

Abstract
Juvenile amyotrophic lateral sclerosis (ALS) with basophilic inclusions is a well‐recognized entity. However, the molecular underpinnings of this devastating disease are poorly understood. Here, we present genetic and neuropathological characterizations in two young women with fatal rapidly progressive ALS with basophilic inclusions. In one case, a germline mutation (P525L) was detected in the fused in sarcoma/translocated in liposarcoma (FUS/TLS) gene, whereas no mutation was identified in the other case. Postmortem examination in both cases revealed severe loss of spinal motor neurons with remaining neurons showing basophilic inclusions that contain abnormal aggregates of FUS proteins and disorganized intracellular organelles, including mitochondria and endoplasmic reticulum. In both patients, the FUS‐positive inclusions were also detected in neurons in layers IV–V of cerebral cortex and several brainstem nuclei. In contrast, spinal motor neurons in patients with late‐onset sporadic ALS showed no evidence of abnormal accumulation of FUS protein. These results underscore the importance of FUS mutations and pathology in rapidly progressive juvenile ALS. Furthermore, our study represents the first detailed characterizations of neuropathological findings in rapidly progressive juvenile ALS patients with a mutation in the FUS/TLS gene.
Keywords: ALS, electron microscopy, FUS, immunohistochemistry, mutation
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that affects the motor neurons in the brain and spinal cord. The majority of cases occur in the older population, with a peak incidence in the sixth decade of life. Approximately 10% of ALS cases are familial, and more than 20% of these cases carry mutations in the Cu/Zn superoxide dismutase (SOD1) gene 3, 19. An important pathological hallmark of ALS is the accumulation of misfolded proteins in affected neurons and glial cells. Recent studies have shown that the affected neurons in sporadic ALS cases contain ubiquitinated TAR‐DNA binding protein (TDP‐43), which is involved in mRNA processing and has also been implicated in the pathogenesis of frontotemporal lobar dementia with ubiquitinated inclusions (FTLD‐U) 5, 18. Interestingly, several groups have reported dominant mutations in the TARDBP gene, which encodes TDP‐43, as the primary cause in patients with late‐onset sporadic or familial ALS 4, 10, 11, 21, 24, 27. In addition, several recent studies have identified mutations involving the fused in sarcoma/translocated in liposarcoma (FUS/TLS) gene as the second most common mutation in familial forms of ALS 2, 13, 14, 26, 28.
Most of the pathological features of ALS have been reported in patients with late‐onset, sporadic ALS and familial cases with mutations in SOD1. However, huge gaps remain in the understanding of the pathogenesis of ALS and correlations between genetic mutations and the pathological features of ALS. For instance, despite the well‐characterized genetic evidence, there have been very few reports on the detailed neuropathological features of ALS patients with FUS/TLS mutations 13, 26, 28. In addition, a juvenile variant of ALS is well known, generally with slow progression. In recent years, several cases of rapidly progressive juvenile ALS with lower motor neuron predominance have been reported 1, 17, 22. Most of these cases show basophilic inclusions in the remaining spinal motor neurons. However, the characteristics of these basophilic inclusions and their role in the pathogenesis of the disease process are poorly understood.
Here we present two cases of rapidly progressive juvenile ALS with robust FUS pathology in spinal motor neurons and other central nervous system (CNS) neurons. We provide discussions regarding the disease progression, mutation in the FUS/TLS gene and the unique neuropathological features.
MATERIALS AND METHODS
Patients
Informed consent was obtained from the legal guardians for DNA and neuropathological examinations. Two patients with rapidly progressive juvenile ALS underwent complete autopsy, including examinations of the brain and spinal cord. Detailed descriptions of the clinical history for both patients are provided in the Results section. To determine if abnormal aggregates of FUS protein can be detected in sporadic ALS cases, we performed immunohistochemistry (IHC) staining for FUS in 10 cases of late‐onset sporadic ALS patients. The age of these 10 patients ranged from 46 to 65, with a median age of 56. Three of these 10 sporadic ALS patients had evidence of dementia during the clinical follow‐up periods. Clinical diagnoses and detailed cognitive evaluations of the 10 patients with late‐onset sporadic ALS have been previously reported 8, 9.
Mutation screen and detection in FUS/TLS gene
The exons and flanking intronic sequence of the FUS/TLS gene were screened by direct sequencing of polymerase chain reaction products amplified from genomic DNA using procedures described by Kwiatkowski and colleagues (13). The amplicons of all 15 FUS/TLS exons, including exon‐intron boundaries, were amplified and sequenced using primers described in our previous study (13). As previously indicated, at least 1446 control DNA samples were analyzed and found no similar mutations (13).
IHC
Diagnostic blocks from frontal lobe, parietal lobe, temporal lobe, hippocampus, midbrain, pons, medulla oblongata, and cervical, thoracic and lumbar spinal cord were paraffin‐embedded, serially sectioned at 5 µm thickness, and stained with hematoxylin and eosin, Luxol Fast Blue‐PAS (LFB‐PAS), and antibodies for FUS (1:200, Novus Biologicals, Littleton, CO, USA, and 1:450, Sigma‐Aldrich, St. Louis, MO, USA), ubiquitin (1:100, Abcam, Cambridge, MA, USA), TDP‐43 (1:500, Proteintech, Chicago, IL, USA), neurofilament (NF) (1:500, Chemicon, Temecula, CA, USA), GFAP (1:3,000, Dako, Carpinteria, CA, USA), CD3 (1:400, Dako), CD8 (1:320, Dako), and CD68 (1:4,000, Dako). The slides were prepared using the microwave antigen retrieval methods, and the immune reaction was detected by the Vectastain kit (Vector Laboratories, Burlingame, CA, USA) using diaminobenzidine as the chromogen. The images were captured using a BX41 Olympus microscope and an Olympus DP70 CCD camera (Olympus USA, Center Valley, PA, USA).
Electron microscopy
Ultrastructural examinations of the basophilic inclusions in spinal motor neurons and cortical neurons of both patients with juvenile ALS were performed using tissues obtained from the formalin‐fixed cervical spinal cord and frontal cortex. Tissues were fixed in 2.5% glutaraldehyde/1% paraformaldehyde and embedded in Pelco Eponate (Ted Pella, Redding CA, USA) as previously described (15). Thin sections were collected on copper grids and examined using a FEI TECNAI 10 electron microscope (Hillsboro, OR, USA). For immunogold EM, tissues were fixed in 0.1% glutaraldehyde/2% paraformaldehyde and was not treated with osmium to preserve antigenicity. The tissues were then incubated with a primary antibody for FUS (1:20, Novus Biologicals) or ubiquitin (1:10, Abcam), and the goat antirabbit secondary antibody conjugated with 15‐nm gold particles (1:20, Ted Pella). Adjacent sections were used for staining for FUS and ubiquitin antibodies, and negative controls were performed using only secondary antibodies to show that the staining for FUS and ubiquitin was specific.
RESULTS
Clinical history
Case 1
A 13‐year‐old girl fell while jumping a low hurdle in June 2007. After initial improvement, her left leg weakened. She was wheelchair‐bound by March 2008. There was no upper motor neuron, bulbar, cognitive or behavioral involvement. Initial electromyography (EMG) suggested a demyelinating polyneuropathy with diffuse active and chronic denervation. Sural nerve biopsy showed no demyelination or inflammation. Lumbar puncture showed slightly elevated protein, normal glucose and no cells. A maternal grandmother had autopsy‐proven progressive supranuclear palsy (neuropathology examinations performed by D.W.D. at the Mayo Clinic). Parents and an older paternal half brother have no neurologic disorders, with no known consanguinity. She had mild gross motor delay as a child, with speech delay and a mild learning disability. However, the mental status of the patient was normal, with respect to attention, memory, orientation, language and fund of knowledge.
She presented to the University of California San Francisco (UCSF) ALS Center in April 2008. Forced vital capacity (FVC) was 54% predicted, and mean inspiratory flow was −30. Cranial nerves were intact. Motor tone was normal to decreased. There was moderate weakness of neck flexion and extension as well as the left arm, and severe weakness of the right arm and both legs. Reflexes were absent, with crossed adductors and mute toes. Weakness progressed despite treatments with intravenous immunoglobulins (IVIg), plasmapheresis, high‐dose steroids and cytoxan. There was no response to Riluzole, Lithium, insulin‐like growth factor 1 or ceftriaxone. By November 2008 she required daily bilevel positive airway pressure (BiPAP) and transitioned to a portable ventilator via a mask. After two apneic episodes she died of respiratory failure 20 months after disease onset.
Case 2
A 21‐year‐old woman tripped and fell, hitting her head in April 2008. Two weeks later she noticed bilateral shoulder weakness, followed by arm and neck weakness over several days. Examination showed severe weakness of neck extensors and flexors and proximal more than distal arms, and mild ankle dorsiflexor weakness. Reflexes were brisk throughout. FVC was 29% predicted. EMG showed active and chronic denervation in the arms and cervical paraspinal muscles. Cerebrospinal fluid and cervical spine magnetic resonance imaging were normal. She quickly developed slurred speech and trouble swallowing. She had no response to IVIg. She refused percutaneous endoscopic gastrostomy tube and BiPAP and died 4 months after initial symptoms. Five siblings and both parents have no neurological disorders, with no known consanguinity. She had gross hypotonia at birth, requiring physical and occupational therapy in early childhood. Similar to Case #1, there was no evidence of cognitive, language or memory impairment in this patient.
Genetic analyses
Genetic tests for hereditary neuropathy with liability to pressure palsies, Charcot‐Marie tooth disease, SOD‐1, ALS2 (Alsin), ALS4 (Senataxin), ALS8 (vesicle‐associated membrane protein B, or VAPB) and spinal muscular atrophy in Case 1 were normal, except the patient had only two copies of the survival of motor neuron‐2 (SMN2) gene. Sequencing of FUS/TLS gene revealed a single C→T point mutation in base pair 1574 on exon 15 in Case 1 (13). This mutation changed amino acid #525 from Proline to Leucine (P525L) and was located in a region that is highly evolutionarily conserved in humans, chimpanzees, Rhesus monkeys, cows, horses, rats and mice (Figure 1). Neither parent of Case 1 carried a mutation in the FUS/TLS gene. In contrast to Case 1, no mutation in the coding sequence of the FUS/TLS gene was identified in Case 2.
Figure 1.
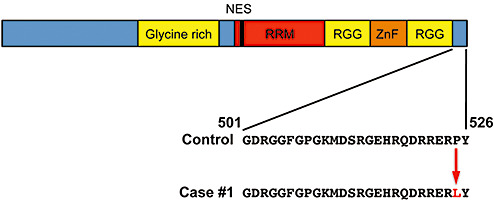
A schematic diagram of the fused in sarcoma/translocated in liposarcoma protein and the location of the P525L mutation in Case 1. Abbreviations: RRM = RNA recognition motif = RGG = arginine‐glycine rich region; ZnF = zinc finger domain.
Neuropathology
Both of the juvenile ALS cases showed a severe loss of motor neurons in the spinal cord. Many remaining spinal motor neurons contained intracytoplasmic basophilic inclusions (Figure 2A–H). IHC showed that the basophilic inclusions were strongly positive for FUS, whereas no FUS‐positive inclusion was identified in the glial cells (Figure 2B, F). The staining results for ubiquitin were quite variable, with few neurons showing light ubiquitin‐positive granules in the cytoplasm (Figure 2C, G). In contrast, IHC for TDP‐43, hyperphosphorylated Tau (CP‐13), α‐synuclein, α‐1‐internexin and NF was negative (Figure 2D, H, and data not shown) 9, 13, 16, 28. To compare and contrast the immunohistochemical profiles of juvenile ALS and late‐onset sporadic ALS, similar IHC tests were performed in 10 cases of sporadic ALS. The results indicated that none of the 10 late‐onset sporadic ALS cases showed FUS‐positive inclusions in the cytoplasm of spinal motor neurons (Figure 2I–J). In contrast, many spinal motor neurons in patients with sporadic ALS showed prominent TDP‐43‐ and ubiquitin‐positive protein aggregates (Figure 2K, L).
Figure 2.
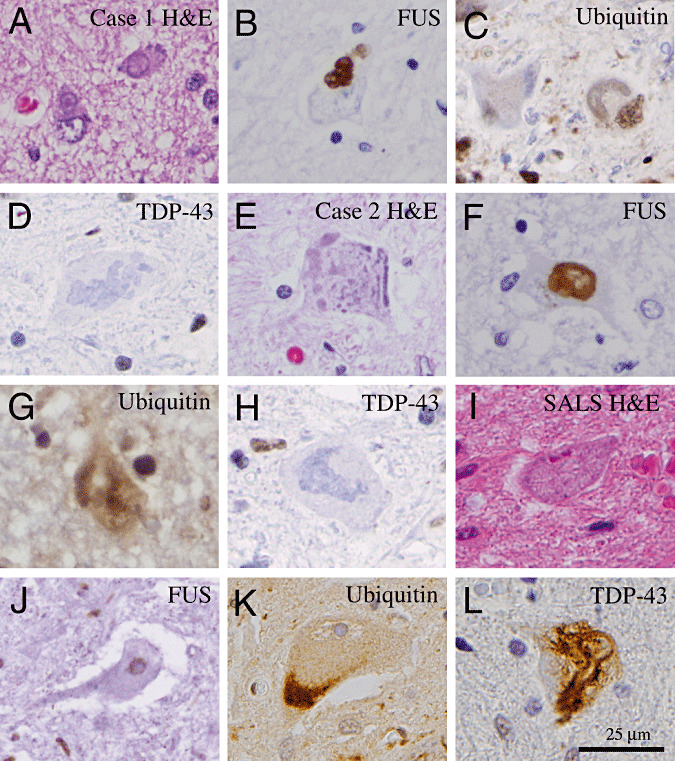
Histopathology of juvenile amyotrophic lateral sclerosis (ALS) with basophilic inclusions in comparison with that of late‐onset sporadic ALS. (A,E) Hematoxylin and eosin (H&E) sections of the spinal cord show the presence of basophilic inclusions in the remaining motor neurons in Cases 1 and 2. (B,F) Immunohistochemical staining shows that the basophilic inclusions are positive for FUS proteins. (C,G) Furthermore, these inclusions are only weakly positive for ubiquitin but negative for TDP‐43 (D,H). (I) In contrast, spinal motor neurons in patients with late‐onset sporadic ALS show no evidence of basophilic inclusions on H&E‐stained section. (J) Furthermore, immunohistochemistry shows the presence of FUS protein in the nucleus of remaining spinal motor neurons. (K,L) Many of the spinal motor neurons in late‐onset sporadic ALS patients show abnormal accumulation of ubiquitinated proteins (K) and TDP‐43 proteins (L). Scale bar in L is 25 µm and applies to all panels.
To further characterize the ultrastructural features of the basophilic inclusions in our juvenile ALS cases, we performed electron microscopic (EM) examinations of the cervical spinal cord using primary antibodies specific for FUS or ubiquitin, and appropriate secondary antibodies conjugated with 15‐nm gold particles. At least 10 inclusions were sampled from the spinal cord for each juvenile ALS case with all inclusions showing essentially identical characteristics. In both juvenile ALS cases, the inclusions in the spinal motor neurons contained multiple filamentous structures with amorphous electron dense material, disorganized rough endoplasmic reticulum (ER) and mitochondria (M) (Figure 3). Figure 3A showed a low magnification image of a representative inclusion in spinal motor neurons, in which three rectangles were used to highlight areas with higher magnifications in Figure 3B–D. The disorganized ER and M were frequently identified at the periphery of the inclusion (Figure 3B, D), whereas the filamentous aggregates were located primarily inside the inclusions, with rather uniform size that measured 15–20 nm in diameter (Figure 3C). Immunogold EM using an FUS‐specific antibody showed that the majority of the abnormal FUS proteins were primarily associated with the filamentous structures, with a minority of the FUS‐positive gold particles found in the rough ER and the amorphous materials (Figure 3E). Consistent with the ubiquitin IHC results, immunogold EM showed very few gold particles that were detected by the ubiquitin antibody (Figure 3F). In contrast, negative controls using no primary antibodies show no gold particles labeling the filamentous structures, supporting the specificity of the staining (data not shown).
Figure 3.
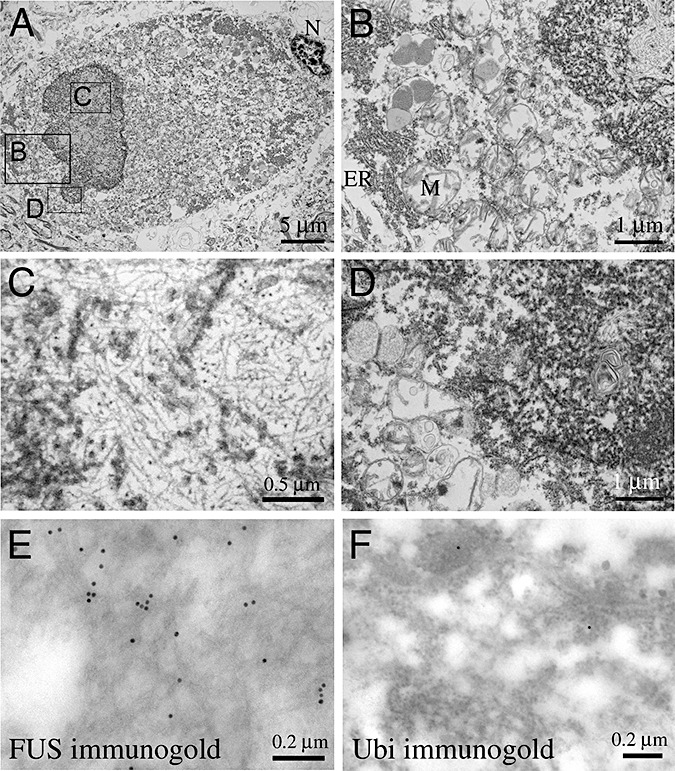
Ultrastructural analyses of basophilic inclusions in juvenile amyotrophic lateral sclerosis (ALS). (A) A low magnification electron micrograph of the intracytoplasmic inclusion in a spinal motor neuron. The three rectangles in panel A highlight areas of higher magnifications shown in panels B, C and D. (B–D) Higher magnification image at the edge and the center of the inclusion. Both panels B and D show aggregates of disorganized endoplasmic reticulum and mitochondria. (C) In contrast, the center of the inclusion contains filamentous structures that measure 15–20 nm in diameter. (E,F) Immunogold EM using FUS‐specific antibody shows that the majority of FUS proteins are associated with the filamentous structures (E), whereas the immunogold staining for ubiquitin show very few positive gold particles.
Similar basophilic inclusions were also identified in the frontal cortex (cingulate gyrus) and sensorimotor cortex, red nucleus, nucleus ambiguus, and the reticular formation in the medulla oblongata (Figure 4). The morphology of the basophilic inclusions in these locations was similar to those in the spinal motor neurons (Figure 4A). The basophilic inclusions in cortical neurons can also be detected in layers IV and V of the cerebral cortex. Similar to the basophilic inclusions in the spinal motor neurons, all inclusions in the cortical neurons showed intense positive staining for FUS (Figure 4B). Unlike the spinal cord, however, there was no detectable loss of neurons in these locations. Similar inclusions were identified in the reticular formation of the medulla oblongata and the red nucleus showed similar morphology and staining results for FUS (Figure 4C–E). In contrast, no basophilic inclusion or FUS‐positive inclusions were identified in neurons of the hypoglossal nucleus (Figure 4F).
Figure 4.
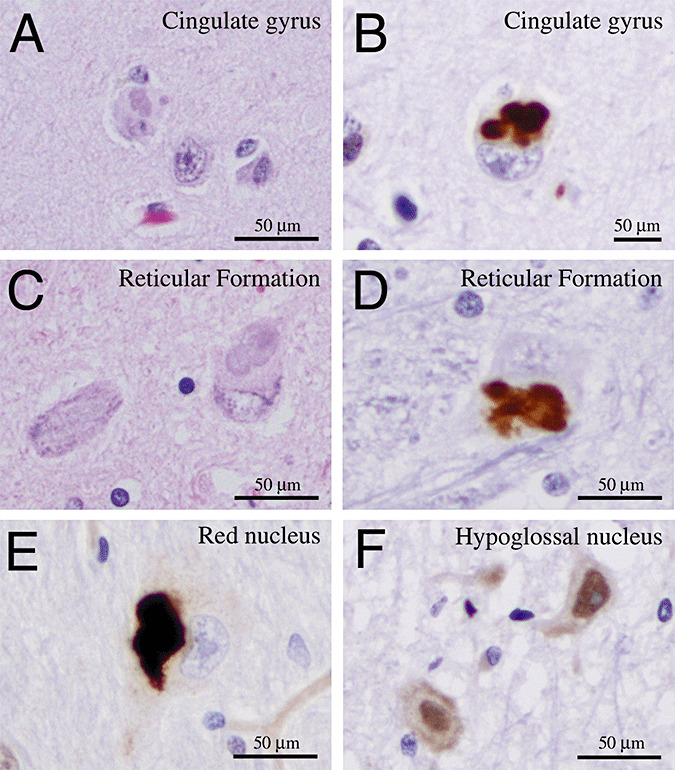
Basophilic inclusions can be detected in cerebral cortex and brainstem nuclei in patients with juvenile ALS. (A,B) A hematoxylin and eosin section shows the presence of basophilic inclusion in the cytoplasm of a neuron in the cingulate gyrus (A). Immunohistochemistry using FUS antibody confirms that these inclusions are positive for FUS protein (B). (C–E) The basophilic and FUS‐positive inclusions can also be identified in neurons in the reticular formation in the medulla oblongata (C,D) and the red nucleus (E). (F) In contrast, neurons in the hypoglossal nucleus in both juvenile ALS patients show staining of FUS proteins in the nuclei.
In addition to the FUS pathology, Case 1 also showed white matter pathology, characterized by loss of myelinated axons, diffuse macrophage infiltrates, and astrogliosis involving the anterior and lateral columns of the spinal cord (Figure 5A, B, arrowheads highlighted the white matter pallor and loss of myelination in the anterior column). The macrophage infiltrates were also found in the anterior horn of the spinal cord. In contrast, the posterior columns of the spinal cord showed no evidence of a diminution of myelinated axons or macrophage infiltrates (Figure 5A, B). Similar pathological features were also detected in Case 2, although the severity and the extent of involvement were not as prominent as in Case 1. An LFB‐PAS stain confirmed the loss of myelinated axons in the lateral and anterior columns with extension into the ventral nerve root (Figure 5B, C). IHC for NF showed a loss of NF positive axons in the regions with reduced LFB‐PAS staining in the spinal cord (Figure 5D). In contrast, the myelination pattern in the posterior column showed no detectable abnormality in either patient (Figure 5E). Furthermore, the axons in the posterior column also showed normal density with uniform diameter (Figure 5F). IHC for CD68 and CD3 confirmed the presence of abundant macrophages and scattered T lymphocytes, respectively (Figure 5G, H). Many of the T lymphocytes also stained positive for CD8, but not CD4, indicating that they were cytotoxic T cells, whereas IHC for LCA and CD20 showed no evidence of infiltration by neutrophils or B lymphocytes (not shown).
Figure 5.
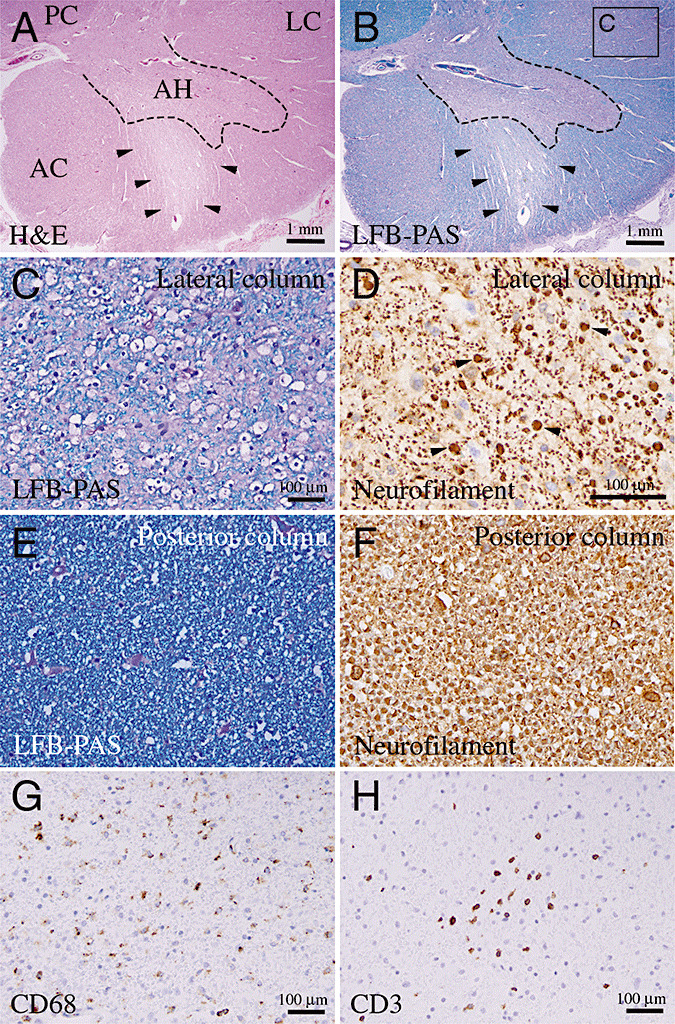
White matter pathology in juvenile amyotrophic lateral sclerosis with FUS‐positive inclusions. (A–C) Hematoxylin‐eosin and Luxol Fast Blue‐PAS (LFB‐PAS) stains of cervical spinal cord show extensive demyelination in the lateral column (LC), but not in posterior column (PC). Arrowheads in panels A and B indicate the motor nerve roots in the anterior horn with severe pallor and loss of myelination. Scale bar in panel C is 100 µm. (D) The density of axons, highlighted by IHC for neurofilament (NF), is slightly reduced in the demyelinated area. Note that many of the axon cylinders show increased diameter (highlighted by arrowheads). Scale bar in panel C is 100 µm. (E,F) In contrast to the lateral column, the myelination in the posterior column in the same patient shows normal density in LFB‐PAS (E), and the axons in the posterior column show uniform diameter (F). The magnifications for panel E and F are the same as in panel C and D, respectively. (G,H) IHC for CD68 and CD3 show extensive infiltration of macrophages and T lymphocytes in the white matter of the spinal cord. Scale bars in panels G and H are 100 µm.
DISCUSSION
ALS with basophilic inclusions has been reported in juvenile and adult onset patients 1, 12, 17. Although several previous studies have attempted to characterize these inclusions, the underlying causes for their formation remain poorly understood 6, 7, 23. In this study, we report two patients with juvenile ALS whose pathology, genetics and clinical presentations provide important insights into the pathogenesis of this devastating disease. The most striking finding in our study is the robust FUS pathology in both patients, which underscores the critical role of abnormal FUS protein accumulations in juvenile ALS (Figure 2). Ultrastructural analyses indicate that the majority of FUS proteins are present in filamentous structures, with a minority of them associated with amorphous material and disorganized rough ER (Figure 3). While the ultrastructural characteristics of the basophilic inclusions are distinctively different from the TDP‐43‐positive inclusions in patients with FTLD‐U and late‐onset sporadic ALS, the majority of FUS proteins in juvenile ALS are identified in the filamentous aggregates in a manner similar to those seen in TDP‐43‐positive inclusion (15). Interestingly, the FUS‐positive inclusions in both our cases do not show immunoreactivity for TDP‐43, whereas late‐onset sporadic ALS cases showed intense TDP‐43, but not FUS, staining in spinal motor neurons (Figure 2). These results raise the interesting possibility that the immunohistochemical profiles for TDP‐43 and FUS may serve as effective markers to distinguish different subtypes of ALS. Future large‐scale studies will provide more definitive assessments for the feasibility of this approach. In addition to the FUS pathology in spinal motor neurons, both cases of juvenile ALS also show FUS‐positive basophilic inclusions in cortical neurons and neurons in the red nucleus, nucleus ambiguus, and the reticular formation of the medulla oblongata (Figure 4). Unlike the spinal motor neurons, however, there is no detectable loss of neurons in these locations.
The results from this study support the notion that the underlying pathogenesis of basophilic inclusions in juvenile ALS may be different from the late‐onset sporadic form of ALS. Indeed, the immunohistochemical profiles of the protein aggregates in juvenile ALS cases are distinctly different from those in patients with late‐onset sporadic ALS (Figure 2). Consistent with this notion, recent studies have provided compelling evidence that accumulation of TDP‐43 is a key pathological feature in patients with sporadic ALS, but not in patients with familial ALS with mutations in the SOD1 gene or transgenic mice the express the mutant SOD1 gene 16, 20, 25.
A series of recent studies have indicated that mutations in the FUS/TLS gene can be detected in patients with familial ALS, but not in a large number of control samples 2, 13, 26, 28. However, there have been very few reports on the detailed neuropathology features in these patients. As summarized in Table 1, perhaps the most striking difference is the age of onset of patients with different mutation in the FUS/TLS gene. Specifically, the age of onset for patients with R521H or R521C mutation tends to be in the late 30s and early 40s, whereas P525L mutation tends to affect patients of much younger age. Indeed, this mutation has been reported once before in a 22‐year‐old woman with rapidly progressive ALS (13). Among the reported cases, there appears to be a spectrum of neuropathological features in patients with FUS/TLS mutations (Table 1). For instance, the distribution of the mutant FUS protein appears diffuse in both the nuclear and cytoplasmic compartments in patients with R521G mutation (Table 1) (13). In patients with R521C mutation, however, the FUS proteins can be detected in the cytoplasmic inclusion (Table 1) (28). Interestingly, the recent report on a case of familial ALS in Japan with R521C mutation in the FUS/TLS gene shows a much more severe and widespread degeneration in CNS (26). Many of the remaining neurons and glial cells in this patient contain basophilic inclusions that are positive for FUS. These results suggest that the FUS pathology can be quite variable even in patients with the same mutation (Table 1). Whether the variations of ubiquitin and TDP‐43 IHC have any pathogenic implications will depend on a more concerted effort to correlate these features with clinical presentations and progression.
Table 1.
Comparisons of age onset and neuropathological features of FUS‐immunostaining in familial ALS cases with FUS/TLS mutations. Abbreviations: Nucl = nuclear; Cyto = cytoplasmic; MΦ = macrophage infiltration; N/A = not available.
| R521G (13) | R521H/R521C (28) | R521C (2) | R521C (26) | P525L† | |
|---|---|---|---|---|---|
| Age of onset | 39.6 ± 13.3 | N/A | 42.6 ± 15.4 | 40.7 ± 13.8 | 13 |
| Immunohistochemistry (IHC) | |||||
| FUS | |||||
| – Spinal motor neurons | Nucl/Cyto (diffuse) | Cyto (inclusion) | N/A | Cyto (inclusion) | Cyto (inclusion) |
| – Cortical neurons | Nucl/Cyto (diffuse) | N/A | N/A | N/A | Cyto (inclusion) |
| Ubiquitin | |||||
| – Spinal motor neurons | Nucl | Rare | Cyto | Cyto (inclusion) | Cyto (diffuse) |
| – Cortical neurons | Nucl | Rare | Cyto | N/A | Cyto (diffuse) |
| TDP‐43 | |||||
| – Spinal motor neurons | N/A | Absent | Nucl | N/A | Nucl |
| – Cortical neurons | N/A | Absent | Nucl | N/A | Nucl |
| Spinal cord white matter | |||||
| pallor | + | + | + | +++ | ++ |
This study.
One important finding in our study is the identification of a P525L mutation in the FUS/TLS gene in Case 1. Hence, the characterizations of Case 1 in our study provide important insights to the neuropathological features that one might detect in familial ALS with a P525L mutation in the FUS/TLS gene. As indicated above, an important characteristic of P525L mutation is that the age of onset appears to be much younger than other mutations. Compared to the previously reported cases, Case 1 in our current study is the youngest ALS case with a well‐delineated mutation in the FUS/TLS gene 2, 13, 28. While our clinical experience with this mutation remains limited, it is tempting to speculate that this particular mutation might confer a rapidly progressive juvenile presentation (Table 1). In this regard, it is also interesting to note that both of our cases had early motor delay, which may be an early manifestation of the disease.
Most mutations in the FUS/TLS gene are identified in the C‐terminus of the FUS/TLS gene, which presumably lead to mislocalization of mutant FUS proteins in the cytoplasm 2, 13, 26, 28. While it remains unclear how mutations in the FUS/TLS gene directly contribute to the pathogenesis of ALS, it is possible that these mutations may result in a loss of function in wild‐type FUS protein or a toxic gain of function in the mutant proteins, leading to neuronal cell death (14). There are several key features in familial ALS patients with FUS/TLS mutations. For instance, the majority of patients tend to be older than 40 years of age (Table 1). Only a very small number of patients have disease onset in their late 20s. Once the disease process is initiated, however, it almost invariably follows a rapid clinical progression leading to death within 2–3 years. Despite the similar pathology in both of our patients, no mutation is found in the FUS/TLS coding sequence of Case 2. These results suggest that a mutation in FUS/TLS may not be necessary for developing FUS pathology. Alternatively, it is possible that Case 2 may have a mutation in the noncoding sequence of the FUS/TLS gene, which may affect alternative splicing. Further examination of the FUS/TLS gene in Case 2 will likely provide definitive answers to the cause–effect relationship between the FUS/TLS mutations and FUS pathology. In addition to the FUS pathology, both of our cases show infiltration of scattered cytotoxic T lymphocytes in the white matter of the spinal cord. A comparison of the familial ALS with FUS/TLS mutations shows that white matter pathology, such as myelin pallor, can be detected in these patients (Table 1). While changes in the spinal cord white matter may be a manifestation of the disease process in rapidly progressive ALS, it is unclear if additional factors can influence its severity and clinical progression. Future studies to compare and contrast the white matter pathology in other juvenile ALS with FUS pathology will be critical in further determining the nature of these features.
In summary, the results in this study underscore the important role of FUS pathology and mutation in rapidly progressive juvenile ALS with basophilic inclusions. These results also indicate that screening for FUS pathology and mutations in the FUS/TLS gene should become an integral component in the standard diagnostics and care for patients with juvenile ALS.
ACKNOWLEDGMENTS
This work has been supported by NIH grants (R01 NS048393 and K26 RR024858) and VA Merit Review Award (to E.J.H.), and NIH grant AG10124 (to J.Q.T.). We thank our colleagues, Drs. Andrew Bollen and Stephen DeArmond, for discussions, and Terry Schuck, Ivy Hsieh, and Sherry Kamiya for technical supports. Special thanks go to the parents of our patients for their unwavering supports of our study. The authors declare that they have no conflict of interest.
REFERENCES
- 1. Aizawa H, Kimura T, Hashimoto K, Yahara O, Okamoto K, Kikuchi K (2000) Basophilic cytoplasmic inclusions in a case of sporadic juvenile amyotrophic lateral sclerosis. J Neurol Sci 176:109–113. [DOI] [PubMed] [Google Scholar]
- 2. Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, Manavis J et al (2010) FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry 81:639–645. [DOI] [PubMed] [Google Scholar]
- 3. Boillee S, Vande Velde C, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59. [DOI] [PubMed] [Google Scholar]
- 4. Dion PA, Daoud H, Rouleau GA (2009) Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet 10:769–782. [DOI] [PubMed] [Google Scholar]
- 5. Forman MS, Trojanowski JQ, Lee VM (2007) TDP‐43: a novel neurodegenerative proteinopathy. Curr Opin Neurobiol 17:548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fujita K, Ito H, Nakano S, Kinoshita Y, Wate R, Kusaka H (2008) Immunohistochemical identification of messenger RNA‐related proteins in basophilic inclusions of adult‐onset atypical motor neuron disease. Acta Neuropathol 116:439–445. [DOI] [PubMed] [Google Scholar]
- 7. Fujita Y, Okamoto K, Sakurai A, Kusaka H, Aizawa H, Mihara B, Gonatas NK (2002) The Golgi apparatus is fragmented in spinal cord motor neurons of amyotrophic lateral sclerosis with basophilic inclusions. Acta Neuropathol 103:243–247. [DOI] [PubMed] [Google Scholar]
- 8. Geser F, Brandmeir NJ, Kwong LK, Martinez‐Lage M, Elman L, McCluskey L et al (2008) Evidence of multisystem disorder in whole‐brain map of pathological TDP‐43 in amyotrophic lateral sclerosis. Arch Neurol 65:636–641. [DOI] [PubMed] [Google Scholar]
- 9. Geser F, Martinez‐Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ et al (2009) Clinical and pathological continuum of multisystem TDP‐43 proteinopathies. Arch Neurol 66:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D et al (2008) TDP‐43 A315T mutation in familial motor neuron disease. Ann Neurol 63:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574. [DOI] [PubMed] [Google Scholar]
- 12. Kusaka H, Matsumoto S, Imai T (1993) Adult‐onset motor neuron disease with basophilic intraneuronal inclusion bodies. Clin Neuropathol 12:215–218. [PubMed] [Google Scholar]
- 13. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 14. Lagier‐Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP‐43. Cell 136:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin WL, Dickson DW (2008) Ultrastructural localization of TDP‐43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol 116:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ et al (2007) Pathological TDP‐43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434. [DOI] [PubMed] [Google Scholar]
- 17. Matsumoto S, Kusaka H, Murakami N, Hashizume Y, Okazaki H, Hirano A (1992) Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol 83:579–583. [DOI] [PubMed] [Google Scholar]
- 18. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 19. Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723. [DOI] [PubMed] [Google Scholar]
- 20. Robertson J, Sanelli T, Xiao S, Yang W, Horne P, Hammond R et al (2007) Lack of TDP‐43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett 420:128–132. [DOI] [PubMed] [Google Scholar]
- 21. Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF et al (2008) Novel mutations in TARDBP (TDP‐43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet 4:e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sabatelli M, Madia F, Conte A, Luigetti M, Zollino M, Mancuso I et al (2008) Natural history of young‐adult amyotrophic lateral sclerosis. Neurology 71:876–881. [DOI] [PubMed] [Google Scholar]
- 23. Sasaki S, Toi S, Shirata A, Yamane K, Sakuma H, Iwata M (2001) Immunohistochemical and ultrastructural study of basophilic inclusions in adult‐onset motor neuron disease. Acta Neuropathol 102:200–206. [DOI] [PubMed] [Google Scholar]
- 24. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al (2008) TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A et al (2007) TDP‐43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113:535–542. [DOI] [PubMed] [Google Scholar]
- 26. Tateishi T, Hokonohara T, Yamasaki R, Miura S, Kikuchi H, Iwaki A et al (2010) Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol 119:355–364. [DOI] [PubMed] [Google Scholar]
- 27. Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB et al (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP‐43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]