

Abstract
We describe the interactions of two benzimidazole derivatives, astemizole (AST) and lansoprazole (LNS), with anomalous aggregates of tau protein (neurofibrillary tangles). Interestingly, these compounds, with important medical applications in the treatment of allergies and gastrointestinal disorders respectively, specifically bind to aggregated variants of tau protein and to paired helical filaments isolated from brains of Alzheimer's disease (AD) patients. These ligands appear to be a powerful tool to tag brain-isolated tau-aggregates and heparin-induced polymers of recombinant tau. The interactions of AST and LNS with tau aggregates were assessed by classical radioligand assays, surface plasmon resonance, and bioinformatic approaches. The affinity of AST and LNS for tau aggregates was comparatively higher than that for amyloid-β polymers according to our data. This is relevant since senile plaques are also abundant but are not pathognomonic in AD patients. Immunochemical studies on paired helical filaments from brains of AD patients and surface plasmon resonance studies confirm these findings. The capacity of these drugs to penetrate the blood-brain barrier was evaluated: i) in vitro by parallel artificial membrane permeability assay followed by experimental Log P determinations; and ii) in vivo by pharmacokinetic studies comparing distribution profiles in blood and brain of mice using HPLC/UV. Importantly, our studies indicate that the brain/blood concentration ratios for these compounds were suitable for their use as PET radiotracers. Since neurofibrillary tangles are positively correlated with cognitive impairment, we concluded that LNS and AST have a great potential in PET neuroimaing for in vivo early detection of AD and in reducing the formation of neurofibrillary tangles.
Keywords: Alzheimer's disease, benzimidazoles, neurofibrillary tangles, neuroimaging, radiotracers, surface plasmon resonance, tangles, tau aggregates
Introduction
Alzheimer's disease (AD) and multi-infarct dementia are the two most common causes of dementia in the middle-aged and elderly people around the world. The fact that the world population is living longer due to the advances in medical treatment and disease prevention has dramatically increased the incidence of these types of dementia. It is estimated that AD accounts for up to 75% of all dementia cases in the US and Europe. In the US, about five million people are affected by this disease, and mortality is nearly 100,000 per year [1,2]. AD constitutes one of the major health problems in the world (projections for year 2010 indicate that over 30 million people will have AD if no cure is found). In developed countries, the impact of this type of disease on the economy is enormous. Direct plus indirect costs to the US economy for AD is over 174 billion USD per year [3]. Thus, it is evident that scientific efforts in translating neuroscience knowledge into reliable and non-invasive diagnosis tools are highly valuable.
Presently, diagnosis of AD is made according to the NIH-ADAD criteria [4] by the application of several neuropsychological tests that exclude other potential causes of dementia. The degree of accuracy for this method ranges from 50 to 90%. As such, clinical diagnosis of AD can only be truly confirmed histopathologically, by the observation of a large amount of neurofibrillary tangles (NFTs) and neuritic plaques in the neocortex of postmortem brain tissue [5]. NFTs, originally visualized in 1907 by Alois Alzheimer, provided a pivotal impetus for the study of their molecular substrate. “Paired helical filaments” (PHFs) formed by hyperphosphorylated forms of the tau protein are the major components of NFTs [6–8]. Studies have confirmed that, during the course of AD, NFTs in the hippocampus and enthorinal cortex are positively correlated with the cognitive decline. Clinical manifestations of AD are preceded by a series of brain pathophysiological events that eventually lead to the aggregation of hyperphophorylated tau protein. Based on the correlations of these events and cognitive decline, several markers have been proposed for the early diagnosis of AD, including levels of amyloid-β (Aβ) peptide fragments in serum and blood cells [9,10], decrease in Aβ1–42 in cerebrospinal fluid (CSF) [11,12], and phosphorylated isoforms of tau in the CSF that positively correlates with cognitive impairment [12–14]. However, none of these markers has been able to discriminate early AD from other types of dementia, or they require invasive procedures such as lumbar puncture to obtain CSF samples, making their use as routine diagnostic/monitoring techniques more difficult. Recently, the potential roles of peripheral inflammatory markers [15–17], altered lipid metabolism and oxidative stress [18–20], or redox iron [20] in the subclinical stages of AD have been suggested as potential biological sensors for AD. This has opened interesting avenues to address the question of how early changes in the levels of peripheral metabolic biomarkers can contribute to the diagnosis of AD.
Tau protein is the most important microtubule associated protein (MAP) in neuronal axons and plays critical physiological roles in stabilizing microtubules and inducing their assembly [21]. However, under pathological conditions, tau self-aggregates into PHFs, which turn into NFTs during the course of AD. Important advances toward our understanding of in vitro tau polymerization have been provided by several studies, including those from our laboratory [7,22,24, 25]. However, the mechanisms underlying the structural transition from an innocuous, natively unfolded tau to its neurotoxic polymers remain unknown, as is the detailed structural features of these macromolecular aggregates.
According to the neuroimmunomodulation theory on AD [17,26,27], a chronic-asymptomatic inflammatory process of the central nervous system (CNS) is responsible for the earliest changes that precede AD clinical onset in the vast majority of sporadic AD cases, including the formation of tau oligomers in the transenthorinal cortex. In this neuroinflammatory process, abnormal phosphorylation of tau and long-term activation of the innate immune system occurs, leading to cytoskeletal alterations such as tau protein aggregation and the formation of PHFs. Several factors combine to trigger innate immune system alarm mechanisms resulting in the overproduction of cytokines such as tumor necrosis factor-α (TNF-α) and others [16,17,20,28,29] associated with progressive cognitive decline. In this context, innovative diagnostic approaches for AD must be based not only on the determination of hyperphosphorylated tau, or redox iron levels [20], but also on correlations with the apolipoprotein E4 allele [12] and, more importantly, on the visualization of early pre-tangles of tau as a more accurate way to assess AD pathological process.
Elucidating tau structure and its conformational changes upon ligand interaction is critical for future investigations on high affinity neuroimaging tracers for tau aggregates as putative pathognomonic markers for the early diagnosis of AD [30]. Some ligands for tau aggregates have been discovered, e.g., thioflavine derivatives, that bind to brain isolated PHFs [31]. Furthermore, it is known that Thioflavine S (ThS) binds to PHFs with a higher affinity than that to straight filaments found in AD [32]. The anticancer drug estramustine-P interact with tau as well as with MAP-2 [33], and some benzimidazoles, benzothiazoles, and quinolines have affinity for tau protein in vitro [34, 35]. The development of specific radio-ligands designed to obtain in vivo images of Aβ-plaques and NFTs, whether by means of positron emission tomography (PET) or single photon emission computed tomography (SPECT), is a very active area [36]. These technologies will allow clinical and basic researchers to evaluate the effects of a variety of anti-tau therapies, currently being developed by the pharmaceutical industry. In addition to the early diagnosis of AD, this type of technology will provide information on subjects at high risk of developing AD or another specific neurodegenerative disorder.
Different neuroimaging approaches have been investigated as a diagnostic tool and as an evaluator of the temporal course of AD. One of the comparative advantages of neuroimaging over other markers is that is non-invasive; the imaging can be repeated in series, and at different time intervals, to evaluate longitudinal changes and to identify brain anomalies before the clinical symptoms can be diagnosed. This makes the application of preventive therapies, such as cognitive rehabilitation and others, feasible [37–39]. Here, we describe that benzimidazolic drugs, such as astemizole (AST) and lansoprazole (LNS), bind aggregated tau variants and may be potentially clinically useful as radiotracers for neuroimaging in AD diagnosis [40].
Material and Methods
Chemicals and reagents
LNS and AST were purchased from Sigma Aldrich (catalog #L8533 and A2861, respectively). Solvents and reagents were of HPLC grade and were obtained from E. Merck. Heparin was from Sigma Aldrich (catalog #H4784). 6E10 monoclonal antibody was purchased from Covance (catalog #SIG-39300).
Animals
All experimental protocols involving animals were reviewed and approved by the Ethic Committee of the Faculty of Sciences of the University of Chile. Male C2 mice, 30 g average weight, were used. The animals had free access to food and water until 12 h prior to be used, at which time only food was removed. Mice were injected with 8 mg/Kg of AST and 16 mg/Kg of LNS in 100 μL injection volume, and sacrificed by administration of ketamine (100 mg/kg, i.p.). At different time intervals (0,30, 60, 90, and 180 min) after drug administration, at least 500 μL of intra-cardiac blood and brain were taken for determination of AST and LNS by HPLC. Blood was collected in heparinized tubes and treated immediately after collection.
Protein and PHF isolation
Isolated preparations of PHFs were obtained according to the procedures described by Jicha et al. [41], with minor modifications, from brains of patients with clinical diagnosis of AD. Postmortem brains from 65 years old patients diagnosed with probable AD and confirmed by neuropathology were from the Brain Bank of the Albert Einstein College of Medicine. Brains were selected on the basis of Western blot screening with monoclonal antibody PHF-1, that epitope-mapped to 396 phosphorylation of tau. Brain extracts were made by homogenizing 78–80 g of selected cortical grey matter in TBS buffer (10 mM Tris and 150 mM NaCl, pH7.4) to yield a 0.1 g/mL mixture containing 1 mM phenyl-methylsulphonyl fluoride. For this procedure, we used a Power Gen 125 homogenizer (Fisher Scientific) on maximal setting. All human tissues were frozen < 7 h after death. Homogenates were centrifuged at 27,000 g for 20 min and the supernatants were decanted, the pellets were re-homogenized in half of the original volume and recentrifuged. The supernatants then were pooled and filtered through a 3.7 cm-high, 2.7 cm-diameter Sepharose 400 superfine to prevent particulate matter from accumulating at the top of the column. The final extract were eluted through a MC-1 column at a flow rate of 50 mL/h for up to 36 h, after which the column was washed with TBS at a flow rate of 100 mL/h for at least 24 h. The MC-1 antibody/affi-gel matrix was poured into a column of 35.0 × 2.0 cm and washed with 50 volumes of Tris buffer saline. Before PHFs purification columns were washed with 2 volumes of 3M KSCN, followed with 5 volumes of TBS. Bound antigen was eluted with 3 M KSCN and fractions were analyzed for protein concentration. Samples containing at least 50 μg of protein/mL were pooled and dialyzed overnight at 4°C against TBS buffer and PHF were characterized by SDS PAGE and western blot using PHF-1 mAb. Additionally, the polymeric form of tau protein isolated from brains was confirmed by fluorescence spectroscopy using the highly fluorescent dye ThS in a Spectramax5 fluorometer, since ThS emission at 570 nm increases in the presence of PHFs. The MC-1 columns were reused for at least six times before replacing the gel. MC-1 is a conformational dependent monoclonal antibody epitop-mapped to 5-14/312-322, specific pathological folding of tau protein within the PHFs. DA9 is a sequence antibody raised against 102 and 150 residues of tau protein. PHF-1 is a sequence antibody raised against the 396, 404-P tau residues.
Preparation of heparin-induced tau filaments (HITF) and Aβ peptide polymers
The human tau protein expressed from cDNA clone of htau441 in E. coli was purchased from Sigma. The recombinant tau (1 mg/mL) was incubated with 200 μg/mL heparin in 30 mM MOPS, 1 mM 4-(2-aminethyl) benzene sulphonylfluoride) pH 7.4 at 30 °C for 21 days. 1 μL samples were taken from the mixture and analyzed by western blot at 0, 3, 22, and 45 h. Samples of tau aggregates were further analyzed by transmission electron microscopy (TEM) to observe their morphology at day 21. Aβ1–42 (molecular mass 4514.1 g/mol) ultra pure hexafluoro-2-propanol(HFIP) was used. HFIP was evaporated by lyophilization and Aβ dissolved in 50 mM potassium phosphate buffer, pH 7.4 to a final concentration of 1.0 mg/ml. Tau protein (1 mg/ml) was incubated in the presence of 200 μM heparin for 21 days with continuous shaking and polymerization was verified by TEM analysis and western blots.
Transmission electron microscopy studies
We used negative staining and 600-mesh carbon-coated copper grids that were glow-discharged twice (CTA 010, Balzers Union). The grids were placed on a drop of protein solution, incubated for 5 sec, then washed with two drops of water, placed on a drop of 2% uranyl acetate (pH 4.5), and incubated for 5 sec. Staining and washing solutions were first filtered through a 0.2 μm membrane. 2% uranyl acetate solution was freshly prepared every time. The replicas were examined at 100 kV in a JeolX100 electron microscope.
In vitro binding assays
The dissociation constant (Kd) and maximum specific binding of AST were determined by standard Scatchard analysis as described [34]. Briefly, 3H-AST solutions of 0-2.5-5.0-7.5-10.5-12.5 nM were incubated independently with either PHFs (40 μg/mL) or with heparin-induced polymers of recombinant tau protein (0.01 mg/mL) under darkness for 4 h at room temperature. Then, the mixture was cooled on ice to avoid disruption of the equilibrium, the cold mixture was vacuum filtered with wathman G/SD filters, and the reactivity retained in the filter counted in the presence of 2 mL of scintillation liquid in a TRICARB 2100 TR Liquid Scinitilation Analyzer. The scintillation signal displayed by each sample was expressed in cpm and registered for 120 sec after 10 min of previous incubation in darkness. Bound and free 3H-AST was calculated from the cpm values obtained from each sample by using a calibration curve constructed by incubating a wide range of concentrations of [3H]-AST with 2 mL of scintillation liquid for 10 min and reading cpm under the same conditions of the samples. For the inhibition assays 3H-AST was incubated in darkness at room temperature for 4 h in the presence of different concentrations, in the range of 3 nM to 9.0 μM of the unlabeled molecule. The mixture was vacuum filtered with Wathman G/SD filter and the reactivity retained in the filter was counted in the presence of 2 mL of scintillation liquid. Values for half-maximal inhibitory concentration (IC50) were determined from displacement curves of three independent experiments using GraphPad Prism software (GraphPad Software, San Diego, CA), and those for inhibition constants (Ki) were determined using the Cheng–Prusoff equation: Ki = (IC50/(1 + D/Kd), where IC50 is the concentration that inhibits 50% percent of the binding of [3H]-AST, D is the final concentration of the 3H-AST, and Kd is the dissociation constant obtained from the Scatchard analysis.
Docking studies with AutoDock
In molecular docking, we attempted to predict the structure (or structures) of the intermolecular complex formed between a domain of tau and both LNS and AST. Docking is widely used to suggest the binding modes of protein inhibitors. Most docking algorithms are able to generate a large number of possible structures, and so they also require a means to score each structure to identify those of most interest. In general, the “docking problem” is concerned with the generation and evaluation of plausible structures of intermolecular complexes. 3D structures of AST and LNS were built with the molecular visualization software MOLDEN [42] and were further geometrically optimized using the semi-empirical quantum chemical method AM1 implemented in the MOPAC 7.0 computer software package (MOPAC, version 7, U.S. Air Force Academy: Colorado Springs, CO). Partial atomic charges were computed using MOPAC applying the AM1 Hamiltonian. Atomic coordinates of the tau fragment 386TDHGAE391 were extracted from the Protein Data Bank (PDB code: 2v17). The fragment protein structure were visualized and built with the software MOLDEN. The atoms belonging to antibody MN423 and all water molecules were removed from the protein structure, and polar hydrogenatoms were added assuming a physiological pH value of 7.0. The CVFF force field was used [43] to assign the partial atomic charges to the tau protein fragment. After that, docking of the two drugs mentioned above into the tau protein fragment was carried out using the AutoDock4 [44] program. The protein fragment and drug structures were opened and prepared with the AUTODOCKTOOLS version 1.4.4. Non-polar hydrogen atoms were merged, for the drug structures, within the united atom approach using the program AUTOTORS, which is part of the AutoDock4 program package. This program also defines which bonds are allowed to rotate freely during the automated docking process and saves the drugs in the PDBQ format. All rotatable drug bonds defined by default for the program were allowed to rotate during the automated docking process. The solvation parameters were added and the prepared protein fragment structure was saved in the pdbqs-format suitable for calculating energy grid maps using the program AUTOGRID. A grid size of 100 × 100 × 100 points with a spacing of 0.375 Å was applied, which corresponds to a cube with an edge length of 37.5 Å. The center of the grid box was defined as the atomic coordinates of the alpha carbon belonging to the residue His388 in the modeled tau fragment. Docking calculations were performed with a maximum number of 300 optimization steps in the local search. For the docking runs employing the standard optimization algorithms incorporated in AutoDock4, the population size was set to 150 individuals and iterated through 2,500.000 energy evaluations. Other parameters were set to their default values. Accessibility of the ligands were analogous to the procedure described for other docking software, with the difference being that only the most favorably bound ligand-protein configuration was presented for each drug.
Experimental octanol/water partition coefficients
PBS buffer was saturated with PBS and 1-octanol before use. Test compounds were dissolved in 1-octanol and shaken with equal amounts of PBS for 30 min at room temperature. After centrifugation at 2,000 per min for 15 min, the absorbency of 1-octanol layer was measured at the peak wavelength of the absorbance spectrum of each compound using single cubette spectrophotometer. The octanol/water partition coefficients were determined by comparing the absorbency with that before shaking with PBS [34]. Each data point was performed in triplicate.
Parallel artificial membrane permeability assay (PAMPA) methods
A modification of the UV-based PAMPA method was used in this study, with data collected at room temperature (25–28°C), following the normal double-chamber PAMPA protocol [45]. The pH of the donor solutions was varied, while the receiver solutions had the same pH 7.4. The PAMPA sandwich was assembled, loaded into the controlled environment chamber, and allowed to incubate for 16 h for all the molecules, as they are mainly lipophylic compounds (octanol–water log 0.9–2.8) with magnetic stirring. The equation used to calculate the membrane, aqueous boundary layer, and intrinsic permeability was: Pe= −218.3/t * log [1–2Ca(t)/Cd(t0)]* 10−6 cm/s [45].
Fluorescent spectroscopy analysis
PHF and ThS fluorescence were measured using a SpectraMax5, Molecular Devices microplate spec-trofluorometer (Molecular Devices, Sunnyvale, CA) using 96-well Maxcsisorp microwell SH plates. Final volume for all the measurements was 100 μL. Optimal exitation and emission wavelengths for the tau-ThS mixture were 360 and 460 nm respectively.
High performance liquid chromatography analysis
The mobile phase was comprised of ammonium acetate 50 mM and the flow-rate of the mobile phase was 0.2 ml/min, run under isocratic. The buffer was filtered through a Millipore 0.22 mm filter and degassed prior to use. Samples were separated using a reverse-phase C18 column 125 × 4 mm (5 μm particle size). The optimal UV detection for LNS was done at the wavelength of 284 nm and at 277 nm for AST. Output data from the detector was integrated via an LaChrom chromatographic software. In these experimental conditions the retention time for AST was 5.1 min and for LNS the retention time was 10.5 min. Blood sample were extracted by classic liquid-liquid extraction with 500 μL of a mixture of ethyl ether/chloroform 70/30. Brain samples were homogenized in TBS buffer in 500 μL of TBS buffer and extracted with the same procedure of blood samples. LNS and AST recovery percentages for brain and blood were an average of 85%. The organic fraction was further evaporated under nitrogen and the remaining pellet was dissolved in 100 μL volume of mobile phase, ammonium acetate 50 mM. The samples were placed in an automatic auto sampler and 20 μL of each sample was injected to the HPLC system.
Surface plasmon resonance (SPR)
AD-PHF and polymers of Aβ peptide were immobilized on separate flow cells of a CM5 sensor chip by standard amine chemistry reactions. TBS buffer was injected to the surface at a flow of 100 μL/min. AD-PHFs were injected at a concentration 50 μg/mL after that the remaining active sities of the C5 Chip were inactivated by a consecutive injection of ethanolamine 1.0 M pH 8.5. The density of proteins on the amyloid channel was 6227 RU (resonance units) and in the AD-PHFs channel was 8822 RU. One RU represents approximately 1pg protein/mm3 of chip surface. It has been established that at least 50 pg/mm3 of protein must be coupled to the chip surface in order reach sufficient protein for binding studies [46]. All the solutions and the running buffer had 4% DMSO. For these experiments BIA Evaluation Software was used to make kinetic evaluations and sensograms. For all final measurements, reference and blank data along with solvent values were subtracted from sample lectures. The flow cell temperature was 25°C. The dissociation was monitored for 40 min for all the drugs and monoclonal antibodies. At the end of every experiment, monoclonal antibodies against phosphorylated tau (PHF-1) and Aβ peptide (6E10) were injected to confirm the integrity of the active binding surface containing PHFs and amyloid (Fig. 3A, C). The polymeric forms of both tau protein and amyloid peptide were confirmed by the injection of ThS (Fig. 3B). The surface was regenerated by injecting 10 μL of 10mM KOH after injection of each sample. In our SPR experimental conditions we decided to use potassium salts instead of sodium salts for all our buffers.
Fig. 3.
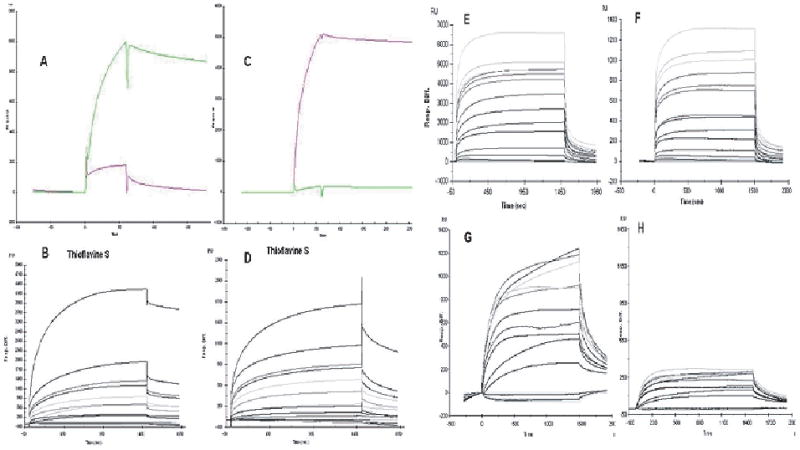
Binding data obtained by surface plasmon resonance (SPR). Sensograms of aggregated amyloid peptide and AD-PHFs covalently inmobilized on a C5 biosensor surface. PHF-1 antibody (A) and 6E10 antibody (C) were injected into the chip to verify the presence of AD-PHFs and amyloid aggregates. Both amyloid (B) and PHF (D) surfaces were also reactive to the injection of ThS, which corroborates the presence of polymeric forms of the amyloid and tau protein immobilized on the chip surface. AST displayed higher signals in its interactions with AD-PHFs (E) than with amyloid (F). LNS differential interaction with AD-PHFs (G) and aggregated amyloid (H) was also verified by SPR.
Results
Characterization of in vitro formation of tau filaments and PHFs by trasmission electron microscopy
Human recombinant tau protein (441aa) was incubated with 200 μM heparin and the resulting filaments (HITF) of 15–20 nm diameter average were monitored by TEM (Fig. 1A). PHFs containing tau of AD type (AD-PHF) were purified from tissue samples from the temporal cortex of postmortem brains clinically diagnosed with AD and analyzed by TEM (Fig. 1B). Im-munoaffinity purification of AD-PHF was performed using MC-1, a conformation-dependent monoclonal antibody. The most reactive PHFs samples to PHF1 mAb, which recognizes tau phosphorylated on Ser-ine396, were used. Interestingly, filaments isolated from AD cases (B) were shorter and twice the diameter than those formed in vitro and displayed significant more turns than the heparin-induced filaments. Thus the AD-PHF preparations have mainly helical filaments of tau, while heparin-induced filaments samples are mainly composed of straight filaments. Senile plaques are abundant in the brain of AD patients. These preparations were used to carry out binding studies of AST and LNS to these tau aggregate preparations. In addition, we decided to study the affinity of benzimidazoles for aggregates of Aβ.
Fig. 1.
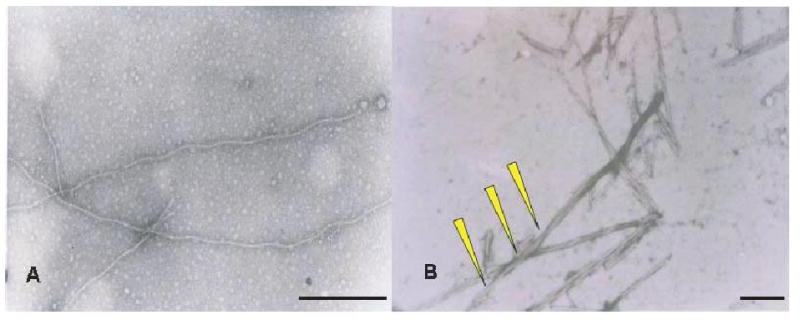
Characterization of AD-PHF and tau filaments by TEM. Filaments obtained from recombinant tau protein induced by 200 μM heparin (A) and AD-PHF from AD patients (B) were characterized by transmission electronic microscopy (TEM). In these macrographs, yellow arrowheads point torsion of the polymers. In the preparations obtained from AD patients, single straight filaments were predominant as compared with the PHF preparation where PHF are predominant (Bars = 80 nm).
Scatchard analysis of labeled benzimidazoles
The structure of AST and LNS are shown below. The Scatchard analyses of [3H]-AST as the labeled benzimidazole with either synthetic Aβ peptide (Fig. 2A), in vitro tau filaments (Fig. 2B), and AD-PHF purified preparations (Fig. 2C) are shown.
Fig. 2.
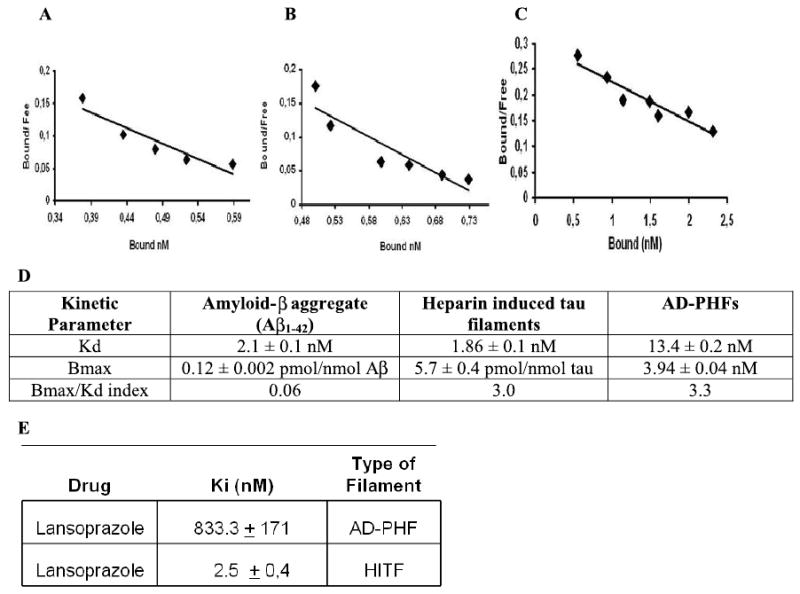
Astemizole displays differential affinities for tau aggregates and Aβ peptide aggregates. Scatchard analyses of [3H]-AST were performed in order to determine Bmax/Kd index. The data shows that atemizole displays differential affinities for amyloid aggregates (A), heparine induced tau filaments (B), and AD-PHF (C). Each point of the curve is the mean of three replicas. Kinetic data is summarized in Table D. Inhibition studies with non labeled LNS and [3H]-AST showed high affinity of LNS for aggregated forms of tau (E).
These experiments showed that [3H]-AST binds with higher affinity to tau filaments than to Aβ peptide aggregates. This was clearly demonstrated by higher values of the ratio Bmax/Kd, which was 0.06 for amyloid aggregates and 3.0 and 3.36 for tau filaments. According to the abundance of these molecular targets in the brain, this parameter is considered suitable for new radiotracers for brain imaging [36]. The kinetic parameters of AST binding with the three types of filaments is shown in Fig. 2D. A strong binding affinity, indicated by a relative low Ki within the series, was displayed by LNS (Fig. 2E) in their interaction with HITF. The results of the inhibition assays performed with PHFs and heparin-induced tau polymers yielded different results, as summarized in Fig. 2E. Thus, AST and LNS are good candidates for developing brain/blood ratio pharmacokinetic studies.
Surface Plasmon Resonance analysis
In order to provide more consistent evidence on the direct binding of AST and LNS to the surface of AD-PHFs and amyloid polymers, we performed comparative binding studies by SPR with the polymers coupled to a C5 Chip. SPR analysis demonstrated that the binding affinity of AST to PHFs surface is comparable to ThS (Fig. 3E, F). In these experiments, increasing concentrations of AST, LNS, and ThS were injected into PHFs and amyloid polymers immobilized in a CM5 chip. The measurements were done in a Biacore3000 optical biosensor equipped with research grade CM5 sensor chips. The AST and ThS binding data was comparable for AD-PHFs coupled to the Chip surface. The analysis of these experiments also allowed us to corroborate the higher affinity of AST and LNS for AD-PHFs than for aggregated amyloid, as the response curves (RU) were at least five times higher for AST and LNS when injected into the AD-PHFs surface as compared to those obtained from amyloid surface (Fig. 3E, F, G, H). Several kinetic analysis were performed with the binding data of AST, LNS, and ThS, but apparently the kinetic characteristics of these interactions does not fit the classical one-site or two-sites binding models. However, it was clearly a dose-dependent interaction. In order to compare the binding affinities of AST, LNS, and ThS for PHFs from AD patients, we analyzed the binding response relative to the molecular weight, which is an index for estimating the interaction of a compound with molecules sited on a biosensor chip [46]. For this analysis, the binding response was obtained from the equilibrium response (Req) value or the maximum response value in the sensorgram obtained after the injection of 150 μM of each drug and divided by the corresponding molecular weight [46]. For this analysis, the binding responses (RU/Da) were normalized against ThS. The binding affinity expressed as RU/Da and normalized against ThS were 1.0, 2.6, and 0.7 for ThS, AST, and LNS. According to this analysis, AST and LNS have high affinity for pathological aggregates of tau protein immobilized on the sensor surface. Remarkably, AST showed at least 2.6 folds the affinity of ThS, which is a widely used for pathological staining for tau aggregates.
Structural studies on the binding of AST and LNS to tau: docking studies
The results for the bioinformatical studies of the affinity of AST for the pronase resistant domain of tau 386TDHGAE391 demonstrated that this drug binds, with very high affinity, to this segment of tau protein (Fig. 4). It is important to mention that tau is natively a random-coil, unfolded protein. Therefore, it has not been possible to obtain the exact crystalline structure of the entire protein. This is a limitation for the systematic evaluation of potential binding sites by bioinformatical methods. Therefore, to gain the most accurate insight to tau structure we modeled the only fragment that has been crystallized and inmuno-isolated from AD-PHFs preparations [47,48]. The binding energies we report here were obtained by using the Autodock® program. In these methodology, the lower values of binding energies, the higher affinity for tau protein (Table 1). According to our results, AST displayed a very high affinity for this fragment of tau ligands. Conversely, metronidazole displayed a low affinity for this domain of tau. It is relevant to mention that 386TDHGAE391 is a very good candidate to be a binding site for these potential radiotracers, as it is surface-exposed in the PHFs of AD patients [48]. All these results support our previous findings for AST by the SPR and Scatchard analysis (Fig. 2) and for metronidazole (data not shown) on inhibition assays.
Fig. 4.
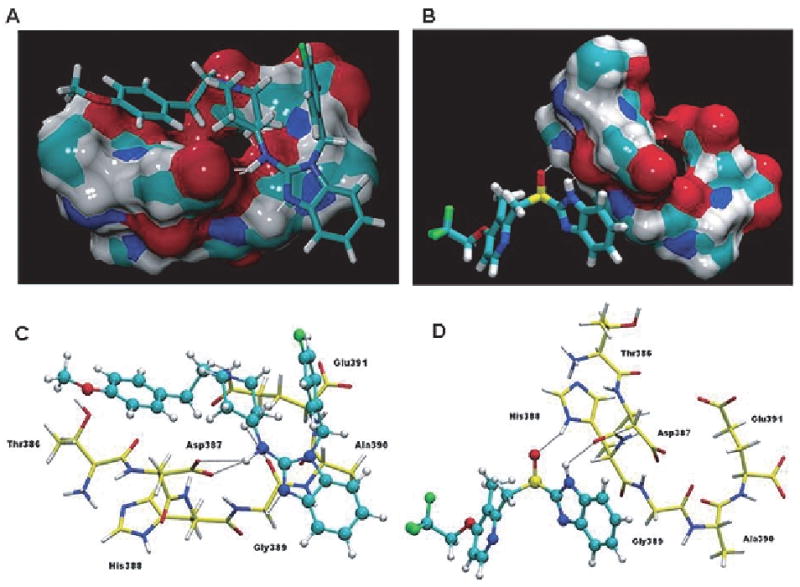
Structures of the drugs astemizole and lansoprazole and their binding interactions with the C-terminal hexapeptide 386TDHGAE391 of PHF core. (A) The interactions of the drugs with the core PHF-tau fragment drawn as an isosurface. The interaction of AST with PHF fragment shows a high fitting on the tau-PHF surface when it is compared with LNS. The H-bonds are drawn in white dashed lines. (B) Detailed view of the interactions between PHF-tau fragment and the drugs studied by docking experiments. Residues of the PHF fragment are shown in licorice representation and drugs are presented in ball-and-stick representation, with carbon atoms colored in yellow (PHF fragment) and cyan (drugs). The hydrogen bonds are depicted as black dashed lines. The same orientation has been used for the two snapshots (A,C and B,D) for the sake of clarity.
Table 1.
Detailed description of the interactions analyzed by Autodock4 between the core386TDHGAE391 of tau and astemizole and lansoprazole. These data corresponds to the most favorable localization of each drug in its interaction with the386TDHGAE391 domain of tau. The Autodock4 binding energies were −3.02 and −1.27 kcal/mol, for AST and LNS respectively
| Drug | Comment | ||
|---|---|---|---|
| Astemizole | Lansoprazole | ||
| Interaction 1 | astemizole-NH–Asp387 | --- | Bifurcated and strong hydrogen bond interaction between the OD1 and OD2 oxygen atoms in the side chain of Asp387 and the amine group that bridges the benzimidazole and piperidine rings in astemizole. The H-bond distances are 2.23 and 2.52 Å, respectively. |
| Interaction 2 | astemizole-N–Gly389 | --- | Hydrogen bond, of the type C-H—N (2.46 Å), between the side chain of residue Gly389 and the N atom in the benzimidaloze ring. |
| Interaction 3 | astemizole-CH2—O=C-Gly389 | --- | Hydrogen bond interaction, of the type C-H—O (2.24 Å), is formed between one of the CH2 groups in the piperidine ring and the oxygen atom in the backbone of residue Gly389. |
| Interaction 4 | astemizole-CH2—O=C-Asp391 | --- | Hydrogen bond interaction is established between the ethyl group, which bridges the methoxyphenyl and piperidine groups, with one of the oxygen atoms in the side chain of residue Glu391. The distance for this interaction is 2.49 Å. |
| Interaction 5 | --- | Iansoprazole-NH–Asp387 | Strong hydrogen bond interaction is formed between the oxygen atom in the backbone of residue Asp387 and the NH group in the benzimidazole ring of Lansoprazole. The distance for this interaction is 2.08 Å. |
| Interaction 6 | --- | Iansoprazole-S=O—His388 | Strong hydrogen bond interaction, of the type S=O—H-N (1.89 Å), is established between the oxygen atom in the sulfinyl group of lansoprazole and the NH group in the side chain ring of the His388 residue. |
Staining of PHF with ThS
As we described in the experimental section, we worked with PHFs from tau protein isolated from brain of AD cases according to the protocol described in the methods section. PHFs were stained according to the protocol described by Santa-Maria et al. [32]. Then, we compared the staining patterns of benzimidazoles with the staining of thioflavine-S and PHF-1 immunostaining. Brains were obtained from autopsy cases of patients with clinical diagnosis of AD. We selected the tissues that were positive for at least two neuropathological criteria for AD diagnosis; the presence of i) amyloid plaques and ii) NFTs of tau protein. Final selection of the cases was done based on the reactivity to PHF-1 monoclonal antibody. The colocalization of the signals from LNS and PHF-1 mAb corroborates the affinity of LNS for AD-PHF. AST displayed very high affinity for tau, but is virtually non-fluorescent.
Experimental LogP and in vitro blood-brain barrier penetration
In these experiments we assessed the capacity of each compound to penetrate the blood-brain barrier (BBB) by two different models. According to what is described in Material and Methods, we used partition coefficient determination (LogP) and PAMPA. Table 2 summarizes the results of both methods. Brain permeability of these compounds was compared to clidinium bromide, which does not penetrate the BBB, and to sodium thiopental, which is a lipophylic drug that easily penetrates the BBB by passive diffusion mechanisms. Here we demonstrated that AST and LNS pass the BBB by passive mechanisms and would reach suitable brain/blood concentration ratios for imaging radiotracers. The results of the binding studies demonstrated that both compounds AST and LNS exhibit a high CNS permeability as evaluated by LogP, and using thiopental as a positive control and clinidium bromide as negative control. Considering the relevance of the permeability results and the affinity displayed by all these drugs for PHFs, we decided to perform pharmacokinetic studies with LNS and AST, which have both high affinity for PHFs and are clinically approved by FDA for other uses in humans. Brain/blood concentration ratios for AST and LNS are not completely clear. This is important in the context of a brain radiotracer, which should reach the brain within the first 60 min after administration, and should not remain in the CNS for a long period of time. This is relevant, considering that a useful PET radiotracer must reach the brain before the half-life of the radioisotope, which is 120 min in the case of 18F used for PET tracers.
Table 2.
Brain penetration as analyzed by LogP and PAMPA assays for astemizole and lansoprazole
| Drug | PAMPA (Pe) (cm/s) | PAMPA CNS permeability | Log P (oct/PBS pH7,4) | Log P CNS permeability |
|---|---|---|---|---|
| astemizole | 1.447* 10−4 | + | 5.57 | ++ |
| lansoprazole | 3.63* 10−5 | + | 1.47 | ++ |
| Clidinium bromide | 0.0029* 10−5 | − | −0.092 | −− |
| Tiopenthal | 1.7* 10−5 | + | 1.4 | + |
Pharmacokinetic studies
AST was analyzed by HPLC/UV in blood and brain at different time points. The results (Fig. 6) show that AST reaches the maximum blood concentration at 28 min and the maximum concentration in brain was reached at 55 min. This is compatible with a radiotracer use [49]. Another finding of these studies is that after the first 60 min, the brain/blood concentration ratio tends to stabilized, which means that AST somehow is not cleared from the brain with a first order kinetic, as it for the rapid clearance occurred in peripheral blood. The pharmacokinetic profile of LNS is different from AST, as it reaches higher concentrations in the brain than AST and the time to maximum concentration in blood is almost equivalent. However, the time at which the concentration is maximum in the brain is completely different, as LNS reached the brain at 37.4 min and AST reached the brain at 55 min. Altogether, the data on the pharmacokinetic profiles for AST and LNS show that both molecules reach the brain with in the first 60 min, and brain/blood concentration ratios are increased and tend to stabilize within this timeframe.
Fig. 6.
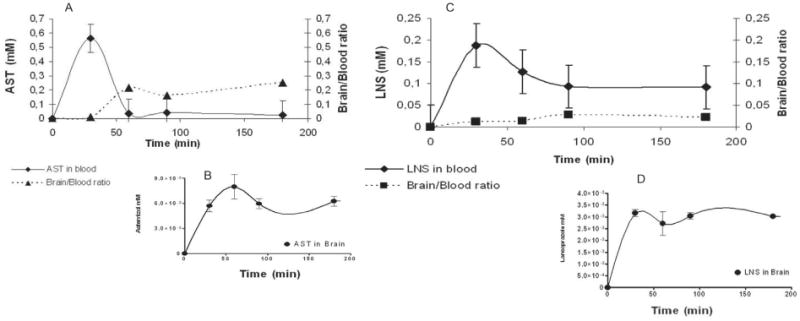
Comparative pharmacokinetic analysis of astemizole and lansoprazole. The concentration of LNS and AST was measured in blood (A, C) and brain (B, D) at different time points (0, 30, 60, 90, 180 min) after intravenous injection. Brain/blood concentration ratio (A, C) were also determined. The results are the mean of three independent experiments.
Discussion
Tau from human brain and obtained in vitro as targets for new radiotracers
Formation of NFTs during the course of AD, and its correlation with cognitive decline, has opened interesting avenues for the potential role of tau oligomers in early stages of disease. In fact, granular tau oligomers with a pre-filamentous structure are present in the frontal cortex of asymptomatic patients displaying Braak-stage I neuropathology. This suggests that tau oligomerization occurs before NFT formation and before expression of clinical symptoms of AD. In this context, the search for a ligand with high affinity for tau polymers or aggregates may provide a promising avenue for future studies on PET radiotracers for imagining of tau pathology. NFTs are mainly brain pathological structures. Conversely, senile plaques, mainly composed of aggregated Aβ1–42 peptide, are found in both cerebrum and cerebellum of AD patients and cognitively healthy individuals [50,51]. This explains why the PIB-compound, the first radiotracer with high affinity for senile plaques, has not been widely accepted as a pathognomonic biomarker after six years following its first clinical trial [36]. Thus, it becomes evident that NFTs of hyperphosphorylated tau protein are considered to be the major defining neuropathological feature of AD and of other tauopthies [2,27,50]. NFTs are a target for specific radioimaging technologies as they are highly correlated with the degree of cognitive impairment [52]. The sequential molecular events and detailed structural modifications of tau that eventually lead to the formation of NFTs are far from being completely elucidated. However, the search for ligands that bind to NFTs with high affinity offers the possibility to understand structural aspects of these aggregates and new pathways to monitor the formation of tau aggregates in the course of AD.
Imaging technologies are other important biomarkers for AD. Important advances have been reached in the past few decades in the neuroimaging technology, toward diagnosis of several neurological disorders. These advances have relied on the development of powerful computing applications and sophisticated software for image processing, along with increasing knowledge of the biology of neurological diseases. At present, these different neuroimaging techniques provide precise information on structural and functional aspects of the brain, but do not provide information on the type of specific pathology and the structural alterations that are pathognomonic of these diseases.
We realized the need to identify novel tau binding molecules that could serve as radiotracers for in vivo detection of NFTs in AD patients and as a way to monitor the progress of AD [18]. Therefore, we investigated the interaction of a family of benzimidazoles with tau aggregates. The latter were obtained in vitro by heparin-induced assembly and from tau polymers retrieved from AD patients, which we have named AD-PHFs. In this context, results showed that heparin-induced tau polymers (HITP) were morphologically different from those obtained from AD patients, as evidenced by TEM images. HITP preparations are predominantly composed by “straight filaments”, structures of several micrometers in length and containing very few helical filaments. Filaments formed from in vitro phosphorylated tau were also straight but significantly shorter as compared with HITF. Filaments of recombinant tau formed in vitro frequently established lateral interactions to form bundles of PHF-like structures, with a wide region of about 5–15 nm, and a narrow region of 2 nm and occasional twists every 40–100 nm. HITF have a 5–10 nm diameter average. AD-PHFs have a wide region of 20–25 nm, a narrow region of 6–8 nm, and a twist every 80–90 nm. The average diameter of HITF was 15 nm with occasional PHF-like formations that were thinner than AD-PHFs. Tau self-assembly is highly dependent on its degree of phosphorylation [21]. This could partially explain the morphological differences that we found between AD-PHFs and HITF.
As for our experimental conditions, HITF were not prepared under hyperphosphorylation conditions. It has been suggested that the incubation time is also a relevant aspect for the morphology of the tau filaments, meaning that straight filaments would be an early step in the transition from soluble tau to PHF. We hypothesize that in vitro polymerization conditions lower the degree of polymerization with respect to polymerization achieved in AD. The polymerization process in vivo takes several decades to occur and is strongly favored by the hyperphosphorylation of tau. Then, it could be expected to yield thicker helical filaments at advanced stages of the disease. The morphology of AD-PHFs is similar to the one displayed by filaments composed of a mixture of the six tau isoforms, as found in PHFs [47]. On the other hand, during early stages of AD it is known that thinner, non-helical filaments are formed inside the neurons. Thus, the evaluation of potential radiotracers with high affinity for the so called “straight filaments” is highly valuable, as they may have a potential diagnostic use in the pre-clinical stages of the disease when tau filament formation is just starting. The present findings are relevant to the future research in this field, as the first screening studies to search for ligands of tau filaments are performed with in vitro filaments [34,35]. To the best of our knowledge, ours is the first report of differential affinities of ligands for in vitro and in vivo tau polymers. The incubation of recombinant tau with GSK-3β produced 90% shorter filaments than those isolated from AD; the incubation of tau with other polymers such as DNA-induced did not produced filaments after 21 days of incubation at 37°C. Therefore we decide to use both HITF and AD-PHFs for our studies and found that the affinity of benzimidazoles for PHFs and HITF followed different patterns.
Different affinities of benzimidazole for PHFs and HITF
Inhibition assays demonstrated that LNS displayed the highest affinity for HITF. Also AST displayed the highest affinity for AD-PHFs, indicated by the relative lower Ki values within the series. This difference in the affinity of benzimidazoles can be explained by the chemical nature of AD-PHFs and HITF and also by different functional groups coupled to the benzimidazole ring in AST and LNS. HITF are composed only by 4R tau, without hyperphosphorylation or glycated or ubiquitinated residues, while AD-PHFs are a completely different molecular entity. The aggregation is preceded by three major posttranslational modifications: hyperphosphorylation, glycation, and ubiquitination [23,50]. None of these modifications are present in HITF tau, which makes of them a different molecular target. This anomalous intracellular tau is highly neurotoxic and is thought to play an important role in AD pathophysiology. Therefore, it is relevant to search for molecules that dissemble or bind to these tau aggregates [53] in order to protect neurons from injury or to visualize early changes in intracellular organization of tau during the course of AD. Benizimidazole and other molecules that have a planar aromatic ring [35] seem to be promising “lead compounds” from which new molecules can be developed for a potential cure or a diagnosis tool for AD. Altogether, it seems that the presence of an aromatic moiety linked to the 2-position of the benzimidazole ring is relevant for the binding affinity to both types of tau filaments, HITF, and AD-PHFs. Interestingly both AST and LNS displayed high affinity for tau filaments, supporting our hypothesis that the presence of an aromatic or voluminous moiety linked to the 2-position of the benzimidazole ring is important for the binding to tau aggregates.
Further synthesis of new benzimidazoles and in vitro screening against AD-PHFs is necessary to find improved drug candidates for in vivo testing. At this point, it is worth to mention that we also tested some quinoline derivatives that have some structural similarities with benzimidazoles, mainly their planar structure. Some of these quinolines also displayed high affinity for AD-PHFs. This fact led us to think that a perpendicular aromatic ring linked to a planar system, such as benzimidazole or quinoline, could be important for the binding to tau aggregates. Our search for AD tracers has been focused on the study of tau aggregates and small molecules that bind to tau fibrils because it has been demonstrated by several laboratories, including our own [26,50,53], that future highly-specific methods for the diagnosis of AD should be developed based on the detection of NTFs and pre-tangle lesions in specific regions of the brain, mainly in the entorhinal cortex and hippocampus. As mentioned above, NFTs are the major pathological culprit for AD progression, and molecules that disassemble these anomalous aggregates could be used as efficient pharmacological agents to treat of this disease [53].
Why benzimidazoles could be useful as tracers for AD based on tau pathology?
Our finding that benzimidazole compounds with voluminous moities in position 2 display high affinity for tau aggregates (Fig. 4) was a milestone in this research. Another important data is the positive correlation between NFTs propagation in brain and the clinical manifestations of AD [52]. This approach would allow clinical researchers to obtain maps of these brain lesions that are the major trademarks of this disease [52,54]. A new neuropathological classification for the topographical distribution of NFTs in the brain was proposed by these investigators. Nowadays it is difficult to develop innovative treatments for AD based on tau inhibitors, because it is not possible to monitor tau aggregation or formation of NFTs in the brain of AD patients. This limitation is also valid for the accurate diagnosis of AD, as it is not possible to differentiate AD from other type of dementias during the early stages of the disease, and for the establishment of preventive therapies as well. Amyloid deposits are also present in AD patients but without correlation with cognitive decline. An area of intense research is the development of radioligands for Aβ of potential use in PET tomography. Considering that PHFs rather than amyloid plaques are pathognomonics biomarkers for AD, we considered it biomedically relevant to investigate innovative approaches to tag NFTs in AD using the small molecules (< 400 Da) AST and LNS.
Bioinformatical approaches
In order to perform docking studies it is necessary to generate plausible structures of intermolecular complexes based on the structural information of the molecules. For most of the structural proteins and receptors, this information is available at the web or in scientific publications, e.g., in Protein Data Bank. However, this is a serious limitation, since tau is a natively unfolded protein. To solve this problem we used a short fragment of tau that was found to be surface-exposed within the PHFs. This fragment is 386TDHGAE391 [48]. This domain of human tau was first isolated and crystallized from PHFs in a complex with the antibody MN423, originally used for the specific recognition of PHFs but the epitope was not clearly identified until Sevik and coworkers published this information in 2007. For our analysis, all water molecules were removed from the protein structure and polar hydrogen atoms were added assuming a physiological pH value. We obtained favorable binding energies for at least ten different configurations of AST interacting with the tau fragment (Fig. 4) and 3D structures of AST and LNS were built with the molecular visualization software MOLDEN 3. Atomic coordinates of tau fragment were extracted from the Protein Data Bank (PDB code: 2v17). Our data were consistent with interaction patterns of these benzimidazoles with tau, as described above.
Pharmacokinetic evaluation of brain imaging agents
Our data demonstrated that LNS and AST have good brain permeability, at least by passive diffusion mechanisms, suggesting that they could be analyzed as potential radiotracers for brain disorders. However, according to the PAMPA assay and partition coefficient, AST and LNS are good candidates for brain radiotracers, because their LogP value are within the range of 0.9 and 3.0 (except for AST, which was somewhat higher), defined as the ideal LogP for brain tracers [36]. AST and LNS displayed have high affinities for tau aggregates, high lipophilicity, good PAMPA permeability index, and are approved by the FDA for other uses in humans. The results of pharmacokinetics studies demonstrated that LNS reaches the brain within the first 37 min after administration, suitable with the use as PET tracer, as the half life of 18F is 120 min. Furthermore, brain/blood ratio tends to stabilize after the first 60 min, which means that LNS is eliminated from brain at a lower rate than blood. The same phenomenon is seen for AST that reaches the maximum concentration in the brain at 55 min and is slowly eliminated, as it is evidenced by a steady state in the brain/blood ratio. This could be explained by the fact that the brain has a lower metabolic capacity to eliminate and biotransform drugs than does plasma. In our study it was possible to quantitatively measure AST and LNS in brain even after 3 h from the administration.
AST is a second-generation antihistaminic drug that is believed to be lipophobic and for this reason would offer the advantage of a less CNS and cholinergic effects such as sedation, which are commonly seen in first-generation antihistamines. However, there is evidence that AST could pass the BBB [55,56], as it was found useful in patients with chronic vertigo [57]. Mice injected with tritiated AST showed the presence of the drug in the vestibular nerve tissues [57]. AST has demonstrated significant inhibitory effect on vestibular nerve activity at μM concentrations. The exact mechanism of how AST is able to reach nerve synapse and CNS is not clear, but the lipophilic nature of the molecule (LogP=5 at pH 7.4), and its inhibitory effect on vertigo suggest that there must be mechanism passing this drug through the BBB. LNS also belongs to the family of benzimidazoles (omeprazole, lansoprazole, and pantoprazole) which, once trapped and activated in the acid milieu of the gastric glands, potently suppress gastric secretion by inhibiting the H(+)-K+ ATPase. The long duration of action of LNS is not related to prolonged high levels in serum, but rather to the slow turnover of parietal cell H(+)-K+ ATPase molecules [58]. We did not find a comprehensive study describing the brain/blood ratio of LNS at different times. There is only one study describing pharmacokinetic profile of omeprazole [59]. These authors reported that omeprazole reaches the brain, blood, and bile fluid after oral administration in mice. They reported a rapid exchange between the compartments of the peripheral and central nervous systems; the results indicated that omeprazole was able to penetrate the blood-brain barrier and in a similar fashion to LNS, which according to our studies, tends to reach a steady state in brain after 60 min. In our biodistribution studies, the time to reach maximum concentrations in brain for AST and LNS were both within the interval of 40 to 60 min, which is very important for their potential use as PET tracers [36]. As the half-life of 18F is 120 min, time to reach the highest concentration in brain is lower than 120 min to have a sufficient radioactivity in the brain to produce NFTs images.
Biosensors technology: a tool to assess real time interactions between drugs and PHFs
SPR biosensors, such as BIACORE, have become a standard method for the study of biomolecular interactions [60], since they allow the measurement of the interaction between an immobilized protein (or protein aggregates) and a potential non-labeled ligand. We decided to use the biosensor technology based on SPR to study the interaction of AST and LNS with AD-PHFs and amyloid polymers. The advantages of this method with respect to the classical ligand-receptor assays are: i) high quality data in real time; ii) no labeling requirements; iii) low consumption of limiting reagents; and iv) a detection of phenomena missed by other techniques, such as radioligand assays. This was especially relevant for the interaction of AST, which is not fluorescent and therefore it can be analyzed by immuno-histochemistry. We used biosensor technology from BIACORE [60] and AD-PHFs and amyloid polymers coupled to a C5 chip by amine chemistry. After the immobilization reaction, we assessed the presence and integrity of protein aggregates immobilized on the sensor surface by injecting specific antibodies for amyloid peptide (6E10) and for PHFs (PHF-1), which yielded excellent responses. In order to verify the presence of polymeric forms of tau and amyloid on our chips, we tested the response to ThS, which is known to interact only with aggregated forms of the amyloid and tau protein [32]. To the best of our knowledge, this is the first time that potential specific radiotracers for AD are evaluated by biosensor technologies. Another important factor, also important for classical binding assays, is the highly heterogeneous nature of these samples. AD-PHFs from human brains not only have hyperphosphorylated tau, but also ubiquitin, different isoforms of tau protein, mainly 3R and 4R tau [21]. All these conditions for the immobilization reaction were determined empirically. This is a simpler procedure when the protein to be immobilized is pure and the isoelectric point is known.
Fig. 5.
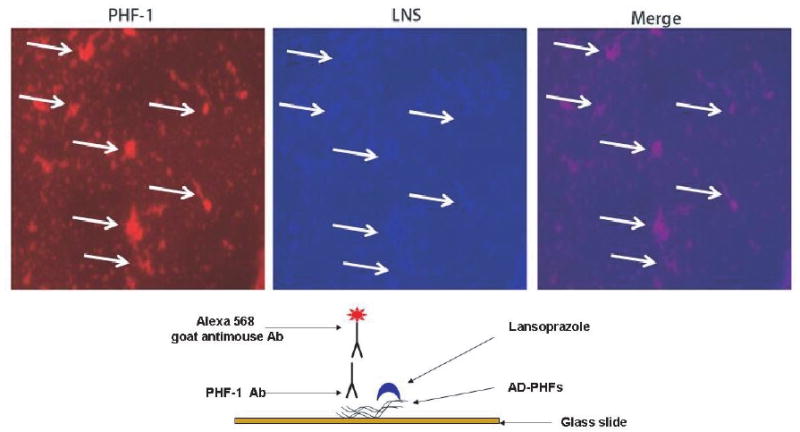
Immunofluorescence characterization of the interaction of AD-PHFs and lansoprazole. A suspension of isolated PHF or tau polymers was fixed to a glass slide (see scheme below) and processed for double staining with immunofluorescence using PHF-1 antibody and LNS 0.1 %.
Footnotes
DISCLOSURE STATEMENT
Authors' disclosures available online (http://www.jalz.com/disclosures/view.php?id=129).
References
- 1.Khachaturian ZS. An overview of Alzheimer's disease research. Am J Med. 1998;104:26S–31S. doi: 10.1016/s0002-9343(98)00026-6. discussion 39S-42S. [DOI] [PubMed] [Google Scholar]
- 2.Terry RD. Where in the brain does Alzheimer's disease begin? Ann Neurol. 2000;47:421. [PubMed] [Google Scholar]
- 3.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 4.Dubois B. ’Prodromal Alzheimer's disease’: a more useful concept than mild cognitive impairment? Curr Opin Neurol. 2000;13:367–369. doi: 10.1097/00019052-200008000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Dawbarn D. Neurobiology of Alzheimer's Disease. BIOS Scientific Publishers Ltd.; Oxford, UK: 1995. [Google Scholar]
- 6.Farias GA, Vial C, Maccioni RB. Specific macromolecular interactions between tau and the microtubule system. Mol Cell Biochem. 1992;112:81–88. doi: 10.1007/BF00229646. [DOI] [PubMed] [Google Scholar]
- 7.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Padovani A, Borroni B, Colciaghi F, Pettenati C, Cottini E, Agosti C, Lenzi GL, Caltagirone C, Trabucchi M, Cattabeni F, Di Luca M. Abnormalities in the pattern of platelet amyloid precursor protein forms in patients with mild cognitive impairment and Alzheimer disease. Arch Neurol. 2002;59:71–75. doi: 10.1001/archneur.59.1.71. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg RN, Baskin F, Fosmire JA, Risser R, Adams P, Svetlik D, Honig LS, Cullum CM, Weiner MF. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch Neurol. 1997;54:139–144. doi: 10.1001/archneur.1997.00550140019007. [DOI] [PubMed] [Google Scholar]
- 11.Hampel H, Teipel SJ. Total and phosphorylated tau proteins: evaluation as core biomarker candidates in frontotem-poral dementia. Dement Geriatr Cogn Disord. 2004;17:350–354. doi: 10.1159/000077170. [DOI] [PubMed] [Google Scholar]
- 12.Lavados M, Farias G, Rothhammer F, Guillon M, Mujica MC, Maccioni C, Maccioni RB. ApoE alleles and tau markers in patients with different levels of cognitive impairment. Arch Med Res. 2005;36:474–479. doi: 10.1016/j.arcmed.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 13.Maccioni RB, Lavados M, Guillon M, Mujica C, Bosch R, Farias G, Fuentes P. Anomalously phosphorylated tau and Abeta fragments in the CSF correlates with cognitive impairment in MCI subjects. Neurobiol Aging. 2006;27:237–244. doi: 10.1016/j.neurobiolaging.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 14.Maccioni RB, Lavados M, Maccioni CB, Mendoza-Naranjo A. Biological markers of Alzheimer's disease and mild cognitive impairment. Curr Alzheimer Res. 2004;1:307–314. doi: 10.2174/1567205043332018. [DOI] [PubMed] [Google Scholar]
- 15.de Leon MJ, Klunk W. Biomarkers for the early diagnosis of Alzheimer's disease. Lancet Neurol. 2006;5:198–199. doi: 10.1016/S1474-4422(06)70357-X. [DOI] [PubMed] [Google Scholar]
- 16.Orellana DI, Quintanilla RA, Maccioni RB. Neuroprotective effect of TNFalpha against the beta-amyloid neurotox-icity mediated by CDK5 kinase. Biochim Biophys Acta. 2007;1773:254–263. doi: 10.1016/j.bbamcr.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Rojo LE, Fernandez JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer's disease. Arch Med Res. 2008;39:1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Rojo L, Sjoberg MK, Hernandez P, Zambrano C, Maccioni RB. Roles of cholesterol and lipids in the etiopathogenesis of Alzheimer's disease. J Biomed Biotechnol. 2006;2006:73976. doi: 10.1155/JBB/2006/73976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sekler A, Jimenez JM, Rojo L, Pastene E, Fuentes P, Slachevsky A, Maccioni RB. Cognitive impairment and Alzheimer's disease: Links with oxidative stress and cholesterol metabolism. Neuropsychiatr Dis Treat. 2008;4:715–722. doi: 10.2147/ndt.s3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavados M, Guillon M, Mujica MC, Rojo LE, Fuentes P, Maccioni RB. Mild cognitive impairment and Alzheimer patients display different levels of redox-active CSF iron. J Alzheimers Dis. 2008;13:225–232. doi: 10.3233/jad-2008-13211. [DOI] [PubMed] [Google Scholar]
- 21.Maccioni RB, Cambiazo V. Role of microtubule-associated proteins in the control of microtubule assembly. Physiol Rev. 1995;75:835–864. doi: 10.1152/physrev.1995.75.4.835. [DOI] [PubMed] [Google Scholar]
- 22.Farias G, Gonzalez-Billault C, Maccioni RB. Immunological characterization of epitopes on tau of Alzheimer's type and chemically modified tau. Mol Cell Biochem. 1997;168:59–66. doi: 10.1023/a:1006838626730. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez C, Farias G, Maccioni RB. Modification of tau to an Alzheimer's type protein interferes with its interaction with microtubules. Cell Mol Biol (Noisy-le-grand) 1998;44:1117–1127. [PubMed] [Google Scholar]
- 24.Lichtenberg-Kraag B, Mandelkow EM, Biernat J, Steiner B, Schroter C, Gustke N, Meyer HE, Mandelkow E. Phosphorylation-dependent epitopes of neurofilament antibodies on tau protein and relationship with Alzheimer tau. Proc Natl Acad Sci U S A. 1992;89:5384–5388. doi: 10.1073/pnas.89.12.5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez M, Arrasate M, Montejo De Garcini E, Munoz V, Avila J. In vitro assembly of tau protein: mapping the regions involved in filament formation. Biochemistry. 2001;40:5983–5991. doi: 10.1021/bi002961w. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez JA, Rojo L, Kuljis RO, Maccioni RB. The damage signals hypothesis of Alzheimer's disease pathogenesis. J Alzheimers Dis. 2008;14:329–333. doi: 10.3233/jad-2008-14307. [DOI] [PubMed] [Google Scholar]
- 27.Maccioni RB, Rojo LE, Fernandez JA, Kuljis RO. The role of neuroimmunomodulation in Alzheimer's disease. Ann N Y Acad Sci. 2009;1153:240–246. doi: 10.1111/j.1749-6632.2008.03972.x. [DOI] [PubMed] [Google Scholar]
- 28.Quintanilla RA, Orellana DI, Gonzalez-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. 2004;295:245–257. doi: 10.1016/j.yexcr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Orellana DI, Q R, Gonzalez-Billault C, Maccioni RB. Role of the JAKs/STATs pathway in the intracellular calcium changes induced by interleukin-6 in hippocampal neurons. Neurotox Res. 2005;8:295–304. doi: 10.1007/BF03033983. [DOI] [PubMed] [Google Scholar]
- 30.von Bergen M, Barghorn S, Muller SA, Pickhardt M, Biernat J, Mandelkow EM, Davies P, Aebi U, Mandelkow E. The core of tau-paired helical filaments studied by scanning transmission electron microscopy and limited proteolysis. Biochemistry. 2006;45:6446–6457. doi: 10.1021/bi052530j. [DOI] [PubMed] [Google Scholar]
- 31.Friedhoff P, Schneider A, Mandelkow EM, Mandelkow E. Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry. 1998;37:10223–10230. doi: 10.1021/bi980537d. [DOI] [PubMed] [Google Scholar]
- 32.Santa-Maria I, Perez M, Hernandez F, Avila J, Moreno FJ. Characteristics of the binding of thioflavin S to tau paired helical filaments. J Alzheimers Dis. 2006;9:279–285. doi: 10.3233/jad-2006-9307. [DOI] [PubMed] [Google Scholar]
- 33.Moraga D, Rivas-Berrios A, Farias G, Wallin M, Maccioni RB. Estramustine-phosphate binds to a tubulin binding domain on microtubule-associated proteins MAP-2 and tau. Biochim Biophys Acta. 1992;1121:97–103. doi: 10.1016/0167-4838(92)90342-b. [DOI] [PubMed] [Google Scholar]
- 34.Okamura N, Suemoto T, Shimadzu H, Suzuki M, Shiomitsu T, Akatsu H, Yamamoto T, Staufenbiel M, Yanai K, Arai H, Sasaki H, Kudo Y, Sawada T. Styrylbenzoxazole derivatives for in vivo imaging of amyloid plaques in the brain. J Neurosci. 2004;24:2535–2541. doi: 10.1523/JNEUROSCI.4456-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okamura N, Suemoto T, Furumoto S, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Fujiwara H, Nemoto M, Maruyama M, Arai H, Yanai K, Sawada T, Kudo Y. Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer's disease. J Neurosci. 2005;25:10857–10862. doi: 10.1523/JNEUROSCI.1738-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, Debnath ML, Holt DP, Huang GF, Shao L, DeKosky ST, Price JC, Mathis CA. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer's disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du AT, Schuff N, Kramer JH, Ganzer S, Zhu XP, Jagust WJ, Miller BL, Reed BR, Mungas D, Yaffe K, Chui HC, Weiner MW. Higher atrophy rate of entorhinal cortex than hippocampus in AD. Neurology. 2004;62:422–427. doi: 10.1212/01.wnl.0000106462.72282.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rusinek H, De Santi S, Frid D, Tsui WH, Tarshish CY, Convit A, de Leon MJ. Regional brain atrophy rate predicts future cognitive decline: 6-year longitudinal MR imaging study of normal aging. Radiology. 2003;229:691–696. doi: 10.1148/radiol.2293021299. [DOI] [PubMed] [Google Scholar]
- 39.Stoub TR, Bulgakova M, Leurgans S, Bennett DA, Fleischman D, Turner DA, deToledo-Morrell L. MRI predictors of risk of incident Alzheimer disease: a longitudinal study. Neurology. 2005;64:1520–1524. doi: 10.1212/01.WNL.0000160089.43264.1A. [DOI] [PubMed] [Google Scholar]
- 40.Rojo L, Chandia M, Becerra R, Avila M, Maccioni RB. Conference on Clinical PET and Molecular Medicine; Bankok, Thailand. 2008. [Google Scholar]
- 41.Jicha GA, O'Donnell A, Weaver C, Angeletti R, Davies P. Hierarchical phosphorylation of recombinant tau by the paired-helical filament-associated protein kinase is dependent on cyclic AMP-dependent protein kinase. J Neurochem. 1999;72:214–224. doi: 10.1046/j.1471-4159.1999.0720214.x. [DOI] [PubMed] [Google Scholar]
- 42.Schaftenaar G, Noordik JH. Molden: a pre- and postprocessing program for molecular and electronic structures. J Comput Aided Mol Des. 2000;14:123–134. doi: 10.1023/a:1008193805436. [DOI] [PubMed] [Google Scholar]
- 43.Osguthorpe JW, Genest M, Hagler AT. Proteins: Structure, Function and Genetics. Wiley-Liss, Inc., A Wiley Company; 1988. [Google Scholar]
- 44.Rosin CD, Belew RK, Morris GM, Olson AJ, Goodsell DS. Computational coevolution of antiviral drug resistance. Artif Life. 1998;4:41–59. doi: 10.1162/106454698568431. [DOI] [PubMed] [Google Scholar]
- 45.Avdeef A, Tsinman O. PAMPA–a drug absorption in vitro model 13. Chemical selectivity due to membrane hydrogen bonding: in combo comparisons of HDM-, DOPC-, and DS-PAMPA models. Eur J Pharm Sci. 2006;28:43–50. doi: 10.1016/j.ejps.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 46.Kawatake S, Nishimura Y, Sakaguchi S, Iwaki T, Doh-ura K. Surface plasmon resonance analysis for the screening of anti-prion compounds. Biol Pharm Bull. 2006;29:927–932. doi: 10.1248/bpb.29.927. [DOI] [PubMed] [Google Scholar]
- 47.Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sevcik J, Skrabana R, Dvorsky R, Csokova N, Iqbal K, Novak M. X-ray structure of the PHF core C-terminus: insight into the folding of the intrinsically disordered protein tau in Alzheimer's disease. FEBS Lett. 2007;581:5872–5878. doi: 10.1016/j.febslet.2007.11.067. [DOI] [PubMed] [Google Scholar]
- 49.Mathis CA, Klunk WE, Price JC, DeKosky ST. Imaging technology for neurodegenerative diseases: progress toward detection of specific pathologies. Arch Neurol. 2005;62:196–200. doi: 10.1001/archneur.62.2.196. [DOI] [PubMed] [Google Scholar]
- 50.Maccioni RB, Munoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Arch Med Res. 2001;32:367–381. doi: 10.1016/s0188-4409(01)00316-2. [DOI] [PubMed] [Google Scholar]
- 51.Mukrasch MD, von Bergen M, Biernat J, Fischer D, Griesinger C, Mandelkow E, Zweckstetter M. The “jaws” of the tau-microtubule interaction. J Biol Chem. 2007;282:12230–12239. doi: 10.1074/jbc.M607159200. [DOI] [PubMed] [Google Scholar]
- 52.Braak H, Braak E. Entorhinal-hippocampal interaction in mnestic disorders. Hippocampus. 1993;3 Spec No:239–246. [PubMed] [Google Scholar]
- 53.Maccioni RB, F G, R LE, Sa M, K R. Hypotheses and Research Milestones in Alzheimer's Disease. Springer-Verlag; New York, New York: 2008. What have we learned from tau hypothesis? [Google Scholar]
- 54.Verhoeff NP, Wilson AA, Takeshita S, Trop L, Hussey D, Singh K, Kung HF, Kung MP, Houle S. In-vivo imaging of Alzheimer disease beta-amyloid with [11C]SB-13 PET. Am J Geriatr Psychiatry. 2004;12:584–595. doi: 10.1176/appi.ajgp.12.6.584. [DOI] [PubMed] [Google Scholar]
- 55.Mattila MJ, Paakkari I. Variations among non-sedating antihistamines: are there real differences? Eur J Clin Pharmacol. 1999;55:85–93. doi: 10.1007/s002280050600. [DOI] [PubMed] [Google Scholar]
- 56.Swiader MJ, Luszczki JJ, Wielosz M, Czuczwar SJ. Effect of histamine receptor antagonists on aminophylline-induced seizures and lethality in mice. Pharmacol Rep. 2005;57:531–535. [PubMed] [Google Scholar]
- 57.Norris CH, Jackson RT. Inhibition by astemizole of activity from the isolated semicircular canal. Arch Otolaryngol Head Neck Surg. 1987;113:981–983. doi: 10.1001/archotol.1987.01860090079024. [DOI] [PubMed] [Google Scholar]
- 58.Hatlebakk JG, Nesje LB, Hausken T, Bang CJ, Berstad A. Lansoprazole capsules and amoxicillin oral suspension in the treatment of peptic ulcer disease. Scand J Gastroenterol. 1995;30:1053–1057. doi: 10.3109/00365529509101606. [DOI] [PubMed] [Google Scholar]
- 59.Cheng FC, Ho YF, Hung LC, Chen CF, Tsai TH. Determination and pharmacokinetic profile of omeprazole in rat blood, brain and bile by microdialysis and high-performance liquid chromatography. J Chromatogr A. 2002;949:35–42. doi: 10.1016/s0021-9673(01)01225-0. [DOI] [PubMed] [Google Scholar]
- 60.Rich RL, Myszka DG. BIACORE J: a new platform for routine biomolecular interaction analysis. J Mol Recognit. 2001;14:223–228. doi: 10.1002/jmr.535. [DOI] [PubMed] [Google Scholar]