

Abstract
Catalytic enantioselective allylic substitution (EAS) reactions, which involve the use of alkyl- or aryl-substituted vinylaluminum reagents and afford 1,4-dienes containing a quaternary carbon stereogenic center at their C-3 site, are disclosed. The C–C bond forming transformations are promoted by 0.5–2.5 mol % of sulfonate bearing chiral bidentate N-heterocyclic carbene (NHC) complexes, furnishing the desired products efficiently (66–97% yield of isolated products) and in high site- (>98% SN2′) and enantioselectivity [up to 99:1 enantiomer ratio (er)]. To the best of our knowledge, the present report puts forward the first cases of allylic substitution reactions that result in the generation of all-carbon quaternary stereogenic centers through the addition of a vinyl unit. The aryl- and vinyl-substituted vinylaluminum reagents, which cannot be prepared in high efficiency through direct reaction with diisobutylaluminum hydride, are accessed through a recently introduced Ni-catalyzed reaction of the corresponding terminal alkynes with the same inexpensive metal-hydride agent. Sequential Ni-catalyzed hydrometallations and Cu-catalyzed C–C bond forming reactions allow for efficient and selective synthesis of a range of enantiomerically enriched EAS products, which cannot cannot be accessed by previously disclosed strategies (due to inefficient vinylmetal synthesis or low reactivity and/or selectivity with Si-substituted derivatives). The utility of the protocols developed is demonstrated through a concise enantioselective synthesis of natural product bakuchiol.
Introduction
A significant challenge in chemical synthesis concerns the development of efficient catalytic enantioselective reactions that furnish all-carbon quaternary stereogenic centers.1 One general strategy for promoting such processes concerns additions of C-based nucleophiles to electrophilic carbon sites. The corresponding transformations with vinylmetal reagents represent an attractive option: the products, bearing a quaternary carbon stereogenic center at the allylic position, can be functionalized and/or utilized in synthesis of natural products. Nonetheless, catalytic enantioselective protocols delivering quaternary carbon stereogenic centers by additions of a vinylmetal are scarce.2-3 There has been only one method developed in which an alkene-based electrophile is utilized in conjunction with a vinylboronic acid;4 related catalytic enantioselective allylic substitution (EAS) reactions that generate quaternary carbons5,6,7 and involve addition of a vinyl group8 have not yet been introduced (eq 1). Herein, we report the first examples of catalytic EAS processes with alkyl- or aryl-substituted vinylaluminums, affording 1,4-dienes that contain a quaternary carbon stereogenic center at their C-3 site. Transformations are promoted by 0.5–2.5 mol % of chiral bidentate N-heterocyclic carbene (NHC) complexes, furnishing the desired products efficiently and in high site- (>98% SN2′) and enantioselectivity [up to 99:1 enantiomer ratio (er)]. Whereas alkyl-substituted vinylaluminums are obtained through reaction of the corresponding alkynes with diisobutylaluminum hydride (dibal–H),8a the aryl- or vinyl-substituted vinylmetal variants are accessed through a recently introduced Ni-catalyzed hydroalumination.9,10 The sequential Ni- and Cu-catalyzed processes allow for efficient and selective synthesis of a range of enantiomerically enriched EAS products, which cannot be accessed by previously disclosed strategies (due to inefficient vinylmetal synthesis or low reactivity and/or selectivity with Si-substituted derivatives).
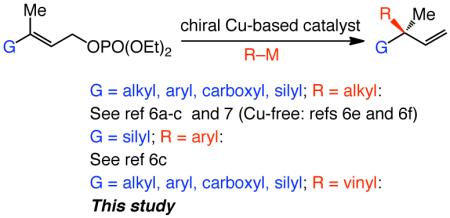 |
(1) |
We have designed methods for enantioselective formation of tertiary C–C bonds through additions of vinylaluminums to allylic phosphates, catalyzed by chiral bidentate NHC–Cu complexes.6,8 The requisite alkyl-substituted vinylmetals were accessed by hydroalumination of terminal alkynes with dibal–H.8a We later addressed the problem of inefficiency in preparing aryl-substituted vinylaluminums through hydrometallations of the derived silyl-containing aryl alkynes;8b desilylation with a protic acid subsequent to Cu-catalyzed alkylation delivers the desired enantiomerically enriched 1,4-diene. However, the more sterically demanding Si-containing aryl-substituted vinylaluminums cannot be utilized effectively in synthesis of the more congested quaternary carbon centers. An alternative approach for efficient preparation of aryl-substituted vinylaluminums, as will be described below, had to be introduced. From inception, we considered Cu-catalyzed EAS reactions of aryl-substituted vinylmetals to be a key component of the our investigations; the present studies were partly driven by the question as to whether a concise synthesis of enantiomerically enriched bakuchiol and related natural products might be devised by the use of the targeted class of reactions (Scheme 1).11
Scheme 1.

Proposed Synthesis of Bakuchiol through Site- and Enantioselective NHC–Cu-Catalyzed Allylic Substitution with a Vinylmetal Reagent
Results and Discussion
1. NHC–Cu-Catalyzed Enantioselective Allylic Substitution (EAS) Reactions with Vinylaluminum Reagents Derived from Alkyl-Substituted Alkynes
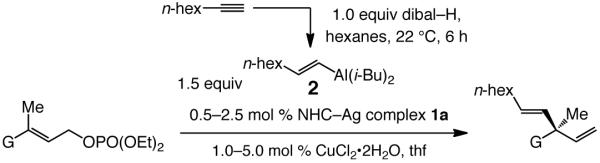
We first established the feasibility of the proposed catalytic transformations. We focused our attention on alkyl-substituted vinylaluminums, since such entities can be easily accessed through site-selective hydroalumination with dibal–H. Initial screening pointed to Cu complexes derived from sulfonate-containing bidentate Ag carbenes 1a–c12 (Scheme 2) as optimal (see below for further discussion).
Scheme 2.

Chiral Bidentate NHC–Ag(I) Complexes Used in This Studya
aMes = 2,4,6-Me3C6H3.
A considerable range of allylic phosphates undergo facile EAS reactions in the presence of 0.5–2.5 mol % 1a13 with n-hexyl-substituted vinylaluminum 2 to furnish the desired 3,3-disubstituted 1,4-dienes in 90:10–98:2 er and 77–97% yield (Table 1). Aryl-substituted allylic phosphates (entries 1–7) bearing electron donating (entry 5) and withdrawing substituents (entries 2-4 and 7), as well as those that carry an alkyl group (entries 8–9) serve as effective substrates. Reactions with aryl-substituted allylic phosphates are sufficiently selective to be performed at −15 °C–22 °C and are complete in three hours. Transformations that involve alkyl-substituted substrates (entries 8–9, Table 1) must be performed at −50 °C for maximum enantioselectivity14 and might require longer reaction times (6–24 h vs. 10 min–3 h), unless higher catalyst loading is used (e.g., 2 mol %, −50 °C, 6 h, entry 8, Table 1). The Cu-catalyzed process can be used to access 1,4-dienes containing a carboxylic ester or silyl-substituted quaternary carbon stereogenic center at the C3 position (entries 10–11).
Table 1.
NHC–Cu-Catalyzed Additions of an Alkyl-Substituted Vinylaluminum Reagent to Allylic Phosphates Bearing a Trisubstituted Alkenea
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate (G) | mol % 1a | temp (°C); time | SN2′ (%)b | yield (%)c | erb |
| 1 | Ph | 0.5 | −15; 3 h | >98 | 82 | 93.5:6.5 |
| 2 | o-BrC6H4 | 1.0 | 22; 10 min | >98 | 92 | 96.5:3.5 |
| 3 | o-CF3C6H4 | 2.5 | −15; 3 h | >98 | 96 | 98:2 |
| 4 | o-NO2C6H4 | 2.0 | 22; 10 min | >98 | 89 | 96.5:3.5 |
| 5 | o-MeOC6H4 | 0.5 | 22; 30 min | >98 | 83 | 97.5:2.5 |
| 6 | o-MeC6H4 | 2.0 | −15; 3 h | >98 | 87 | 96.5:3.5 |
| 7 | p-NO2C6H4 | 0.5 | 22; 10 min | >98 | 91 | 94.5:5.5 |
| 8 | Cy | 2.0 | −50; 6 h | >98 | 91 | 95:5 |
| 9 | Me2CCH(CH2)2 | 1.0 | −50; 24 h | >98 | 77 | 92.5:7.5 |
| 10 | CO2t-Bu | 0.5 | −15; 3 h | >98 | 97 | 90:10 |
| 11 | SiMe2Ph | 0.5 | −15; 3 h | >98 | 85 | 95.5:4.5 |
Reactions were performed under N2 atmosphere; >98% conversion in all cases, except 91% conv. in entry 9; >98% E product isomer in all cases.
Determined by analysis of 400 MHz 1H NMR of unpurified mixtures.
Yields of isolated and purified products.
Determined by HPLC analysis; see the Supporting Information for details.
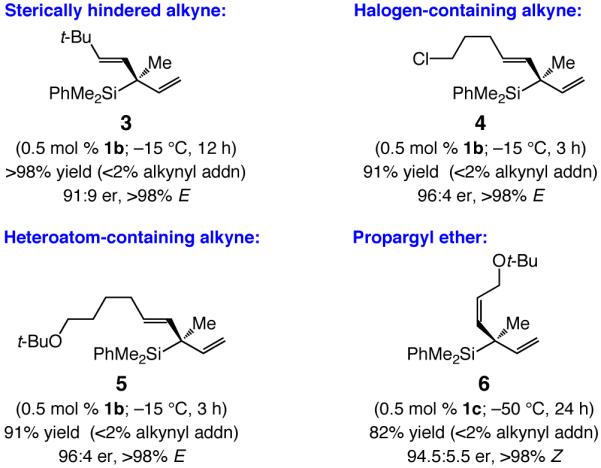
As illustrated through Cu-catalyzed synthesis of 1,4-dienes 3–6 (Scheme 3), a variety of vinylaluminum reagents, including one that contains a sterically demanding tert-butyl substituent (3), a halide (4) or heteroatom units (5–6),15 can be used. Such transformations proceed with similarly high efficiency and stereoselectivity (single olefin isomer and 91:9–96:4 er) as observed in processes that involve alkyl-substituted alkynes (Table 1).
Scheme 3.
2. NHC–Cu-Catalyzed Enantioselective Allylic Substitution (EAS) Reactions with Vinylaluminum Reagents Derived from Vinyl or Aryl-Substituted Alkynes
i. Inefficient synthesis of vinylaluminum reagents
In contrast to NHC–Cu-catalyzed reactions that involve alkyl-substituted vinylaluminum reagents, when the vinylmetal is derived from hydrometallation of an enyne or an aryl alkyne (e.g., 7 and 8, Scheme 4), although high er values are obtained, product mixtures are contaminated with alkynyl addition products (up to 45%).16 Presumably, during hydroalumination, the vinylmetal serves as an efficient base to deprotonate the starting alkyne, affording alkynylaluminum and the corresponding protonated alkene. The above complication is exacerbated by reaction of the alkynylaluminum, which appears to be somewhat more efficient relative to addition of the vinylmetal reagent. Control experiments indicate that whereas 25% alkyne deprotonation occurs in hydroalumination of phenylacetylene, 45% alkynyl addition product17 is generated along with 1,4-diene 8 (1.5 equiv hydroalumination product mixture used; see Scheme 4).
Scheme 4.

NHC–Cu-Catalyzed EAS with Aryl Alkynes or Enynes Leads to Significant Amounts of Alkynyl Addition Products
ii. The first approach: Hydroalumination of silyl-substituted alkynes
Our first attempt to bypass the inefficiency of the aforementioned hydroaluminations led us to examine the formerly reported catalytic EAS reactions with silyl-substituted vinylaluminum;8b the outcome of these investigations is shown in Scheme 5. In a Cu-catalyzed process with allylic phosphate 10, applicable to enantioselective synthesis of bakuchiol (Scheme 1), use of trisubstituted vinylaluminum 9, generated from reaction of the corresponding trimethylsilylethynyl-4-anisole with dibal–H, gives rise to substantial amounts of i-Bu addition product and furnishes 1,4-diene 11 with low enantioselectivity (52.5:47.5–61:39 er). Stabilization of electron density at the vinylic carbon by the silyl substituent likely retards the rate of vinyl transfer, resulting in the formation of 17–45% of 12 in the product mixture.
Scheme 5.
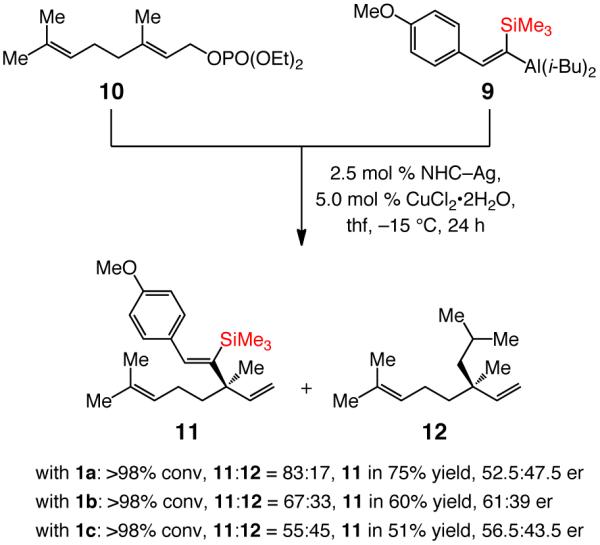
Use of a Si-Substituted Vinylaluminum Reagent Derived from an Aryl Alkyne in NHC–Cu-Catalyzed EAS
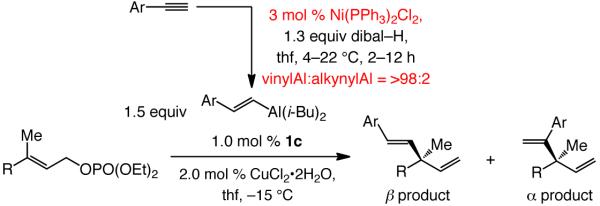
iii. The second approach: Efficient Ni-catalyzed hydroalumination of vinyl- or aryl-substituted terminal alkynes
In the course of searching for an effective solution to the problem of efficient vinylaluminum synthesis with minimal alkynylaluminum contamination, we discovered that in the presence of 3.0 mol % of commercially available Ni(PPh3)2Cl2, reaction of dibal–H with enynes or aryl-substituted alkynes proceeds efficiently (within 2–12 hours at 4–22 °C) to afford the desired vinylaluminum with high selectivity [85% to >98% terminal:internal (β:α) vinylaluminum] and with <5% alkyne deprotonation. Thus, as depicted in Scheme 6, 1,4-diene 7 can be obtained in 89% yield when the Ni-catalyzed hydroalumination is utilized in conjunction with the NHC–Cu-catalyzed process (vs. 66% yield with uncatalyzed hydroalumination in Scheme 4); the amount of alkynyl addition side product is reduced to 5% (vs 29%). Formation of the desired 1,4-diene (7) in 99:1 er indicates that the presence of the Ni salt in the reaction mixture does not have an adverse effect on the EAS process; this contention is further substantiated by reactions of aryl alkynes, described below.
Scheme 6.

Ni-Catalyzed Hydroalumination of an Enyne and In Situ Use in an NHC–Cu-Catalyzed EAS Reaction
As the data in Table 2 illustrate, an assortment of aryl-substituted alkynes is readily and selectively converted to the derived β-vinylaluminums, which are then used in situ for highly efficient as well as site- and enantioselective EAS reactions (78–92% yield of pure β product, >98% SN2′, 87:13–98:2 er). In stark contrast to the process involving uncatalyzed hydroalumination (45% alkynyl addition; Scheme 3), the transformation illustrated in entry 5 of Table 2 does not afford any products derived from alkynylaluminum addition (<2% by 400 MHz 1H NMR analysis of the unpurified mixture). Only in cases where the aryl unit of the alkyne bears an electron-withdrawing p-CF3 substituent is >10% of the α-vinylaluminum adduct observed; otherwise, the desired β-substituted 1,4-diene products are obtained in ≥92% selectivity. It is worthy of note that the presence of the Ni catalyst enhances the rate of hydroalumination such that higher conversion is attained for the overall process (vinylaluminum formation/EAS). As an example, only 72% conversion is achieved in formation of 8 under the conditions shown in Scheme 4, largely due to lower amounts of available vinylaluminum reagent, whereas complete consumption of the allylic phosphate is observed in reaction in entry 5 of Table 2. In cases where the alkyne substrate contains a sterically demanding o-substituted aryl group (entries 2 and 11, Table 2), ~10% of the alkynyl addition product is formed, likely as a result of diminution in the rate of hydroalumination.
Table 2.
NHC–Cu-Catalyzed Additions of Aryl-substituted Vinylaluminum Reagents to Allylic Phosphates Containing a Trisubstituted Alkenea
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | substrate (R) | alkyne (Ar) | time (h) | β:αb | SN2′:SN2b | yield (%)c | erd |
| 1 | Ph | Ph | 3 | 95:5 | >98 | 78 | 96:4 |
| 2 | Ph | o-MeC6H4 | 6 | >98:2 | >98 | 88e | 95:5 |
| 3 | Ph | p-MeOC6H4 | 6 | >98:2 | >98 | 84 | 96.5:3.5 |
| 4 | Ph | p-CF3C6H4 | 3 | 87:13 | >98 | 82 | 94:6 |
| 5 | o-BrC6H4 | Ph | 3 | 96:4 | >98 | 91 | 98:2 |
| 6 | o-NO2C6H2 | Ph | 24 | >98:2 | >98 | 92 | 98:2 |
| 7 | o-MeC6H4 | p-MeOC6H4 | 3 | >98:2 | >98 | 82 | 98:2 |
| 8 | p-NO2C6H4 | Ph | 3 | 92:8 | >98 | 84 | 93:7 |
| 9 | p-CF3C6H4 | p-MeOC6H4 | 3 | >98:2 | >98 | 89 | 94:6 |
| 10 | Me2CCH(CH2)2 | Ph | 3 | 93:7 | >98 | 81 | 90:10 |
| 11 | Me2CCH(CH2)2 | o-MeC6H4 | 6 | >98:2 | >98 | 85e | 91:9 |
| 12 | Me2CCH(CH2)2 | p-MeOC6H4 | 6 | >98:2 | >98 | 90 | 91:9 |
| 13 | Me2CCH(CH2)2 | p-CF3C6H4 | 3 | 85:15 | >98 | 79 | 87:13 |
Reactions were performed under N2 atmosphere; >98% conversion in all cases.
Determined by analysis of 400 MHz 1H NMR spectra of the unpurified mixtures.
Yields of isolated and purified β products.
Enantiomer ratio of the β product, determined by HPLC analysis; see the Supporting Information for details.
Alkynyl product (~10%) present in the product mixture.
The utility of the sequential β-selective Ni-catalyzed terminal alkyne hydroalumination/NHC–Cu-catalyzed EAS is highlighted in the synthesis of enantiomerically enriched bakuchiol shown in Scheme 7. The three-vessel process, involving geraniol and a readily available terminal alkyne as starting materials, proceeds in 72% overall yield. The route depicted in Scheme 7 is substantially more concise than the most efficient of the previously reported approaches, the shortest of which requires ten steps and delivers the target in 49% yield.11b
Scheme 7.

Application of Sequential Ni-Catalyzed Alkyne Hydroalumination/NHC–Cu-Catalyzed EAS to Enantioselective Synthesis of Bakuchiol
3. Mechanistic Considerations and the Significance of Various Structural Features of Bidentate Sulfonate-Containing NHC–Cu Complexes
The efficiency and high site- and enantioselectivities in the catalytic EAS reactions described above arise from unique catalytic abilities of sulfonate-based bidentate NHC–Cu complexes 1a–c;18 as shown in Scheme 8, phenoxy-bridged or monodentate NHC–Cu variants are ineffective catalysts. The above findings can be explained through mode of reaction I (Scheme 8). The syn relationship between the phenyl backbone of the NHC and the sulfonate, which is in contrast to the anti stereochemistry in the phenoxy-bridged systems, has been substantiated by X-ray structures and rationalized in a previous report regarding the corresponding Zn and Al complexes.6f The above stereochemical attribute allows the vinyl unit to be positioned properly for SN2′ addition to the coordinated alkene,19 while the equatorially positioned sulfonate oxygen facilitates addition by Lewis acid activation of the allylic phosphate.
Scheme 8.

Proposed Mechanistic Model and Key Features of the Chiral Bidentate NHC–Cu Complexa
 |
(2) |
Such features are absent in phenoxy-bridged or monodentate NHC–Cu complexes. High enantioselectivity likely arises, since mode of addition II engenders unfavorable steric interactions, as shown in Scheme 8; furthermore, examination of molecular models indicate that the aforementioned Lewis acid activation involving the phosphate group (cf. I) cannot be easily assisted by the sulfonate unit in II. Consistent with the proposed model, when the corresponding Z-trisubstituted allylic phosphates are used, the opposite product enantiomer is generated predominantly but in low enantioselectivity, since reaction via I would lead to unfavorable steric interactions shown in II; the example in eq 2 is representative.
Conclusions
Among various types of C-based nucleophiles used in C–C bond forming reactions, products arising from transformations with vinylmetal reagents are among the most versatile and useful. Nonetheless, related catalytic enantioselective protocols20,21,22 – particularly those that furnish quaternary carbon stereogenic centers2,4 – continue to be relatively uncommon. In addition to the challenges associated with the design of effective chiral catalysts that promote vinylmetal additions to a sterically congested electrophilic site, identification of a class of vinylmetal reagents, which can be easily and efficiently accessed and are only sufficiently nucleophilic to allow the catalytic process to predominate, has been a longstanding complication. The present investigations put forth protocols that address both of the aforementioned challenges: a Ni-catalyzed reaction involving readily available alkynes and inexpensive dibal–H, generating vinylaluminums that are used in situ for efficient and highly site- and enantioselective EAS reactions promoted by chiral NHC–Cu complexes.
Together with previous disclosures regarding metal-catalyzed EAS as well as conjugate additions involving alkyl- and aryl-substituted zinc and aluminum reagents,12 and the more recent examples delivering boron-substituted quaternary carbon stereogenic centers,23 the present findings further underline the unique catalytic activity of sulfonate-based bidentate chiral NHC–Cu complexes. Design of additional classes of NHC-based catalysts that exhibit exceptional reactivity and deliver high selectivity, and development of additional catalytic enantioselective protocols, including those that involve various vinyl units, are in progress and will be disclosed in due course.
Supplementary Material
Acknowledgments
The NIH (GM-47480) provided financial support. Y. L. is grateful for an AstraZeneca graduate fellowship. We thank Tricia L. May for helpful discussions. Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available. Experimental procedures and spectral data for substrates and products (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes and References
- (1).(a) Christophers J, Baro A, editors. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley–VCH; Weinheim: 2006. [Google Scholar]; (b) Douglas CJ, Overman LE. Proc. Nat. Acad. Sci. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cozzi PG, Hilgraf R, Zimmermann N. Eur. J. Org. Chem. 2007:5969–5994. [Google Scholar]
- (2).For Ti-catalyzed enantioselective additions of vinylzinc reagents to ketones, see: Li H, Walsh PJ. J. Am. Chem. Soc. 2004;126:6538–6539. doi: 10.1021/ja049206g. Li H, Walsh PJ. J. Am. Chem. Soc. 2005;127:8355–8361. doi: 10.1021/ja0425740. For Ti-catalyzed enantioselective additions of vinylaluminums to aryl ketones, see: Biradar DB, Gau H-M. Org. Lett. 2009;11:499–502. doi: 10.1021/ol801999u.
- (3).Molecules that bear a benzylic vinyl unit at an all-carbon quaternary stereogenic center have been synthesized enantioselectively through Ni-catalyzed reactions of styrene precursors with ethylene. See: Shi W-J, Zhang Q, Xie J-H, Zhu S-F, Hou G-H, Zhou Q-L. J. Am. Chem. Soc. 2006;128:2780–2781. doi: 10.1021/ja057654y. Zhang A, RajanBabu TV. J. Am. Chem. Soc. 2006;128:5620–5621. doi: 10.1021/ja060999b. Smith CR, Lim HJ, Zhang A, RajanBabu TV. Synthesis. 2009:2089–2900. doi: 10.1055/s-0029-1216826.
- (4).For Rh-catalyzed additions of vinylboronic acids to α,β-unsaturated pyridylsulfones, see: Mauleón P, Carretero JC. Chem. Commun. 2005:4961–4963. doi: 10.1039/b508142d. In addition, two isolated cases of alkyl-containing vinylaluminum conjugate additions to β-substituted cyclic enones, promoted by Feringa-type chiral phosphoramidite ligands and which proceed in 75:25 and 86.5:13.5 er, have been reported. See: Vuagnoux-d’Augustin M, Alexakis A. Chem. Eur. J. 2007;13:9647–9662. doi: 10.1002/chem.200701001. Palais L, Alexakis A. Chem. Eur. J. 2009;15:10473–10485. doi: 10.1002/chem.200901577.
- (5).For reviews on catalytic allylic alkylation reactions with “hard” C-based nucleophilic reagents, see: Hoveyda AH, Hird AW, Kacprzynski MA. Chem. Commun. 2004:1779–1785. doi: 10.1039/b401123f. Yorimitsu H, Oshima K. Angew. Chem. Int. Ed. 2005;44:4435–4439. doi: 10.1002/anie.200500653. Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M. Chem. Rev. 2008;108:2796–2823. doi: 10.1021/cr0683515.
- (6).For catalytic enantioselective allylic substitutions promoted by chiral bidentate NHC–Cu complexes developed in these laboratories, see: Larsen AO, Leu W, Nieto Oberhuber C, Campbell JE, Hoveyda AH. J. Am. Chem. Soc. 2004;126:11130–11131. doi: 10.1021/ja046245j. Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J. Am. Chem. Soc. 2005;127:6877–6882. doi: 10.1021/ja050179j. Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew. Chem. Int. Ed. 2007;46:4554–4558. doi: 10.1002/anie.200700841. For application of such transformations to natural product synthesis, see: Gillingham DG, Hoveyda AH. Angew. Chem. Int. Ed. 2007;46:3860–3864. doi: 10.1002/anie.200700501. For reactions promoted by the corresponding Mg(II)-based complexes, see: Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2006;128:15604–15605. doi: 10.1021/ja067456m. For transformations promoted by the corresponding Zn(II)- and Al(III)-based complexes, see: Lee Y, Li B, Hoveyda AH. J. Am. Chem. Soc. 2009;131:11625–11633. doi: 10.1021/ja904654j. For EAS reactions involving aryl- and hetero-arylmetal reagents and promoted by the same class of NHC–Cu complexes, see: Gao F, Lee Y, Mandai K, Hoveyda AH. Angew. Chem., Int. Ed. 2010;49 doi: 10.1002/anie.201005124. in press.
- (7).For Cu-catalyzed EAS reactions involving amino acid-based ligands developed in these laboratories, see: Luchaco-Cullis CA, Mizutani H, Murphy KE, Hoveyda AH. Angew. Chem. Int. Ed. Engl. 2001;40:1456–1460. doi: 10.1002/1521-3773(20010417)40:8<1456::AID-ANIE1456>3.0.CO;2-T. Kacprzynski MA, Hoveyda AH. J. Am. Chem. Soc. 2004;126:10676–10681. doi: 10.1021/ja0478779. Murphy KE, Hoveyda AH. Org. Lett. 2005;7:1255–1258. doi: 10.1021/ol047331r.
- (8).For Cu-catalyzed EAS reactions with vinylaluminum reagents that afford tertiary C–C bonds and are promoted by sulfonate-based bidentate chiral NHC–Cu complexes, see: Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. J. Am. Chem. Soc. 2008;130:446–447. doi: 10.1021/ja0782192. Akiyama K, Gao F, Hoveyda AH. Angew. Chem. Int. Ed. 2010;49:419–423. doi: 10.1002/anie.200905223.
- (9).Gao F, Hoveyda AH. J. Am. Chem. Soc. 2010;132:10961–10963. doi: 10.1021/ja104896b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For a review on hydroaluminations of alkynes and alkenes, see: Eisch JJ. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Schreiber SL, editors. Vol. 8. Pergamon; Oxford: 1991. pp. 733–761.
- (11).For previous enantioselective syntheses of bakuchiol, see: Takano S, Shimazaki Y, Ogasawara K. Tetrahedron Lett. 1990;31:3325–3326. Du X-L, Chen H-L, Feng H-J, Li Y-C. Helv. Chim. Acta. 2008;91:371–378. Esumi T, Shimizu H, Kashiyama A, Sasaki C, Toyota M, Fukuyama Y. Tetrahedron Lett. 2008;49:6846–6849. Bequette JP, Jungong CS, Novikov AV. Tetrahedron Lett. 2009;50:6963–6964. doi: 10.1016/j.tetlet.2009.09.147.
- (12).For initial reports on sulfonate-based bidentate NHC–Cu complexes, see: Brown MK, May TL, Baxter CA, Hoveyda AH. Angew. Chem. Int. Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511. May TL, Brown MK, Hoveyda AH. Angew. Chem. Int. Ed. 2008;47:7358–7362. doi: 10.1002/anie.200802910. For applications in complex molecule synthesis, see: (c) Ref 6d. Brown MK, Hoveyda AH. J. Am. Chem. Soc. 2008;130:12904–12906. doi: 10.1021/ja8058414. Peese KM, Gin DY. Chem. Eur. J. 2008;14:1654–1665. doi: 10.1002/chem.200701290.
- (13).Similar efficiencies and selectivities are observed with complexes derived from 1b and 1c. Use of the NHC–Cu derived from 1a is due to the slightly shorter route used for the preparation of the latter Ag complex.
- (14).For example, reaction in entry 9 of Table 1, when performed at 22 °C, affords the desired product in 88.5:11.5 er.
- (15).For directed (Z-selective) hydroalumination of terminal propargyl ethers (cf. 6), see: Alexakis A, Duffault JM. Tetrahedron Lett. 1988;29:6243–6246.
- (16).The ability of NHC–Cu complexes to promote addition of an alkyne unit is noteworthy. Development of the corresponding enantioselective variants is in progress and will be disclosed shortly.
- (17).The higher preponderance of alkynyl product (45%) versus alkyne deprotonation (25%) is because 1.5 equivalents of alkyne substrate (and 1.5 equiv of dibal–H) are used, which indicates that Cu-catalyzed addition of alkynylaluminum is faster than that of the vinylaluminum reagent.
- (18).Cu complexes derived from NHC–Ag complexes 1a-c afford the desired EAS products in similar yields (±5%) and enantioselectivities (±3%).
- (19).An NHC–Cu-catalyzed EAS process might proceed through a Cu(I) mechanism (direct transfer of the vinyl unit) or a pathway that involves a Cu(III) complex (cuprate addition followed by alkyl-vinyl reductive elimination). Either scenario benefits from the Lewis acid activation proposed, whereas the possibility of situating the vinyl group vis-à-vis the coordinated substrate in the manner shown in I, would facilitate the Cu(I) pathway (proper alignment of C–Cu and alkene π*). It is not clear at present which pathway is energetically preferred. Although previous mechanistic studies point to Cu(III) mechanism being operative, such investigations were in connection to alkyl- or allylcopper complexes, considered allyl halides as substrates, and did not involve a catalyst. The more polarized nature of a Cu–C bond in a vinylmetal complex, particularly a strongly Lewis base-activated NHC–Cu-vinyl complex (see ref. 6f), and the associated steric demands of forming a Cu(III)-substituted quaternary carbon, could favor the Cu(I) pathway. For recent reports regarding the mechanism of non-catalytic allylic substitution reactions with alkyl- and allylcopper reagents, see: Sofia A, Karlström E, Bäckvall J-E. Chem. Eur. J. 2001;7:1981–1989. doi: 10.1002/1521-3765(20010504)7:9<1981::aid-chem1981>3.0.co;2-c. Yoshikai N, Zhang S-L, Nakamura E. J. Am. Chem. Soc. 2008;130:12862–12863. doi: 10.1021/ja804682r. Bartholomew ER, Bertz SH, Cope S, Murphy M, Ogle CA. J. Am. Chem. Soc. 2008;130:11244–11245. doi: 10.1021/ja801186c.
- (20).For examples of catalytic enantioselective vinyl conjugate additions to unsaturated carbonyls, affording tertiary C–C bonds, see: Oi S, Taira A, Honma Y, Inoue Y. Org. Lett. 2003;5:97–99. doi: 10.1021/ol0272904. Oi S, Sato T, Inoue Y. Tetrahedron Lett. 2004;45:5051–5055. Otomaru Y, Hayashi T. Tetrahedron: Asymmetry. 2004;15:2647–2651. Nicolaou KC, Tang W, Dagneau P, Faraoni R. Angew. Chem. Int. Ed. 2005;44:3874–3879. doi: 10.1002/anie.200500789. Nakao Y, Chen J, Imanaka H, Hiyama T, Ichikawa Y, Duan W-L, Shintani R, Hayashi T. J. Am. Chem. Soc. 2007;129:9137–9143. doi: 10.1021/ja071969r. Lee K-s., Hoveyda AH. J. Org. Chem. 2009;74:4455–4462. doi: 10.1021/jo900589x.
- (21).For examples of catalytic enantioselective vinyl additions to carbonyls, affording tertiary C–C bonds, see: Oppolzer W, Radinov RN. J. Am. Chem. Soc. 1993;115:1593–1594. Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. Tomita D, Wada R, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2005;127:4138–4139. doi: 10.1021/ja0507362. Yang Y, Zhu S-F, Zhou C-Y, Zhou Q-L. J. Am. Chem. Soc. 2008;130:14052–14053. doi: 10.1021/ja805296k. Kerrigan MH, Jeon S-J, Chen YK, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2009;131:8434–8445. doi: 10.1021/ja809821x.
- (22).For examples of catalytic enantioselective vinyl additions to aldimines, affording tertiary C–C bonds, see: Patel SJ, Jamison TF. Angew. Chem. Int. Ed. 2004;43:3941–3944. doi: 10.1002/anie.200460044. Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;127:11269–11276. doi: 10.1021/ja051104i. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:12644–12645. doi: 10.1021/ja075438e. Lou S, Schaus SE. J. Am. Chem. Soc. 2008;130:6922–6923. doi: 10.1021/ja8018934. Nakao Y, Takeda M, Chen J, Salvi L, Hiyama T, Ichikawa Y, Shintani R, Hayashi T. Chem. Lett. 2008;37:290–291.
- (23).Guzman-Martinez A, Hoveyda AH. J. Am. Chem. Soc. 2010;132:10634–10637. doi: 10.1021/ja104254d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.