;2:676–683. doi: 10.1161/CIRCHEARTFAILURE.109.869909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sweeney G. Cardiovascular effects of leptin. Nat Rev Cardiol. 2010:22–29. doi: 10.1038/nrcardio.2009.224. [DOI] [PubMed] [Google Scholar]
- 17.Gates PE, Strain WD, Shore AC. Human endothelial function and microvascular ageing. Exp Physiol. 2009;94:311–316. doi: 10.1113/expphysiol.2008.043349. [DOI] [PubMed] [Google Scholar]
- 18.Brasier AR. The nuclear factor-κB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res. 2010;86:211–218. doi: 10.1093/cvr/cvq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaziano TA, Bitton A, Anand S, Abrahams-Gessel S, Murphy A. Growing epidemic of coronary heart disease in low- and middle-income countries. Curr Probl Cardiol. 2010;35:72–115. doi: 10.1016/j.cpcardiol.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zannad F, Agrinier N, Alla F. Heart failure burden and therapy. Europace. 2009;11 (Suppl 5):v1–9. doi: 10.1093/europace/eup304. [DOI] [PubMed] [Google Scholar]
- 21.Pepine CJ. Residual risk for secondary ischemic events in patients with atherothrombotic disease: opportunity for future improvements in patient care. Ann Med. 2010;42:19–35. doi: 10.3109/07853890903260898. [DOI] [PubMed] [Google Scholar]
- 22.Tarride JE, Lim M, DesMeules M, Luo W, Burke N, O’Reilly D, Bowen J, Goeree R. A review of the cost of cardiovascular disease. Can J Cardiol. 2009;25:e195–202. doi: 10.1016/s0828-282x(09)70098-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Zhang W, Crisostomo P, Markel T, Meldrum KK, Fu XY, Meldrum DR. Endothelial STAT3 plays a critical role in generalized myocardial proinflammatory and proapoptotic signaling. Am J Physiol Heart Circ Physiol. 2007;293:H2101–8. doi: 10.1152/ajpheart.00125.2007. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovacic JC, Gupta R, Lee AC, Ma M, Fang F, Tolbert CN, Walts AD, Beltran LE, San H, Chen G, St Hilaire C, Boehm M. Stat3-dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J Clin Invest. 2010;120:303–314. doi: 10.1172/JCI40364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida Y, Kumar A, Koyama Y, Peng H, Arman A, Boch JA, Auron PE. Interleukin 1 activates STAT3/nuclear factor-κB cross-talk via a unique TRAF6- and p65-dependent mechanism. J Biol Chem. 2004;279:1768–1776. doi: 10.1074/jbc.M311498200. [DOI] [PubMed] [Google Scholar]
- 27.Agrawal A, Cha-Molstad H, Samols D, Kushner I. Overexpressed nuclear factor-κB can participate in endogenous C-reactive protein induction, and enhances the effects of C/EBPβ and signal transducer and activator of transcription-3. Immunology. 2003;108:539–547. doi: 10.1046/j.1365-2567.2003.01608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagihara K, Nishikawa T, Sugamata Y, Song J, Isobe T, Taga T, Yoshizaki K. Essential role of STAT3 in cytokine-driven NF-κB-mediated serum amyloid A gene expression. Genes Cells. 2005;10:1051–1063. doi: 10.1111/j.1365-2443.2005.00900.x. [DOI] [PubMed] [Google Scholar]
- 29.Uskoković A, Dinić S, Mihailović M, Grigorov I, Ivanović-Matić S, Bogojević D, Grdović N, Arambasić J, Vidaković M, Martinović V, Petrović M, Poznanović G. STAT3/NFκB interplay in the regulation of α2-macroglobulin gene expression during rat liver development and the acute phase response. IUBMB Life. 2007;59:170–178. doi: 10.1080/15216540701272612. [DOI] [PubMed] [Google Scholar]
- 30.Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor κB. Biochem J. 2002;367:97–105. doi: 10.1042/BJ20020588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan NY, Khachigian LM. Sp1 phosphorylation and its regulation of gene transcription. Mol Cell Biol. 2009;29:2483–2488. doi: 10.1128/MCB.01828-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu S, Ferro TJ. Sp1: regulation of gene expression by phosphorylation. Gene. 2005;348:1–11. doi: 10.1016/j.gene.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Liao M, Dufau ML. Unlocking repression of the human luteinizing hormone receptor gene by trichostatin A-induced cell-specific phosphatase release. J Biol Chem. 2008;283:24039–24046. doi: 10.1074/jbc.M801878200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu S, Cockrell CA, Ferro TJ. Expression of α-ENaC2 is dependent on an upstream Sp1 binding motif and is modulated by protein phosphatase 1 in lung epithelial cells. Biochem Biophys Res Commun. 2003;303:1159–1168. doi: 10.1016/s0006-291x(03)00497-2. [DOI] [PubMed] [Google Scholar]
- 35.Vicart A, Lefebvre T, Imbert J, Fernandez A, Kahn-Perlès B. Increased chromatin association of Sp1 in interphase cells by PP2A-mediated dephosphorylations. J Mol Biol. 2006;364:897–908. doi: 10.1016/j.jmb.2006.09.036. [DOI] [PubMed] [Google Scholar]
- 36.Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 37.Hamanaka R, Kohno K, Seguchi T, Okamura K, Morimoto A, Ono M, Ogata J, Kuwano M. Induction of low density lipoprotein receptor and a transcription factor SP-1 by tumor necrosis factor in human microvascular endothelial cells. J Biol Chem. 1992;267:13160–13165. [PubMed] [Google Scholar]
- 38.Yang XP, Irani K, Mattagajasingh S, Dipaula A, Khanday F, Ozaki M, Fox-Talbot K, Baldwin WM, 3rd, Becker LC. Signal transducer and activator of transcription 3α and specificity protein 1 interact to upregulate intercellular adhesion molecule-1 in ischemic-reperfused myocardium and vascular endothelium. Arterioscler Thromb Vasc Biol. 2005;25:1395–1400. doi: 10.1161/01.ATV.0000168428.96177.24. [DOI] [PubMed] [Google Scholar]
- 39.Loeffler S, Fayard B, Weis J, Weissenberger J. Interleukin-6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp1. Int J Cancer. 2005;115:202–213. doi: 10.1002/ijc.20871. [DOI] [PubMed] [Google Scholar]
- 40.Su HW, Wang SW, Ghishan FK, Kiela PR, Tang MJ. Cell confluency-induced Stat3 activation regulates NHE3 expression by recruiting Sp1 and Sp3 to the proximal NHE3 promoter region during epithelial dome formation. Am J Physiol Cell Physiol. 2009;296:C13–24. doi: 10.1152/ajpcell.00263.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang G, Lim CY, Li C, Xiao X, Radda GK, Li C, Cao X, Han W. FoxO1 inhibits leptin regulation of pro-opiomelanocortin promoter activity by blocking STAT3 interaction with specificity protein 1. J Biol Chem. 2009;284:3719–3727. doi: 10.1074/jbc.M804965200. [DOI] [PubMed] [Google Scholar]
- 42.Kiryu-Seo S, Kato R, Ogawa T, Nakagomi S, Nagata K, Kiyama H. Neuronal injury-inducible gene is synergistically regulated by ATF3, c-Jun, and STAT3 through the interaction with Sp1 in damaged neurons. J Biol Chem. 2008;283:6988–6996. doi: 10.1074/jbc.M707514200. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Sif S, DeWille J. The mouse C/EBPdelta gene promoter is regulated by STAT3 and Sp1 transcriptional activators, chromatin remodeling and c-Myc repression. J Cell Biochem. 2007;102:1256–1270. doi: 10.1002/jcb.21356. [DOI] [PubMed] [Google Scholar]
- 44.Lin S, Saxena NK, Ding X, Stein LL, Anania FA. Leptin increases tissue inhibitor of metalloproteinase I (TIMP-1) gene expression by a specificity protein 1/signal transducer and activator of transcription 3 mechanism. Mol Endocrinol. 2006;20:3376–3388. doi: 10.1210/me.2006-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mangan JK, Tantravahi RV, Rane SG, Reddy EP. Granulocyte colony-stimulating factor-induced upregulation of Jak3 transcription during granulocytic differentiation is mediated by the cooperative action of Sp1 and Stat3. Oncogene. 2006;25:2489–2499. doi: 10.1038/sj.onc.1209280. [DOI] [PubMed] [Google Scholar]