

Abstract
Water-protein interactions dictate many processes crucial to protein function including folding, dynamics, interactions with other biomolecules, and enzymatic catalysis. Here we examine the effect of surface fluorination on water-protein interactions. Modification of designed coiled-coil proteins by incorporation of 5,5,5-trifluoroleucine or (4S)-2-amino-4-methylhexanoic acid enables systematic examination of the effects of side-chain volume and fluorination on solvation dynamics. Using ultrafast fluorescence spectroscopy, we find that fluorinated side chains exert electrostatic drag on neighboring water molecules, slowing water motion at the protein surface.
Keywords: fluorine, noncanonical amino acids, protein engineering, solvation dynamics, ultrafast hydration
The past decade has witnessed substantial expansion in the number and diversity of noncanonical amino acids that can be incorporated into recombinant proteins expressed in bacterial cells (1–3). Fluorinated amino acids have drawn special attention (4–16) because of the unusual solubility properties of fluorinated hydrocarbons. Several independent studies have shown that fluorination of coiled-coil and helix-bundle proteins leads to enhanced stability with respect to thermal or chemical denaturation (6–12), an effect attributed to the hyper-hydrophobic and fluorophilic character of fluorinated amino acid side chains.
Although both classes of compounds are hydrophobic, hydrocarbons and fluorocarbons differ in important ways (17–22). The high electronegativity of fluorine renders the C-F bond both strongly polar and weakly polarizable (17, 21, 22). The dipole associated with the C-F bond exerts strong inductive effects on neighboring bonds (23) and can form reasonably strong electrostatic interactions with ionic or polar groups when the two moieties are appropriately positioned. The hydrophobic character of fluorinated compounds has been described as “polar hydrophobicity (17),” and is believed to play important roles in organic and medicinal chemistry. Furthermore, the C-F bond is significantly longer than the C-H bond, and the calculated volume of the trifluoromethyl group is about twice that of a methyl group (20). The studies described here constitute an attempt to understand more fully the interaction of water with fluorinated molecular surfaces, and to provide a sound basis for the use of fluorinated amino acids in the engineering of proteins with unique and useful physical properties.
The hydration layer adjacent to protein surfaces exhibits properties different from those of bulk water; the more rigid and denser structure of the hydration layer plays a crucial role in protein structure, folding, dynamics, and function (24–26). Elucidation of the dynamic features of this region, on the time scales of atomic and molecular motion, is essential in understanding protein hydration. In the past decade, the knowledge of hydration on protein surfaces has been extensively expanded by studying the dynamic properties of biological water for various proteins containing tryptophan (Trp) or synthetic fluorescent amino acids as local probes; the results have revealed multicomponent relaxation dynamics spanning a wide range of time scales (25, 27–29). The nature of the protein hydration layer can be affected not only by the topographic and electrostatic properties of the protein surface (24), but also by the physical and chemical properties of individual surface-exposed residues (27, 30). In view of the unique properties of the C-F bond and of fluorocarbon—water interfaces (23, 31), we anticipated that fluorinated amino acid side chains might exhibit unusual hydration behavior. Here we report studies of local hydration dynamics at fluorinated protein surfaces, by monitoring the time-dependent fluorescence Stokes shifts of surface-exposed Trp residues in coiled-coil proteins with 5,5,5-trifluoroleucine (Tfl, 1) residues adjacent to the probe. The results are compared to the hydration dynamics at hydrogenated protein surfaces with Leu (2) or (4S)-2-amino-4-methylhexanoic acid (homoisoleucine, Hil, 3) adjacent to the Trp probe. Hil has approximately the same volume as Tfl (20, 21), and although the shapes of the residues differ, the nearly identical side-chain volumes of Tfl and Hil allow us to differentiate changes due to fluorination from those that result from the increase in side-chain volume that accompanies replacement of Leu (Scheme 1).
Scheme 1.

Results
Coiled-Coil Protein System.
The coiled-coil protein A1 (Fig. 1 A and B) was used as a model system to examine the effects of fluorinated amino acids on local hydration dynamics. The primary structure of A1 contains six copies of a heptad repeat (abcdefg)n, where positions a and d are occupied by hydrophobic amino acids. Self-association of the peptide juxtaposes the a and d positions and results in the formation of a hydrophobic core. Fluorinated Leu analogues have previously been incorporated into the d positions of A1; the resulting proteins exhibited improved resistance to thermal and chemical denaturation with minimal differences in secondary structure (9, 11, 12). In this work, the surface-exposed Asp residue at the f position of the third heptad (position 34) was replaced by Trp, which serves as a fluorescence probe (Fig. 1C). The Trp variant of A1 was designated A1m. In order to examine the effects of fluorinated analogues on the local hydration dynamics, a Leu codon was introduced at one of two positions within A1m. Mutation of a serine residue at the c position of the third heptad (position 31) yielded a variant of A1 designated S31L (Fig. 1D), while replacement of an alanine residue at position b of the fourth heptad (position 37) gave the A1 variant A37L (Fig. 1E). Each protein was expressed in Tfl, Leu, and Hil form, yielding a total of nine different proteins that were examined in detail.*
Fig. 1.

Protein sequence and structure. (A) Helical wheel diagram and (B) amino acid sequence of the A1 protein. The Asp residue at the f position of the third heptad (position 34) was replaced by Trp to yield a variant of A1 designated A1m. A Leu codon was introduced at the c position of the third heptad (position 31, dark blue) or at the b position of the fourth heptad (position 37, dark blue) to yield S31L and A37L, respectively. Side views of A1m (C), S31L (D), and A37L (E).
Characterization of Global Structure.
Analysis of each protein showed that the overall structural properties of the molecules were generally insensitive to genetic mutations and incorporation of noncanonical amino acids. Circular dichroism spectroscopy indicated that all nine proteins were helical, as determined from the molar ellipticity at 222 nanometers (Fig. 2) (32); an analysis with K2D2 software showed that the helicities of individual proteins range between approximately 40% and 48% (33). These results are consistent with the design of the A1 protein (34), in which approximately half of the amino acids are located within the heptad repeats expected to form α-helical secondary structure. The oligomerization states of the protein samples were determined by sedimentation velocity analysis (Fig. S1). Although A1 forms dimers and tetramers at neutral pH (11), the variants examined in this study form trimers or hexamers under mildly acidic conditions (pH 4). We suggest that protonation of Glu side chains at the e and g positions (Fig. 1A) of the proteins decreases the density of negative charges adjacent to the hydrophobic core and promotes formation of larger helical aggregates at pH 4. A1m, in which the single Trp residue occupies a surface-exposed position, is predominantly trimeric in Leu-, Tfl-, and Hil-forms, with a small fraction of hexamers (see Fig. S1). The majority of the S31L samples are present as hexamers, while the A37L samples appear to contain mixtures of trimers and hexamers.
Fig. 2.

Circular dichroism. Wavelength scans of A1m and variant proteins at 25 °C. The protein samples were prepared at 20 μM concentration in 10 mM acetate (pH 4)/100 mM NaCl solutions.
Characterization of Local Structure.
The steady-state fluorescence emission spectrum of Trp depends on the extent of exposure of the Trp side chain to water (35). All nine protein samples showed emission maxima between 349 nanometers and 352 nanometers, close to that of free Trp at 353 nanometers (Table 1 and Fig. S2). These observations indicate that the Trp residues are exposed to the aqueous environment (consistent with the original design), and not involved in oligomerization of the proteins. In addition, the steady-state UV-visible absorption and steady-state fluorescence emission spectra of each mutant containing Leu were nearly identical to the spectra of the corresponding mutant when it contained Tfl or Hil (Fig. S2), further confirming that perturbation of the protein structure upon replacement of Leu by Tfl or Hil was minimal.
Table 1.
Fluorescence emission maxima (λmax), hydration-correlated energy relaxation [ΔEs(t)], and depolarization dynamics [r(t)]
| Sample |
λmax, nanometers |
ΔEs(t) * |
r(t) † |
||||||
| τ1, ps | τ2, ps | τ3, ps | E1, cm-1 | E2, cm-1 | E3, cm-1 | rTrp | θ , ° | ||
| Trp | 353 | 0.30 | 1.5 | 13 | 883 | 682 | 18 | 0.196 | |
| A1m-L | 352 | 0.30 | 2.1 | 31 | 610 | 646 | 171 | 0.056 | 21 |
| A1m-T | 352 | 0.28 | 2.5 | 31 | 877 | 568 | 161 | 0.049 | 20 |
| A1m-H | 352 | 1.9 | 34 | 0 | 2138 | 500 | 0.058 | 33 | |
| S31L-L | 349 | 0.53 | 3.6 | 40 | 580 | 450 | 194 | 0.030 | 18 |
| S31L-T | 349 | 0.79 | 3.0 | 48 | 607 | 375 | 333 | 0.044 | 19 |
| S31L-H | 349 | 0.21 | 1.4 | 10 | 308 | 492 | 344 | 0.023 | 18 |
| A37L-L | 352 | 0.31 | 1.7 | 13 | 685 | 522 | 157 | 0.055 | 20 |
| A37L-T | 350 | 0.56 | 6.1 | 61 | 685 | 324 | 108 | 0.030 | 17 |
| A37L-H | 349 | 0.34 | 2.2 | 21 | 443 | 625 | 128 | 0.034 | 17 |
*All hydration-correlated energy relaxation dynamics were fitted to ΔEs(t) = E1 exp(−t/τ1) + E2 exp(−t/τ2) + E3 exp(−t/τ3).
†Refer to the SI Text for anisotropy analysis detail.
The mobility of the probe residue was explored in each protein by measuring fs-resolved depolarization dynamics (Fig. S3). The anisotropic dynamics were found to consist of three components: ultrafast (≤ 500 fs), intermediate (20–80 ps), and slow (≥2 ns) decays. The ultrafast decays are attributed to fast internal conversion between the first two excited singlet states (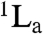 and
and 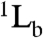 ) of Trp, the intermediate decays to local wobbling motions of Trp, and the slow decays to tumbling motions of the proteins (28, 36). Similar values for the wobbling motions (ϕTrp) and their cone semiangles (θ) were observed for each series of S31L and A37L proteins (Table 1); see SI Text for details.
) of Trp, the intermediate decays to local wobbling motions of Trp, and the slow decays to tumbling motions of the proteins (28, 36). Similar values for the wobbling motions (ϕTrp) and their cone semiangles (θ) were observed for each series of S31L and A37L proteins (Table 1); see SI Text for details.
Both mutation of residues around Trp and fluorination of the protein hydrophobic core can affect the environment of the probe and change the protein structure and/or the dynamic properties of the hydration layer. In many cases these properties are related to one another. The minimal change in the steady-state fluorescence spectrum caused by replacement of Leu by Tfl or Hil suggests similar features of the hydration region probed by Trp (e.g., the effective number of water molecules in the hydration shell). In addition, the similarity of the Trp wobbling angle of the Leu-, Tfl-, and Hil-forms of the proteins suggests similar organization and flexibility of neighboring residues around the probe (28). All these features make it possible to compare the dynamic properties of protein hydration for most of the proteins in the A1m, S31L, and A37L proteins containing Leu, Tfl, and Hil. For A1m-H, we note that the wobbling angle of Trp was found to be 33°, which is significantly higher than the 17–21° wobbling angles determined for all of the other proteins. This result indicates that the organization of local residues or the flexibility of the local Trp environment in A1m-H differs from that in the other proteins, despite the lack of global structural changes observed by circular dichroism or sedimentation velocity measurements. The abnormal behavior of the A1m-H variant is also observed in the fluorescence lifetime measurements. Every protein except A1m-H displayed a short-lifetime component of a few hundred ps, present at all wavelengths. These types of quenching processes have been attributed to Trp interactions with nearby charged residues (37–39), and the absence of such a feature in A1m-H again indicates that this protein has a local structure different from those of the other eight proteins. The perturbation of local structure and, thus, local solvent exposure can result in different hydration dynamics, making it unreliable to compare the dynamics of A1m-H to those of the other A1m proteins. Accordingly, the dynamics obtained for A1m-H were not used in the analysis that follows. Small shifts in the fluorescence emission maximum and Trp wobbling angle were observed for A37L-L as compared to A37L-T and A37L-H (see Table 1 and Fig. S2). These differences may be significant enough to alter the local environment surrounding the Trp probe, potentially complicating assignment of changes observed in the hydration dynamics to a particular effect (e.g., changes in an amino acid close to Trp). Despite these concerns, the dynamics results for A37L-L remain consistent with the conclusions of the paper (see below).
Our stringent standards for comparison of hydration dynamics between modified proteins require that there be (i) no global change in protein structure as measured by circular dichroism spectroscopy and sedimentation velocity measurements; (ii) no change in solvent exposure as measured by steady-state fluorescence maximum (± 1 nm); and (iii) no change in local protein structure or flexibility as measured by fluorescence anisotropy (± 1°). Seven of the nine proteins prepared in this study met all of these criteria, and an additional protein, A37L-L, displayed changes just outside the margin of error. Only one protein, A1m-H, showed changes significant enough to require us to disregard the hydration measurements observed. Given the subtle effects of the chemical environment on hydration dynamics, we will compare hydration results only within protein families. Thus, our strongest conclusions will be drawn from observations made on the S31L protein variants, and the data for A1m and A37L will be used as corroborating evidence.
Ultrafast Hydration Dynamics.
To investigate hydration dynamics at the protein surfaces, we utilized a methodology developed by Zhong and coworkers for the reconstruction of fs-resolved fluorescence spectra (28, 40). As an example, Fig. 3A shows several representative fs-resolved fluorescence transients recorded for A1m-T. The overall decay dynamics is retarded compared to that of free Trp in buffer solution. Details of the results for all the protein samples are presented in Table 1. The hydration dynamics of the proteins were well represented by triple-exponential decays with distinctive time scales of 0.2–0.8, 1.4–6.1, and 10–61 ps. Relaxation occurring on a time scale of a few hundred fs to several ps is attributed to fast librational/rotational motions of bulk-type and local water molecules around Trp. Observation of the fs component suggests that the Trp probe in the test proteins is neither crowded by neighboring residues nor protected from exposure to water (27, 28, 41). The slowest phase of hydration dynamics (on the time scale of tens of ps) is collective water network rearrangement coupled to protein fluctuation dynamics (27, 41–43).
Fig. 3.

Hydration dynamics. Experimental determination of local hydration dynamics at the surface of A1m-T, excited at 295 nanometers. (A) Representative fs-resolved fluorescence upconversion transients. (B) Normalized time-resolved fluorescence spectra at different time delays. The steady-state emission spectrum is also depicted (dotted line). (C) Time-dependent spectral shift of the apparent emission maxima (νs) and the lifetime-associated (population) emission maxima (νl). (Inset) Entire evolution of νs and νl.
Several key features of the results (Fig. 4 and Table 1) are summarized as follows. First, S31L-T and A37L-T, in which Tfl lies close to Trp as well as in the hydrophobic core, showed slower hydration dynamics than their Hil and Leu counterparts, indicating that the fluorinated surface of the protein slows down the hydration dynamics. For S31L-T, the time scales of local and collective water motions (τ2 and τ3, respectively) are increased by 2–5 times (3.0 ps and 48 ps) from those of S31L-H (1.4 ps and 10 ps). The overall solvation of S31L-T is slower than that of S31L-L as well. However, this difference is manifested as an increase in the contribution of the τ3 component to the overall solvation of Trp. The 70% increase of the relaxation energy (333 cm.1) of the slowest component (E3) compared to that of the nonfluorinated S31L-L (194 cm1) is an indication of the dramatic slowing of hydration dynamics near the fluorinated surface. For A37L-T, τ2 and τ3 are also retarded to a greater extent (3–5 times slower) than those for A37L-H. These results suggest that replacement of Leu by Tfl increases the residence time of water molecules near the Trp probe and/or the number of water molecules influenced by the amino acid side chain.
Fig. 4.

Hydration Energy Relaxation. Comparison of the hydration-correlated energy relaxation, ΔEs(t), probed by Trp emission. Top: The solvation-energy relaxation data for A1m proteins. Data for free Trp in the same buffer is also depicted for comparison. Middle: Solvation energy relaxation data for S31L analogs. Bottom: Solvation energy relaxation data for A37L analogs. Insets: Enlargement of the early time hydration behavior.
Second, S31L-H and A37L-H showed similar or even faster hydration than their Leu counterparts. This result indicates that in the comparison between hydrocarbon residues, increasing the hydrophobic surface area results in faster motion of water molecules around the residue. It should be noted that, for the S31L series, hydration is greatly accelerated when Leu is replaced with Hil. This pronounced hydrophobic effect (due to the increase in the size of the residue) on the hydration is counteracted by fluorination of leucine, resulting in slowing dynamics for S31L-T. On the other hand, for A37L, increasing the size of the hydrophobic surface does not appear to affect the hydration dynamics as greatly. Therefore, the retardation of the dynamics upon fluorination is much more pronounced for A37L-T than for the corresponding fluorination of S31L. Finally, A1m-L and A1m-T, which differ from one another only in the nature of their hydrophobic cores, exhibited almost identical hydration dynamics. Fluorination of the hydrophobic core of a helix-bundle protein can affect protein dynamics (44). However, the nearly identical hydration dynamics for A1m-L and A1m-T spanning a few hundred ps indicates that modification of the hydrophobic core of A1m does not affect protein motions that are coupled to local hydration dynamics at the surface of the protein on the time scales examined here.†
Discussion
Protein surfaces are heterogeneous, consisting of polar, hydrophilic, and hydrophobic residues, and it is intriguing to consider how the heterogeneous surface chemistry affects the behavior of water molecules in the protein hydration layer. Head-Gordon and coworkers have reported heterogeneous water dynamics in the first hydration shell of model peptides (N-acetyl-leucine-methylamide and N-acetyl-glycine-methylamide), with faster water motions near the hydrophobic side chains than near the hydrophilic backbone (45, 46). Similar results have been reported for molecular dynamics simulation studies of a folded β-hairpin peptide (30). In addition, Qiu et al. showed that mutation of charged or polar residues of the enzyme staphylococcal nuclease into more hydrophobic residues (Ala), resulted in faster hydration dynamics; this result was attributed to the lack of strong interaction between the charges (or dipoles) of the mutated protein and the surrounding water (27). This observation can be understood in that the elimination of specific interactions between hydrophobic residues and water molecules causes a lower number of hydrogen bonds between water and a hydrophobic surface compared to those near a hydrophilic surface, thus allowing water molecules to reorient more freely. Computational studies have suggested that water layers adjacent to extended hydrophobic surfaces of low curvature are of lower density than those around hydrophilic and small hydrophobic molecules, and are dynamic rather than static (47–54). X-ray reflectivity experiments indicate submonolayer water depletion at hydrocarbon and fluorocarbon surfaces (55). Our observation of accelerated hydration dynamics around larger Hil residues as compared to the smaller Leu is consistent with these experimental and computational results, supporting the idea that water molecules neighboring hydrophobic side chains in the hydration layer of proteins are more dynamic than those around polar or hydrophilic residues.
Even though a simple comparison suggests that Tfl should be more hydrophobic than Leu by virtue of its larger surface area, introduction of a Tfl residue adjacent to the Trp probe caused retardation of the local hydration dynamics, in contrast to the results obtained when Leu was replaced with Hil (Fig. 4). These results suggest that hydration dynamics around fluorinated amino acid side chains cannot be explained exclusively by the increase in residue size. The C-F bond is assumed not to be involved in hydrogen bonding with liquid water, largely because of its low polarizability (17). However, replacement of Leu by Tfl introduces a strong dipole at the fluorinated protein surface. Lee and coworkers have shown that introduction of CF3 groups reduces the contact angle of water on self-assembled alkanethiol monolayers (23), an effect that they attribute to dipolar interactions. Our results suggest that such dipolar interactions can also slow water motions at fluorinated molecular surfaces.
Fluorinated compounds are more hydrophobic than hydrogenated compounds of equal carbon number (4, 5, 17–21), and the increase in hydrophobic character of fluorocarbons has been ascribed to their increased molecular size (18, 56). This interpretation appears to be consistent with the observation that the melting temperature of A1-Tfl is 13 °C higher than that of A1-Leu (11), while Tm for A1-Hil is increased by 17 °C in comparison to A1-Leu. However, the results reported here clearly indicate that the chemical nature of the protein surface dictates the dynamics of solvent-protein interactions, and that size effects alone cannot explain the altered solvation dynamics observed at fluorinated protein surfaces.
Concluding Remarks
The results reported here show that fluorinated amino acids influence hydration dynamics at protein surfaces in a manner quite different from their hydrogenated counterparts. In general, water-protein interactions dictate many processes crucial to protein function including folding, dynamic motions, interactions with other biomolecules, and enzymatic catalysis (26). The slower time scales of hydration dynamics observed near fluorinated residues in proteins suggest that some of the water-mediated processes listed above may be changed upon fluorination. Tailoring the dynamics of protein-water interactions by the introduction of fluorinated residues may yield proteins with functional properties, such as binding, molecular recognition, or catalytic activities, that cannot be achieved with the canonical amino acids. Understanding hydration dynamics at fluorinated molecular surfaces is a critical step toward exploiting the properties of fluorine in biological systems.
Materials and Methods
Protein Expression and Characterization.
A1 variants A1m, S31L, and A37L were expressed in 2xYT medium (which contains Leu) to yield proteins A1m-L, S31L-L, and A37L-L, respectively, in Leu-free M9 minimal medium (12.8 g/L Na2HPO4·7H2O, 3 g/L KH2PO4, 0.5 g/L NaCl, 1 g/L NH4Cl) supplemented with 19 canonical amino acids plus Tfl to give A1m-T, S31L-T, and A37L-T, and in Leu-free M9 medium supplemented with 19 canonical amino acids plus Hil to give A1m-H, S31L-H, and A37L-H. The proteins were purified under denaturing conditions and dialyzed against 10 mM acetate (pH 4)/100 mM NaCl. The extent of replacement of Leu by Tfl in A1m-T, S31L-T, and A37L-T, was determined by amino acid analysis to be 90%–91%. Leu replacement by Hil in A1m-H, S31L-H, and A37L-H was analyzed by liquid chromatography tandem mass spectrometry and determined to be at least 90% (Fig. S4). See SI Text for further details of expression, purification, and incorporation analysis.
Steady-State Measurements.
Circular dichroism spectra were recorded on an Aviv 62DS spectropolarimeter (Lakewood, NJ). Absorption spectra were collected using a Cary 500 UV-Vis spectrophotometer and a 0.05-mm path length cuvette. Steady-state fluorescence emission spectra were measured using a FluoroMax-2 fluorimeter (ISA-Spex).
Time-Resolved Fluorescence Measurement.
The experimental apparatus for time-resolved measurements are detailed in SI Text. All fluorescence spectra and transients were obtained by the excitation of samples (∼550 μM) at 295 nanometers. The lifetime components were obtained by global analysis of fluorescence transients collected using a time-correlated single photon counting spectrometer. All transients show additional multiple-exponential decay (at the blue side) and rise (at the red side) with time constants spanning from a few hundred fs to several tens of ps. In order to extract hydration dynamics precisely, we reconstructed apparent and lifetime-associated time-resolved fluorescence spectra with eight or nine transients at different wavelengths covering the blue and the red sides (Fig. 3B). By fitting these spectra to lognormal functions, we traced the time-dependent apparent emission maxima (νs) and lifetime-associated emission maxima (νl) as plotted in Fig. 3C. Using Δν(t) = νs(t) - νl(t), we correlated the extracted time-dependent spectral shift, Δν(t), to the hydration energy relaxation, ΔEs (Fig. 4).
Supplementary Material
Acknowledgments.
We thank Prof. Thomas Miller for helpful discussion, J.D. Fisk for synthesis of homoisoleucine, and the referees for their thoughtful comments on the original manuscript. This work is supported by National Institutes of Health (NIH) Grant GM62523, National Science Foundation (NSF) Grant DMR-0964886, Office of Naval Research (ONR) Grant N00014-03-1-0793, a Samsung Scholarship (to T.H.Y.), and a National Defense Science and Engineering Graduate Fellowship (to J.A.V.D.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1011569107/-/DCSupplemental.
*Protein variants containing Tfl, Leu, and Hil, are identified by the addition of “-T,” “-L,” and “-H,” respectively. For example, S31L containing Leu is designated “S31L-L.”
†We anticipated that A1m-H would also show hydration dynamics identical to those of A1m-L and A1m-T. However as discussed before, the local protein packing and flexibility near the Trp probe appear to have been altered by substitution of Hil for Leu, reflected in the large Trp wobbling angle and total Stokes shift, ΔES. Because the local structure has been changed, we would expect to observe altered hydration dynamics.
References
- 1.Budisa N. Engineering the genetic code: expanding the amino acid repertoire for the design of novel proteins. Weinheim, Germany: Wiley-VCH; 2006. [Google Scholar]
- 2.Link AJ, Mock ML, Tirrell DA. Non-canonical amino acids in protein engineering. Curr Opin Biotechnol. 2003;14:603–609. doi: 10.1016/j.copbio.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Xie JM, Schultz PG. Innovation: a chemical toolkit for proteins—an expanded genetic code. Nat Rev Mol Cell Biol. 2006;7:775–782. doi: 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- 4.Marsh ENG. Towards the nonstick egg: designing fluorous proteins. Chem Biol. 2000;7:R153–R157. doi: 10.1016/s1074-5521(00)00139-3. [DOI] [PubMed] [Google Scholar]
- 5.Yoder NC, Yuksel D, Dafik L, Kumar K. Bioorthogonal noncovalent chemistry: fluorous phases in chemical biology. Curr Opin Chem Biol. 2006;10:576–583. doi: 10.1016/j.cbpa.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Bilgicer B, Fichera A, Kumar K. A coiled coil with a fluorous core. J Am Chem Soc. 2001;123:4393–4399. doi: 10.1021/ja002961j. [DOI] [PubMed] [Google Scholar]
- 7.Jackel C, Salwiczek M, Koksch B. Fluorine in a native protein environment—how the spatial demand and polarity of fluoroalkyl groups affect protein folding. Angewandte Chemie International Edition. 2006;45:4198–4203. doi: 10.1002/anie.200504387. [DOI] [PubMed] [Google Scholar]
- 8.Lee KH, Lee HY, Slutsky MM, Anderson JT, Marsh ENG. Fluorous effect in proteins: de novo design and characterization of a four-alpha-helix bundle protein containing hexafluoroleucine. Biochemistry. 2004;43:16277–16284. doi: 10.1021/bi049086p. [DOI] [PubMed] [Google Scholar]
- 9.Montclare JK, Son S, Clark GA, Kumar K, Tirrell DA. Biosynthesis and stability of coiled-coil peptides containing (2S,4R)-5,5,5-Trifluoroleucine and (2S,4S)-5,5,5-Trifluoroleucine. ChemBioChem. 2009;10:84–86. doi: 10.1002/cbic.200800164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Son S, Tanrikulu IC, Tirrell DA. Stabilization of bzip peptides through incorporation of fluorinated aliphatic residues. ChemBioChem. 2006;7:1251–1257. doi: 10.1002/cbic.200500420. [DOI] [PubMed] [Google Scholar]
- 11.Tang Y, et al. Fluorinated coiled-coil proteins prepared in vivo display enhanced thermal and chemical stability. Angewandte Chemie International Edition. 2001;40:1494–1496. doi: 10.1002/1521-3773(20010417)40:8<1494::AID-ANIE1494>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 12.Tang Y, Tirrell DA. Biosynthesis of a highly stable coiled-coil protein containing hexafluoroleucine in an engineered bacterial host. J Am Chem Soc. 2001;123:11089–11090. doi: 10.1021/ja016652k. [DOI] [PubMed] [Google Scholar]
- 13.Montclare JK, Tirrell DA. Evolving proteins of novel composition. Angewandte Chemie International Edition. 2006;45:4518–4521. doi: 10.1002/anie.200600088. [DOI] [PubMed] [Google Scholar]
- 14.Wang P, Tang Y, Tirrell DA. Incorporation of trifluoroisoleucine into proteins in vivo. J Am Chem Soc. 2003;125:6900–6906. doi: 10.1021/ja0298287. [DOI] [PubMed] [Google Scholar]
- 15.Yoo TH, Link AJ, Tirrell DA. Evolution of a fluorinated green fluorescent protein. Proc Natl Acad Sci USA. 2007;104:13887–13890. doi: 10.1073/pnas.0701904104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoo TH, Tirrell DA. High-throughput screening for Methionyl-tRNA synthetases that enable residue-specific incorporation of noncanonical amino acids into recombinant proteins in bacterial cells. . Angewandte Chemie International Edition. 2007;46:5340–5343. doi: 10.1002/anie.200700779. [DOI] [PubMed] [Google Scholar]
- 17.Biffinger JC, Kim HW, DiMagno SG. The polar hydrophobicity of fluorinated compounds. ChemBioChem. 2004;5:622–627. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- 18.Gao JM, Qiao S, Whitesides GM. Increasing binding constants of ligands to carbonic-anhydrase by using greasy tails. J Med Chem. 1995;38:2292–2301. doi: 10.1021/jm00013a005. [DOI] [PubMed] [Google Scholar]
- 19.Jackel C, Koksch B. Fluorine in peptide design and protein engineering. Eur J Org Chem. 2005;2005:4483–4503. [Google Scholar]
- 20.Leroux F. Atropisomerism, biphenyls, and fluorine: a comparison of rotational barriers and twist angles. ChemBioChem. 2004;5:644–649. doi: 10.1002/cbic.200300906. [DOI] [PubMed] [Google Scholar]
- 21.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 22.Dunitz JD. Organic fluorine: odd man out. ChemBioChem. 2004;5:614–621. doi: 10.1002/cbic.200300801. [DOI] [PubMed] [Google Scholar]
- 23.Graupe M, Takenaga M, Koini T, Colorado R, Lee TR. Oriented surface dipoles strongly influence interfacial wettabilities. J Am Chem Soc. 1999;121:3222–3223. [Google Scholar]
- 24.Merzel F, Smith JC. Is the first hydration shell of lysozyme of higher density than bulk water? Proc Natl Acad Sci USA. 2002;99:5378–5383. doi: 10.1073/pnas.082335099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pal SK, Zewail AH. Dynamics of water in biological recognition. Chem Rev. 2004;104:2099–2123. doi: 10.1021/cr020689l. [DOI] [PubMed] [Google Scholar]
- 26.Levy Y, Onuchic JN. Water mediation in protein folding and molecular recognition. Annu Rev Biophys Biomol Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 27.Qiu WH, et al. Protein surface hydration mapped by site-specific mutations. Proc Natl Acad Sci USA. 2006;103:13979–13984. doi: 10.1073/pnas.0606235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu WH, et al. Ultrafast solvation dynamics of human serum albumin: correlations with conformational transitions and site-selected recognition. J Phys Chem B. 2006;110:10540–10549. doi: 10.1021/jp055989w. [DOI] [PubMed] [Google Scholar]
- 29.Cohen BE, et al. Probing protein electrostatics with a synthetic fluorescent amino acid. Science. 2002;296:1700–1703. doi: 10.1126/science.1069346. [DOI] [PubMed] [Google Scholar]
- 30.Daidone I, Ulmschneider MB, Di Nola A, Amadei A, Smith JC. Dehydration-driven solvent exposure of hydrophobic surfaces as a driving force in peptide folding. Proc Natl Acad Sci USA. 2007;104:15230–15235. doi: 10.1073/pnas.0701401104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore FG, Richmond GL. Integration or segregation: how do molecules behave at oil/water interfaces? Acc Chem Res. 2008;41:739–748. doi: 10.1021/ar7002732. [DOI] [PubMed] [Google Scholar]
- 32.Chen YH, Yang JT, Martinez HM. Determination of secondary structures of proteins by circular-dichroism and optical rotary dispersion. Biochemistry. 1972;11:4120–4131. doi: 10.1021/bi00772a015. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Iratxeta C, Andrade-Navarro MA. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol. 2008;8:25–29. doi: 10.1186/1472-6807-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Reversible hydrogels from self-assembling artificial proteins. Science. 1998;281:389–392. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 35.Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steiner RF. In: Top. fluoresc. spectrosc. Lakowicz JR, editor. Vol 2. New York: Plenum Press; 1991. pp. 1–52. [Google Scholar]
- 37.Qiu WH, et al. Ultrafast quenching of tryptophan fluorescence in proteins: interresidue and intrahelical electron transfer. Chem Phys. 2008;350:154–164. [Google Scholar]
- 38.Siemiarczuk A, Petersen CE, Ha CE, Yang JS, Bhagavan NV. Analysis of tryptophan fluorescence lifetimes in a series of human serum albumin mutants with substitutions in subdomain 2A. Cell Biochem Biophys. 2004;40:115–122. doi: 10.1385/CBB:40:2:115. [DOI] [PubMed] [Google Scholar]
- 39.Xu JH, Knutson JR. Quasi-static self-quenching of Trp-X and X-Trp dipeptides in water: ultrafast fluorescence decay. J Phys Chem B. 2009;113:12084–12089. doi: 10.1021/jp903078x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu WY, Kim J, Qiu WH, Zhong DP. Femtosecond studies of tryptophan solvation: correlation function and water dynamics at lipid surfaces. Chem Phys Lett. 2004;388:120–126. [Google Scholar]
- 41.Li TP, Hassanali AAP, Kao YT, Zhong DP, Singer SJ. Hydration dynamics and time scales of coupled water-protein fluctuations. J Am Chem Soc. 2007;129:3376–3382. doi: 10.1021/ja0685957. [DOI] [PubMed] [Google Scholar]
- 42.Zhang LY, Yang Y, Kao YT, Wang LJ, Zhong DP. Protein hydration dynamics and molecular mechanism of coupled water-protein fluctuations. J Am Chem Soc. 2009;131:10677–10691. doi: 10.1021/ja902918p. [DOI] [PubMed] [Google Scholar]
- 43.Golosov AA, Karplus M. Probing polar solvation dynamics in proteins: a molecular dynamics simulation analysis. J Phys Chem B. 2007;111:1482–1490. doi: 10.1021/jp065493u. [DOI] [PubMed] [Google Scholar]
- 44.Lee HY, Lee KH, Al-Hashimi HM, Marsh ENG. Modulating protein structure with fluorous amino acids: increased stability and native-like structure conferred on a 4-helix bundle protein by hexafluoroleucine. J Am Chem Soc. 2006;128:337–343. doi: 10.1021/ja0563410. [DOI] [PubMed] [Google Scholar]
- 45.Russo D, Hura G, Head-Gordon T. Hydration dynamics near a model protein surface. Biophys J. 2004;86:1852–1862. doi: 10.1016/S0006-3495(04)74252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russo D, Murarka RK, Copley JRD, Head-Gordon T. Molecular view of water dynamics near model peptides. J Phys Chem B. 2005;109:12966–12975. doi: 10.1021/jp051137k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 48.Richmond GL. Molecular bonding and interactions at aqueous surfaces as probed by vibrational sum frequency spectroscopy. Chem Rev. 2002;102:2693–2724. doi: 10.1021/cr0006876. [DOI] [PubMed] [Google Scholar]
- 49.Shen YR, Ostroverkhov V. Sum-frequency vibrational spectroscopy on water interfaces: polar orientation of water molecules at interfaces. Chem Rev. 2006;106:1140–1154. doi: 10.1021/cr040377d. [DOI] [PubMed] [Google Scholar]
- 50.Miller TF, Vanden-Eijnden E, Chandler D. Solvent coarse-graining and the string method applied to the hydrophobic collapse of a hydrated chain. Proc Natl Acad Sci USA. 2007;104:14559–14564. doi: 10.1073/pnas.0705830104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.ten Wolde PR, Chandler D. Drying-induced hydrophobic polymer collapse. Proc Natl Acad Sci USA. 2002;99:6539–6543. doi: 10.1073/pnas.052153299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashbaugh HS, Paulaitis ME. Effect of solute size and solute-water attractive interactions on hydration water structure around hydrophobic solutes. J Am Chem Soc. 2001;123:10721–10728. doi: 10.1021/ja016324k. [DOI] [PubMed] [Google Scholar]
- 53.Huang DM, Chandler D. The hydrophobic effect and the influence of solute-solvent attractions. J Phys Chem B. 2002;106:2047–2053. [Google Scholar]
- 54.Jensen TR, et al. Water in contact with extended hydrophobic surfaces: direct evidence of weak dewetting. Phys Rev Lett. 2003;90:086101. doi: 10.1103/PhysRevLett.90.086101. [DOI] [PubMed] [Google Scholar]
- 55.Mezger M, et al. On the origin of the hydrophobic water gap: an X-ray reflectivity and MD simulation study. J Am Chem Soc. 2010;132:6735–6741. doi: 10.1021/ja910624j. [DOI] [PubMed] [Google Scholar]
- 56.Rossky PJ, Dalvi VH. Molecular origins of fluorocarbon hydrophobicity. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.0915169107. 10.1073/pnas.0915169107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.