

Abstract
Familial hypertrophic cardiomyopathy (FHC) is caused by mutations in sarcomeric proteins including the myosin regulatory light chain (RLC). Two such FHC mutations, R58Q and N47K, located near the cationic binding site of the RLC, have been identified from population studies. To examine the molecular basis for the observed phenotypes, we exchanged endogenous RLC from native porcine cardiac myosin with recombinant human ventricular wild type (WT) or FHC mutant RLC and examined the ability of the reconstituted myosin to propel actin filament sliding using the in vitro motility assay. We find that, whereas the mutant myosins are indistinguishable from the controls (WT or native myosin) under unloaded conditions, both R58Q- and N47K-exchanged myosins show reductions in force and power output compared with WT or native myosin. We also show that the changes in loaded kinetics are a result of mutation-induced loss of myosin strain sensitivity of ADP affinity. We propose that the R58Q and N47K mutations alter the mechanical properties of the myosin neck region, leading to altered load-dependent kinetics that may explain the observed mutant-induced FHC phenotypes.
Keywords: in vitro motility, load-dependent kinetics, familial hypertrophic cardiomyopathy, R58Q, N47K
Familial hypertrophic cardiomyopathy (FHC), the leading cause of sudden cardiac death in people under 30 (1), is characterized by several changes in cardiac structure including myofibrillar disarray and thickening of the left ventricle, papillary muscles, and/or septum. FHC is a disease of the sarcomere, resulting from mutations of cardiac proteins (reviewed in refs. 1 and 2) including the regulatory (RLC) and essential (ELC) light chains of myosin.
Each myosin molecule is a hexamer composed of two myosin heavy chains (MHC), two RLCs, and two ELCs (3). The α-helical neck region of the MHC has been proposed to act as a lever arm, amplifying small conformational changes that originate at the myosin catalytic site into large movements needed to produce contractile force. The α-helical neck, supported by the two light chains, has been proposed to function as a compliant element (4–6), with the light chains imparting stiffness to the lever arm. Therefore, disruption of the light chain binding to the MHC could conceivably impair force generation as well as the transmission of external loads to the myosin active site, leading to a loss of myosin strain sensitivity.
The RLC is a calmodulin homology protein that contains an EF-hand Ca2+–Mg2+ binding site and also a highly conserved phosphorylatable serine, both sites located at the N terminus of the RLC. It has also been shown that both phosphorylation of the RLC (7, 8) and the presence of bound cation (9) play important roles in the structure and function of myosin, reinforcing the notion that there is a high degree of interdomain communication between these sites.
Given the functional importance of the RLC-containing neck domain of myosin, it is not surprising that mutations of the RLC can cause FHC. Population studies have identified two FHC-associated mutations located near the RLC cation binding site, R58Q and N47K (10, 11), that were shown to disrupt Ca2+ binding to RLC and its phosphorylation (12, 13). Several biochemical, structural, and physiological defects have been shown to correlate with the R58Q and N47K mutations when studied in isolation (12), exchanged in skinned porcine papillary muscles (13) or in transgenic mouse models (14, 15).
Here, we studied the mechanical properties of the R58Q and N47K RLC mutations using RLC mutant-exchanged native porcine cardiac myosin, whose RLC was replaced with the recombinant human cardiac RLC mutants (R58Q or N47K), and examined actin filament sliding using the in vitro motility assay. We found that the principal defects caused by the mutations included a reduction in isometric force, power output, and the load at which peak power was achieved. Furthermore, we found that the reductions in power and force are related to changes in the strain-dependent mechanochemistry of myosin, where the mutations disrupt the ability for the strain to trap ADP in the active site and prevent the rebinding of exogenous ADP. These data suggest that FHC-induced changes in strain sensitivity could directly contribute to the disease phenotype.
Results
RLC Depletion and Reconstitution.
Approximately 85% (86 ± 3%; n = 10) of the endogenous RLC was extracted from native porcine cardiac myosin as described in Fig. S1. Reconstitution of the RLC-depleted native myosin with either human wild type (WT) or the human ventricular RLC mutant was accomplished by incubation with an excess of the recombinant WT, N47K, or R58Q proteins leading to 95 ± 13% (n = 11), 97 ± 8% (n = 10), and 92 ± 9% (n = 9) reconstitution, respectively. Recombinant human cardiac RLC wild-type reconstituted myosin (WT) was used as a control for R58Q or N47K reconstituted myosins. There is ∼96% identity between the native porcine RLC and the human cardiac RLC-WT (Fig. S2).
Unloaded Mechanics.
There were no differences in the measured maximal unloaded actin filament velocity, the unloaded duty cycle, or the regulated thin filament velocity between the WT myosin and either N47K- or R58Q-mutant myosins (Table 1 and Figs. S3 and S4). Thus, it appears that N47K- and R58Q-mutant myosin light chains have little effect on unloaded actomyosin contractility.
Table 1.
Native porcine cardiac myosin and human WT-RLC exchanged and FHC-RLC mutant exchanged myosins have similar unloaded kinetics and mechanics
| Native | WT | N47K | R58Q | |
| Unloaded velocity (μm/s) | 0.80 ± 0.06 | 0.79 ± 0.03 | 0.78 ± 0.04 | 0.80 ± 0.05 |
| Vmax for regulated thin filaments (μm/s) | 1.14 ± 0.04 | 1.08 ± 0.05 | 1.06 ± 0.05 | 1.06 ± 0.05 |
| pCa50 for regulated thin filaments | 6.33 ± 0.04 | 6.19 ± 0.05 | 6.26 ± 0.04 | 6.21 ± 0.05 |
There are no significant differences between mutant and WT exchanged myosins.
Myosin Isometric Force.
Because the heart operates against a load due to mean arterial blood pressure and peripheral resistance, the effects of load on myosin biochemistry must be considered when examining the effects of mutations on myosin contractility. The myosin isometric force was measured using a frictional loading assay (16) where α-actinin, a low-affinity actin binding protein, is added to the motility assay surface. The α-actinin transiently binds to actin, exerting a frictional load that opposes the driving force of the bed of myosin. As can be seen in Fig. 1, more α-actinin is required to slow the WT and native myosin, clearly showing that the mutations reduce force-generating capability.
Fig. 1.

Frictional loading assays measuring the isometric force of the native porcine cardiac, wild-type recombinant human (WT), N47K recombinant human, and R58Q recombinant human myosins. The average actin filament sliding velocity and SEs in the mean sliding velocity are plotted as a function of α-actinin added to the motility assay surface. Shown are native (●, solid line), WT (◇, dotted line), N47K (▼, short-dashed line), and R58Q (□, long-dashed line). Data were fit as described in Materials and Methods. Both R58Q and N47K showed significant reductions in isometric force compared with the WT myosin (Table 2).
The load on an actin filament can be calculated as a function of added α-actinin (16) (Materials and Methods), allowing the determination of myosin isometric force. We observed no difference in the isometric force between native and WT myosins (P = 0.677, Table 2). However, both N47K and R58Q reconstituted myosins show significant reductions in isometric force compared with the WT (P < 0.001, Table 2).
Table 2.
FHC–RLC mutants have altered mechanics and kinetics in the presence of exogenous load
| Native | WT | N47K | R58Q | |
| Motility data | ||||
| Isometric force (pN) frictional loading assay | 2,200 ± 200 | 2,000 ± 100 | 490 ± 40* | 1,070 ± 50* |
| Isometric force (pN) from Hill | 3,500 ± 300 | 2,900 ± 200 | 780 ± 70* | 1,590 ± 60* |
| a (pN) | 3,400 ± 300 | 2,900 ± 300 | 840 ± 90* | 1,580 ± 80* |
| Maximal power (fW) | 0.44 ± 0.07 | 0.37 ± 0.06 | 0.07 ± 0.01* | 0.17 ± 0.01* |
| Load where peak power occurs (pN) | 1,400 ± 100 | 1,210 ± 80 | 330 ± 20* | 660 ± 20* |
| KM of velocity for ATP (μM), unstrained | 100 ± 20 | 70 ± 10 | 120 ± 20 | 130 ± 20* |
| KM of velocity for ATP (μM), strained | 60 ± 10† | 40 ± 5† | 70 ± 20 | 80 ± 20 |
| KI of velocity for ADP (μM), unstrained | 160 ± 10 | 130 ± 20 | 190 ± 10* | 200 ± 20* |
| KI of velocity for ADP (μM), strained | 400 ± 60† | 370 ± 70† | 140 ± 20 | 170 ± 20 |
| Transgenic heart and skinned fiber data | ||||
| Vmax fiber ATPase (s−1) | 5.4 ± 0.2 | 5.6 ± 0.3 | 4.6 ± 0.3* | 5.1 ± 0.3 |
| pCa50 fiber ATPase | 5.27 ± 0.01 | 5.28 ± 0.02 | 5.28 ± 0.02 | 5.41 ± 0.03* |
| Isometric force (105 N/m2) transgenic fiber | 0.63 ± 0.02 | 0.64 ± 0.02 | 0.58 ± 0.03* | 0.57 ± 0.02* |
| Cardiac power (mJ/min) transgenic heart | 84 ± 5 | 72 ± 7 | 53 ± 4* | 44 ± 7* |
Force–Velocity Curves and Power.
Force–velocity curves were constructed for native, WT, N47K, and R58Q myosins by converting the α-actinin concentrations to frictional force (16) (Materials and Methods). These data were then fit to the Hill force–velocity relationship (Fig. 2). Whereas there was no difference in either a (P = 0.23) or Po (P = 0.13) between native and WT myosins, both N47K and R58Q myosins showed significant reductions (P < 0.001) in a and Po (Table 2) compared with the WT. Using these values for a and Po, the power output could be calculated for each of the myosins using the Hill description for the force–velocity relationship. There was no difference in either the maximal power output, Pmax (P = 0.40), or the load at which maximal power output occurred, FPmax, between the WT and native myosins (P = 0.09). However, both of the mutants showed significant reductions in maximal power and the load at which maximal power occurred compared with the WT (P < 0.001, Table 2).
Fig. 2.

Force–velocity curves fit to the Hill equation for mutant and wild-type myosins. Using the data from Fig. 1, α-actinin concentrations added to the flow cell were converted to forces (15) as described in Materials and Methods. From these data, force–velocity curves were generated and the data were fit to the Hill equation (34). The mutants show a clear reduction in power as well as a shift in the load at which power is maximal. Native (●, solid line), WT (◇, dotted line), N47K (▼, short-dashed line), and R58Q (□, long-dashed line) are shown.
Strain-Sensitive Kinetics.
We tested whether the changes in power output and the force–velocity relationship of the mutations could result from changes in strain-dependent kinetics by introducing a frictional load into the in vitro motility assay using α-actinin and varying nucleotide concentration.
ADP Affinity Is Strain Sensitive in Cardiac Muscle Myosin.
Because ADP release is rate limiting for unloaded shortening velocity (17) and is thought to be a strain-dependent state in the actomyosin biochemical cycle (18–21), the ADP-induced inhibition of velocity was measured in the presence and absence of load imposed by α-actinin (Fig. 3). The data were fit to a competitive inhibitor model (Eq. 4), using the ATP affinities measured in the absence of ADP (see below). Whereas there was no difference in the KI between the native and WT myosins (P = 0.12), both N47K and R58Q myosins were slightly less sensitive (P < 0.01) to exogenously added ADP compared with the WT (Fig. 3 and Table 2).
Fig. 3.

Exogenous ADP decreases actin filament velocity. The raw unloaded (A), unnormalized loaded (B), and normalized data (C) are shown with fits to a competitive inhibitor model (Materials and Methods). The inhibition constant is similar for all myosins under unloaded conditions (A) (Table 2). (B and C) Load decreases the sensitivity of actin filament velocity to exogenously added ADP. The unnormalized (B) and normalized (C) data were fit as in A. Loading the myosin causes a significant reduction in the ability of ADP to depress actomyosin sliding for native and WT myosins. Loading both native and WT exchanged myosins causes a lowering of the affinity for exogenously added ADP (Table 2). However, load has minimal effect for the N47K and R58Q mutant myosins with KI values similar to the unloaded case, suggesting a loss of strain sensitivity (Table 2). Native (●, solid line), WT (◇, dotted line), N47K (▽, short-dashed line) and R58Q (□, long-dashed line) are shown.
In the presence of a load, both native and WT myosins showed a significant decrease in the sensitivity to exogenously added ADP compared with the unloaded case (P < 0.001 for both myosins). In contrast, neither N47K nor R58Q demonstrated any significant change in ADP affinity in the presence of load (P > 0.05, Table 2). The effect of strain can be quantified using a strain sensitivity ratio defined as the ratio between the strained and unstrained KI values. The strain sensitivity ratios of native (ψADP = 2.46) and WT (ψADP = 2.85) myosins are greater than unity, suggesting that actomyosin ADP affinity is strain dependent. Both N47K (ψADP = 0.73) and R58Q (ψADP = 0.87) myosins showed strain sensitivity ratios near unity, showing that unlike native and WT myosins, the ADP affinity of the mutant myosins is not strain sensitive.
ATP Binding Is Strain Sensitive in Cardiac Muscle Myosin.
The strain sensitivity of ATP binding was also measured in the absence and presence of a load (Fig. 4). Whereas there was no difference in the KM between the native and WT myosin (P = 0.15), the R58Q-exchanged myosin demonstrated a significantly lower affinity for ATP than the WT under unstrained conditions (P < 0.05, Table 2). In the presence of a load (Fig. 4), both native and WT myosins showed a significant decrease in the KM for ATP compared with the unloaded case (P < 0.05). In contrast, the increase in ATP affinity with load was not significant for either N47K (P = 0.10) or R58Q (P = 0.10, Table 2).
Fig. 4.

Exogenously added ATP increases actin filament velocity. The (A) unnormalized and (B) normalized data are shown with fits to a Michaelis–Menten model. Decreasing the concentration of ATP below 100 μM causes a significant depression in actin filament velocity for both unloaded and loaded myosins. Load increases the sensitivity of actin filament velocity to exogenously added ATP. The KM for unloaded myosin (●, solid line) is significantly higher than that for loaded myosin (○, dashed line; Table 2; P < 0.05). Load was introduced into the motility assay using the low-affinity actin-binding protein, α-actinin. A similar shift was observed for WT myosin (Table 2).
Discussion
In this report, we examined the effects of N47K and R58Q FHC-causing mutations in myosin RLC on the mechanical properties of myosin under both unloaded and loaded conditions. We show that both R58Q- and N47K-exchanged myosins exhibit similar unloaded kinetics and mechanics with velocities, calcium sensitivities, and duty cycles that are indistinguishable from the WT myosin (Table 1 and Figs. S3 and S4). This implies that, unlike the F103L mutation of the RLC in skeletal muscle, which showed a reduction in sliding velocity and step size (22), these parameters are unchanged by FHC RLC mutations.
Similar to what we observed previously using transgenic mouse cardiac myosin harboring the N47K or R58Q mutations in the in vitro motility assay (14) and skinned muscle fibers (15), the mutations caused a reduction in isometric force compared with native and wild-type myosin (Table 2). The mutations also appear to cause a reduction in the peak power output as well as the load at which peak power occurs (Table 2).
The isometric force of myosin can be affected by the number of myosin heads available to interact with actin, the duty cycle, or the unitary force. Thus, the reductions in force seen in these mutants could be due to changes in any of these parameters. It is unlikely that the mutations change the number of heads bound to the surface because neither phosphorylation of the RLC nor depletion of the RLC, both more drastic changes in the myosin structure than these one-amino-acid changes, alters the number of heads on the surface (6, 23, 24). Furthermore, the unloaded velocity and the unloaded duty cycle are statistically indistinguishable, suggesting that the step size of the myosin is unchanged by these mutations. Therefore, the data suggest that there is a change in the stiffness of the RLC or the strain-dependent kinetics of the myosin duty cycle, although these two mechanisms are not mutually exclusive.
Strain Sensitivity of Nucleotide Binding.
The strain-sensitive biochemistry of the native and WT myosins was investigated by examining the sensitivity of myosin sliding velocity to the addition of nucleotides both in the presence and in the absence of load. It is apparent from the relationship between velocity and ATP concentration (Fig. 3) that the application of a load causes a reduction in the KM for sliding velocity (Table 2). A strain-sensitive ATP-binding step for myosin has previously been suggested for muscle (18, 25, 26) and nonmuscle myosins (19, 21).
Whereas the WT and native myosins show significant increases in the KM for ATP in the presence of a load, such a shift was not seen for the mutant myosins. This suggests that the mutations disrupt the strain sensitivity of myosin ATP binding. This shift is probably not physiologically significant, because the concentration of ATP in the heart is much greater than the KM. Therefore, although these results provide insight into the mechanochemistry of actomyosin, the observed changes in power and the force–velocity relationship seen in the mutants are unlikely to stem from alterations in ATP strain sensitivity. Instead, mutation-induced alterations in the strain sensitivity of ADP binding are more plausible and more physiologically relevant.
The data presented here, examining the velocity of the WT and native myosins as a function of ADP concentration in the presence and absence of load, show that load causes a significant reduction in the affinity of myosin for exogenously added ADP. In this scheme, straining the myosin traps ADP from ATP hydrolysis in the myosin active site by increasing the affinity of myosin for ADP so that the ADP from ATP hydrolysis becomes unexchangeable with the medium, making the myosin less sensitive to exogenously added ADP. This is consistent with observations of skeletal muscle myosin under strain (24, 27), which also shows a strain-induced reduction in the affinity for exogenously added ADP. This is also consistent with a kinetic scheme in which there is a strain-dependent isomerization in the ADP-bound state (28). Such an isomerization in the ADP-bound state has been proposed to be the strain-sensitive transition in other myosins (19, 21, 28), suggesting a common mechanism for myosin strain-sensitive mechanochemistry.
Interestingly, whereas the WT and native myosins show significant reductions in the affinity for ADP under load, this behavior is not seen for the mutant myosins. It has been shown that the sliding velocity of myosin correlates with the rate of ADP release (17, 29), implying that changes in the sensitivity to ADP would have major consequences for power output and the force-velocity relationship. Furthermore, the changes in strain sensitivity observed here would likely alter the oscillatory work capacity of the heart, consistent with fiber studies of papillary muscle from mice bearing mutations in the myosin light chains (30).
Structural Role of the RLC in Mediating Strain Sensitivity.
The RLC surrounds and supports the α-helical neck region of myosin, allowing for the transmission of the free energy of ATP hydrolysis from the myosin active site down the neck domain, ultimately leading to force and movement. The transmission of force from the lever arm to the active site is necessary for strain sensing and the degree of force transmission depends on the mechanical properties of the lever arm. It is possible that the R58Q and N47K mutations alter the mechanical properties of the neck region of myosin, leading to altered strain-dependent biochemistry. As mentioned earlier, the observed reduction in force in the mutant myosins is consistent with a mutation-induced reduction in the stiffness of the lever arm. Consistent with the idea that the mutations change the mechanical properties of the lever arm, both R58Q and N47K disrupt cationic binding to the RLC (12, 13). Disruption of the calcium binding site has been shown to cause structural changes in the RLC, possibly impairing its role in mechanically supporting the MHC α-helix (3, 4) and transmitting strain to the active site.
Interestingly, cationic binding to R58Q RLC can be restored with phosphorylation of the RLC (12, 13), raising the intriguing possibility that a phosphorylation-induced stiffening of the myosin lever arm could rescue the mutant phenotypes. Our previous research with skeletal muscle myosin suggests that myosin with phosphorylated RLC exhibits a greater strain sensitivity than dephosphorylated myosin (23, 24). It has been shown that phosphorylation of the mutant light chains can restore calcium binding to the RLC and restore mutation-induced changes in the RLC structure (12). Consistent with the notion that modulation of myosin kinetics by phosphorylation is critical for proper cardiac contractility, it has been shown that the hypertrophic response can be attenuated by RLC phosphorylation (31). Recent studies have shown that hypertrophic human hearts show reductions in the levels of RLC phosphorylation (32). Furthermore, a recent report has shown that R58Q mutant hearts show reduced levels of phosphorylation (33), raising the possibility that RLC phosphorylation could be a drug target for alleviating symptoms in patients with RLC FHC mutations.
Relationship to Physiology and Clinical Perspectives.
Here, we show that N47K and R58Q mutant myosins have significant reductions in isometric force and power output. Furthermore, the load at which maximal power output occurs is shifted toward lower loads. These changes in power are due to disruption in force production and changes in the strain-dependent mechanochemistry of the myosin. The shift in power output is functionally significant, because the heart operates against a load imposed by mean arterial blood pressure and peripheral resistance. These mutations, in addition to reducing the maximal power output, would tend to further reduce cardiac output under physiologically relevant loads, placing an additional energetic load on the myosin, consistent with the decreased cardiac function observed in transgenic mouse hearts bearing these mutations (33) (Table 2). Furthermore, the mismatch between the maximal oscillatory work capacity of the mutant hearts and the physiological load could add to the functional deficit, consistent with previous studies showing that light chain mutations can alter the oscillatory work capacity of the papillary muscles (30).
Whereas both the N47K and R58Q mutations have been shown to cause hypertrophic cardiomyopathy, the R58Q mutation appears to cause a more severe phenotype characterized by a rapidly progressing hypertrophy and several instances of sudden cardiac death (11). On the other hand, the disease progression of patients with the N47K mutation is slower and is not associated with sudden death. In vivo data indicate that the N47K and R58Q mutations affect cardiac power similarly (15, 33), whereas in vitro data suggest that the contractile defect in the mutant myosin is more severe for the N47K mutation (Table 2). Our studies with transgenic mice may shed light on this apparent discrepancy. In the transgenic model, the R58Q mutation causes a reduction in isometric force, less efficient usage of ATP, prolonged calcium transients, and activation at submaximal calcium levels (14, 15, 33) (Table 2). The combination was proposed (14, 15) and later confirmed (33) to lead to diastolic dysfunction. The N47K mutation on the other hand did not show any alterations in myofilament calcium sensitivity but did show reductions in force and velocity (14, 15) as well as compromised cardiac work and cardiac power in isolated perfused hearts from transgenic mice (33) (Table 2). Thus our previous and current results suggest that, although both mutations result in altered load-dependent actomyosin biochemistry, the more severe phenotype of the R58Q mutation in vivo is most likely due to the additional effects of altered RLC phosphorylation (33), calcium handling, and energy usage (14, 33). However, it is also worth noting that there are species-to-species variations in cardiac contractility and MHC isoform distribution that may be important in understanding the observed mutation phenotype.
Taken together, the data presented here suggest that defects in strain-dependent mechanochemistry can contribute to disease. Furthermore, the data suggest that future in vitro and in vivo studies examining mutation-based defects in contractility must not only be characterized under unloaded conditions, but also load-dependent changes in myosin mechanochemistry must be considered.
Materials and Methods
Depletion of Native RLC from Porcine Cardiac Myosin and Reconstitution with Human Cardiac WT or N47K- and R58Q-RLC Mutants.
Endogenous native RLC was depleted from the myosin by treatment with 1% Triton X-100 and 5 mM CDTA, pH 8.5, as described previously (6). Myosin depleted of endogenous RLC was then resuspended in a buffer composed of 0.4 M KCl, 50 mM Mops, pH 7, 2 mM MgCl2, and 1 mM DTT mixed in a 1:3 molar ratio with recombinant human cardiac RLC (WT, N47K, or R58Q) dialyzed into the same buffer and incubated on ice for 2 h with slow stirring. RLC-reconstituted myosin was dialyzed in 5 mM DTT and collected by centrifugation. Myosin pellets were resuspended in myosin buffer composed of 0.4 M KCl, 10 mM Mops, pH 7, and 1 mM DTT. Myosin preparations were clarified by ultracentrifugation at 149,000 × g and 4 °C for 30 min then mixed 1:1 with glycerol and stored at −20 °C until needed for experiments.
Unloaded in Vitro Motility Measurements.
The unloaded in vitro motility assays were performed as previously described (14). The myosin duty cycle, the fraction of the myosin biochemical cycle spent attached to actin, was studied by examining the actin sliding velocity as a function of the concentration of myosin added to the flow cell (Fig. S3).
The procedure for regulated motility assays was similar to that for the unregulated motility assays, except tropomyosin and troponin were added to the actin. The velocity of thin filament sliding was measured as a function of calcium concentration and the Hill equation was fit to the data (Fig. S4).
Frictional Loading Assay.
The procedure for the frictional loading assays was identical to that for the unregulated motility assay except that the initial myosin incubation was done with 100 μg/mL of myosin mixed with an appropriate amount of α-actinin (Cytoskeleton Inc.) in myosin buffer.
The data were fit to a viscoelastic model of frictional loads (16). The velocity (V) dependence of actin filament sliding as a function of α-actinin added ([α]) is given by
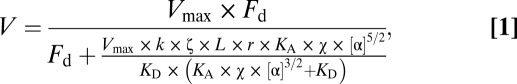 |
where Vmax is the maximal sliding velocity, Fd is the driving force of the bed of myosins, k is the system compliance associated with the α-actinin and the α-actinin linkages, L is the length of a typical actin filament, r is the reach of an α-actinin to bind to an actin filament, KA is the second-order rate constant for α-actinin attachment to actin, KD is the α-actinin detachment rate, and ζ and χ are constants that define the surface geometry of α-actinin on the surface. From these data, the maximal sliding velocity, Vmax, and the driving force of the bed of myosins, Fd, was derived from least-squares fitting in Origin (OriginLab Corp.), using the Levenberg–Marquardt algorithm.
The derivation of Eq. 1, values used for model fitting, and an explanation of model limitations are provided in Greenberg and Moore (16). It is important to note that although this model is based on the physics underlying the basis of the frictional loads in the in vitro motility assay, there is error associated with the absolute values computed with the model due to the fact that several of the physical constants in the model are poorly defined. These errors affect the absolute values obtained from the model but not the conclusions of the data or the comparisons between the different myosins.
Force–Velocity and Power Curves.
The force–α-actinin data were converted to force–velocity curves by considering the viscoelastic nature of the actin–α-actinin interaction. The frictional force, Ffric, is given by
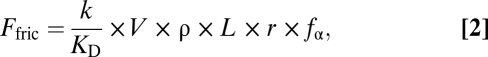 |
where k is the stiffness of an α-actinin molecule, KD is the detachment rate of α-actinin from actin, V is the actin filament sliding velocity, fα is the α-actinin duty cycle, r is the distance over which an α-actinin can reach and attach to actin, ρ is the surface density of α-actinin, and L is the average length of an actin filament (16). Using the converted data, force–velocity curves were constructed. The force–velocity curves were fit to the empirically derived Hill equation
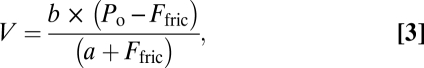 |
where Po is the isometric force and a and b are constants. The power output could then be measured by taking the product of force and velocity. The load at which maximal power output occurs was derived by taking the derivative of the power with respect to load, setting the expression equal to zero, and then solving the expression for the load. This value could then be substituted into the equation for power to find the maximal power output. The errors in the power output and the load at which maximal power output occurs were calculated by propagating the errors from the least-squares fitting.
Strain-Sensitive Kinetics.
The procedure for examining the strain-dependent myosin kinetics is identical to the procedure for a typical motility. The myosin for the motility assay was mixed with 1.75 μg/mL of α-actinin before incubation on the flow cell surface. The motility buffer, with an appropriate amount of ATP and ADP, was designed using Bathe, as was done for the regulated motility experiments. A competitive inhibitor model was fit to the ADP data:
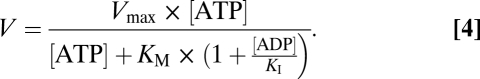 |
Statistics.
At least two different preparations of myosin were used for each experiment. For each actin filament, the average velocity was measured over 5–10 frames. Then, for each flow cell, the velocities of 15–30 actin filaments were averaged together. Each point shown in the data plots represents the average velocity of these 15–30 actin filaments with error bars given by the SE. Data were fit to the appropriate model using a nonlinear least-squares algorithm (SigmaPlot; Systat Software). A two-tailed t test was used to determine significant differences between velocities, using the errors in the fits of the data to the model curve. The P value was calculated from Student's t test distribution and corrected for multiple comparisons using the Holm t test criteria when appropriate.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants HL077280 (to J.R.M.) and R01-HL071778 (to D.S.-C.) and American Heart Association Grant 0815704D (to M.J.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1009619107/-/DCSupplemental.
References
- 1.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115:518–526. doi: 10.1172/JCI200524351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: Linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- 3.Rayment I, et al. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 4.Howard J, Spudich JA. Is the lever arm of myosin a molecular elastic element? Proc Natl Acad Sci USA. 1996;93:4462–4464. [PubMed] [Google Scholar]
- 5.Moore JR, Krementsova EB, Trybus KM, Warshaw DM. Does the myosin V neck region act as a lever? J Muscle Res Cell Motil. 2004;25:29–35. doi: 10.1023/b:jure.0000021394.48560.71. [DOI] [PubMed] [Google Scholar]
- 6.Pant K, et al. Removal of the cardiac myosin regulatory light chain increases isometric force production. FASEB J. 2009;23:3571–3580. doi: 10.1096/fj.08-126672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: Regulation and function. Am J Physiol. 1993;264:C1085–C1095. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- 8.Cooke R. Modulation of the actomyosin interaction during fatigue of skeletal muscle. Muscle Nerve. 2007;36:756–777. doi: 10.1002/mus.20891. [DOI] [PubMed] [Google Scholar]
- 9.Reinach FC, Nagai K, Kendrick-Jones J. Site-directed mutagenesis of the regulatory light-chain Ca2+/Mg2+ binding site and its role in hybrid myosins. Nature. 1986;322:80–83. doi: 10.1038/322080a0. [DOI] [PubMed] [Google Scholar]
- 10.Andersen PS, et al. Myosin light chain mutations in familial hypertrophic cardiomyopathy: Phenotypic presentation and frequency in Danish and South African populations. J Med Genet. 2001;38:E43. doi: 10.1136/jmg.38.12.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flavigny J, et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J Mol Med. 1998;76:208–214. doi: 10.1007/s001090050210. [DOI] [PubMed] [Google Scholar]
- 12.Szczesna D, et al. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J Biol Chem. 2001;276:7086–7092. doi: 10.1074/jbc.M009823200. [DOI] [PubMed] [Google Scholar]
- 13.Szczesna-Cordary D, Guzman G, Ng SS, Zhao J. Familial hypertrophic cardiomyopathy-linked alterations in Ca2+ binding of human cardiac myosin regulatory light chain affect cardiac muscle contraction. J Biol Chem. 2004;279:3535–3542. doi: 10.1074/jbc.M307092200. [DOI] [PubMed] [Google Scholar]
- 14.Greenberg MJ, et al. Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J Mol Cell Cardiol. 2009;46:108–115. doi: 10.1016/j.yjmcc.2008.09.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, et al. Prolonged Ca2+ and force transients in myosin RLC transgenic mouse fibers expressing malignant and benign FHC mutations. J Mol Biol. 2006;361:286–299. doi: 10.1016/j.jmb.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg MJ, Moore JR. The molecular basis of frictional loads in the in vitro motility assay with applications to the study of the loaded mechanochemistry of molecular motors. Cytoskeleton. 2010;67:273–285. doi: 10.1002/cm.20441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siemankowski RF, Wiseman MO, White HD. ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc Natl Acad Sci USA. 1985;82:658–662. doi: 10.1073/pnas.82.3.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veigel C, Molloy JE, Schmitz S, Kendrick-Jones J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nat Cell Biol. 2003;5:980–986. doi: 10.1038/ncb1060. [DOI] [PubMed] [Google Scholar]
- 19.Veigel C, Schmitz S, Wang F, Sellers JR. Load-dependent kinetics of myosin-V can explain its high processivity. Nat Cell Biol. 2005;7:861–869. doi: 10.1038/ncb1287. [DOI] [PubMed] [Google Scholar]
- 20.Kad NM, Patlak JB, Fagnant PM, Trybus KM, Warshaw DM. Mutation of a conserved glycine in the SH1-SH2 helix affects the load-dependent kinetics of myosin. Biophys J. 2007;92:1623–1631. doi: 10.1529/biophysj.106.097618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laakso JM, Lewis JH, Shuman H, Ostap EM. Myosin I can act as a molecular force sensor. Science. 2008;321:133–136. doi: 10.1126/science.1159419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sherwood JJ, Waller GS, Warshaw DM, Lowey S. A point mutation in the regulatory light chain reduces the step size of skeletal muscle myosin. Proc Natl Acad Sci USA. 2004;101:10973–10978. doi: 10.1073/pnas.0401699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenberg MJ, Mealy TR, Jones M, Szczesna-Cordary D, Moore JR. The direct molecular effects of fatigue and myosin regulatory light chain phosphorylation on the actomyosin contractile apparatus. Am J Physiol Regul Integr Comp Physiol. 2010;298:R989–R996. doi: 10.1152/ajpregu.00566.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenberg MJ, et al. The molecular effects of skeletal muscle myosin regulatory light chain phosphorylation. Am J Physiol Regul Integr Comp Physiol. 2009;297:R265–R274. doi: 10.1152/ajpregu.00171.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith DA, Geeves MA. Strain-dependent cross-bridge cycle for muscle. Biophys J. 1995;69:524–537. doi: 10.1016/S0006-3495(95)79926-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith DA, Geeves MA, Sleep J, Mijailovich SM. Towards a unified theory of muscle contraction. I: Foundations. Ann Biomed Eng. 2008;36:1624–1640. doi: 10.1007/s10439-008-9536-6. [DOI] [PubMed] [Google Scholar]
- 27.Capitanio M, et al. Two independent mechanical events in the interaction cycle of skeletal muscle myosin with actin. Proc Natl Acad Sci USA. 2006;103:87–92. doi: 10.1073/pnas.0506830102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nyitrai M, Geeves MA. Adenosine diphosphate and strain sensitivity in myosin motors. Philos Trans R Soc Lond B Biol Sci. 2004;359:1867–1877. doi: 10.1098/rstb.2004.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bloemink MJ, Adamek N, Reggiani C, Geeves MA. Kinetic analysis of the slow skeletal myosin MHC-1 isoform from bovine masseter muscle. J Mol Biol. 2007;373:1184–1197. doi: 10.1016/j.jmb.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vemuri R, et al. The stretch-activation response may be critical to the proper functioning of the mammalian heart. Proc Natl Acad Sci USA. 1999;96:1048–1053. doi: 10.1073/pnas.96.3.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang J, Shelton JM, Richardson JA, Kamm KE, Stull JT. Myosin regulatory light chain phosphorylation attenuates cardiac hypertrophy. J Biol Chem. 2008;283:19748–19756. doi: 10.1074/jbc.M802605200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacques AM, et al. The molecular phenotype of human cardiac myosin associated with hypertrophic obstructive cardiomyopathy. Cardiovasc Res. 2008;79:481–491. doi: 10.1093/cvr/cvn094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham TP, et al. Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice. Cardiovasc Res. 2009;82:84–92. doi: 10.1093/cvr/cvp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill AV. The heat of shortening and the dynamic constants of muscle. Proc R Soc Lond B Biol Sci. 1938;126:136–195. doi: 10.1098/rspb.1949.0019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.