

Abstract
In this issue of Cell Stem Cell, Kiel et al. (2007) demonstrate that N-cadherin is not expressed on repopulating hematopoietic stem cells (HSCs) and that reduction of osteoblasts does not affect HSC frequency, suggesting that other molecular pathways may also modulate the interaction of HSCs with their niches.
Within the bone marrow (BM) micro-environment, hematopoietic stem cells (HSCs) reside in proximity to osteoblastic, vascular, and stromal cells, where they maintain a predominantly quiescent state, undergo self-renewal, and are recruited to reconstitute hematopoiesis (Adams et al., 2006; Calvi et al., 2003; Heissig et al., 2002; Kiel et al., 2005; Zhang et al., 2003). Given the technical hurdles associated with isolating and tracking long-term repopulating HSCs within the BM, the molecular and cellular pathways that mediate interaction of true HSCs with their niches have been difficult to study. Utilizing immunoselection approaches, it has been proposed that homotypic N-cadherin-mediated interactions between phenotypically marked N-cadherin+ HSCs and N-cadherin+ osteoblasts support long-term maintenance of HSCs (Zhang et al., 2003). However, in the current issue, Kiel et al. have used genetic tracking to mark N-cadherin expression and demonstrate that N-cadherin is not expressed on long-term repopulating HSCs. In addition, reduction of osteoblasts in this system does not affect the number of HSCs in BM. These data point to the complexity of deciphering the molecular pathways that regulate interaction of the repopulating HSCs with their niches, and suggest that other as of yet unrecognized adhesion molecules and chemokines may also participate in the interaction of HSCs with their niches.
Reconstitution and maintenance of hematopoiesis are dependent not only on cell intrinsic properties of HSCs, but also on extrinsic, dynamic interactions of HSCs with their niches. A “stem cell niche” is defined as a highly specialized microenvironment that preserves a balance between quiescence and self-renewal of HSCs by interactions with stromal cells, such as endosteal cells (osteoblastic niche) or vascular cells (vascular niche) (Figure 1). However, the precise anatomical location of the HSC niche has dodged precise definition mainly due to technical barriers of working with calcified boney tissues and the non-static nature of the hematopoietic system. Heissig et al. demonstrated that after myelosuppression, hematopoietic recovery was initiated in the osteoblastic followed by vascular niches (Heissig et al., 2002). The delay in hematopoietic recovery of the vascular niche was due to rapid regression of endothelial cells, as these cells are more susceptible to myeloablative insults (Kopp et al., 2005). Subsequently, it was demonstrated that a hormonally induced increase in the number of osteoblasts correlated with a modest increase in the number of HSCs (Calvi et al., 2003). N-cadherin was also shown to be expressed on ~10% of the Sca1+cKit+lin− hematopoietic cells, supporting the notion that homotypic N-cadherin interaction with osteoblasts is critical for maintenance of phenotypically marked HSCs (Zhang et al., 2003). However, from these studies it was unclear whether N-cadherin was indeed expressed on authentic repopulating HSCs or whether N-cadherin is necessary for osteoblastic regulation of HSC homeostasis.
Figure 1. Interaction of HSCs with Osteoblastic and Vascular Niches.
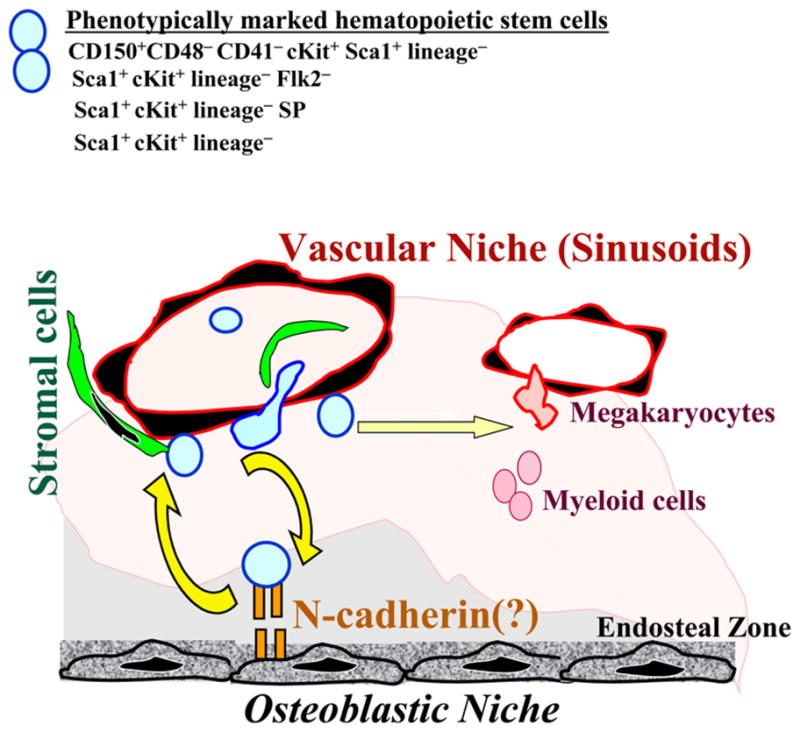
Within the BM microenvironment, phenotypically marked HSCs have been shown to be positioned within the vicinity of the endosteal zone (osteoblastic niche) and sinusoidal endothelial cells (vascular niche). While the osteoblastic niche may provide a safe haven for the maintenance and self-renewal of HSCs, BM’s vascular niche may also set up a cellular platform for the reconstitution of hematopoiesis and directing the trafficking of HSCs and their progeny. However, whether true long-term repopulating HSCs are tethered to the osteoblastic niche through homotypic N-cadherin interaction requires further rigorous analysis. It is possible that under certain physiological conditions subsets of true repopulating HSCs may indeed interact with specific subset osteoblastic cells through an N-cadherin homotypic engagement. The mechanism by which HSCs crosstalk with the vascular niche is also not fully defined. There is no doubt that other as of yet unrecognized cytokines, chemokines, and adhesion molecules contribute to the complex interaction of the true HSCs with their niches.
To answer these questions, Kiel et al. using both immunoselection methods and genetic tracking of N-cadherin+ hematopoietic cells, show that N-cadherin is not expressed on long-term repopulating HSCs. Using commercially available antibodies to N-cadherin, Kiel et al. did not detect HSC activity in the N-cadherin+ fraction of BM cells. Importantly, only N-cadherin− BM populations could reconstitute long-term hematopoiesis in lethally irradiated mice. To eliminate the possibility that N-cadherin expression affects homing in vivo, the authors found that N-cadherin+ fractions had little progenitor capacity in an in vitro colony assay, suggesting that N-cadherin may not mark a population of primitive hematopoietic cells.
More convincingly, Kiel et al. utilized the N-cadherinlacZ gene trap mice, in which the endogenous promoter of N-cadherin drives the expression of lacZ (Luo et al., 2005), in order to formally prove that phenotypically marked Sca1+cKit+lin−Flk2− andCD150+CD48−CD41−cKit+Sca1+lin− HSCs lack the expression of N-cadherin. To address whether N-cadherin expression is upregulated only during physiological stress, Kiel et al. demonstrated that even after myelosuppression, N-cadherin expression was undetectable on mobilized HSCs. It is conceivable that in N-cadherinlacZ gene trap mice the expression of N-cadherin is aberrantly silenced. However, this is unlikely, because Kiel et al. demonstrated that N-cadherin expression follows the reported expression pattern in other tissues.
How, then, can one reconcile the differing findings of the impact of N-cadherin expression on HSC function? Might N-cadherin detection be inconsistent due to technical differences of FDG-dependent cell sorting of N-cadherin-lacZ+ cells, or to antibodies with varying affinities directed against protease-sensitive N-cadherin epitopes? In addition, analysis of diverse populations of HSCs (Sca1+lin−cKit+-SP and Sca1+lin−cKit+Flk2−, versus SLAM+Sca1+lin−cKit+) and technical hurdles associated with the processing of BM (i.e., use of collagenase versus marrow burrowing) may also contribute to the discrepancies in the detection of N-cadherin on true HSCs. Notwithstanding, the ultimate proof of the role of N-cadherin in the regulation of hematopoiesis may emerge from studies in which the expression of N-cadherin is conditionally knocked out in the HSCs, or their progeny, and/or osteoblastic cells. This approach requires generation of a floxed N-cadherin allele, as homozygous N-cadherin-deficient mice do not survive. Until then, the role of N-cadherin homotypic interaction of HSCs with osteoblastic cells necessitates further experimentation, including generation of more specific reagents to detect functional N-cadherin.
Another concept explored by Kiel et al. is the role of osteoblasts in modulating HSC proliferative capacity. An increase in the number of osteoblasts has been shown to augment the number of HSCs (Adams et al., 2006; Calvi et al., 2003). However, as it is technically difficult to isolate putative HSCs directly from the osteoblastic niche and evaluate their stem cell activity, it has been unclear to what extent osteoblasts increase the numbers of true HSCs. Kiel et al. took advantage of a genetic model of biglycan deficiency to reduce osteoblast number and found no effects on HSC pools, suggesting that osteoblasts may not directly influence the size of HSC population. It is possible that in the biglycan-deficient mice, the number of osteoblasts were not sufficiently decreased to affect frequency of the HSCs. Indeed, near complete ablation of osteoblasts in adult mice results in impaired reconstitution of hematopoiesis (Visnjic et al., 2004). However, Kiel et al. show that the majority of phenotypically marked HSCs were not localized to osteoblasts, but nearly 60% of putative HSCs were detected in the vicinity of the BM’s sinusoidal vessels (Kiel et al., 2005). These data suggest that multidimensional interaction of true HSCs with osteoblastic and vascular niches, and perhaps as yet unrecognized stromal cells, may be necessary for the HSC maintenance.
This also raises the intriguing possibility that endosteal cells regulate HSC function indirectly by regulating the integrity of the vasculature in the BM and vice versa. To this end, development of models to selectively target osteoblastic, vascular, or BM stromal cells is essential to interrogate the autonomous role of these complex niches in the regulation trafficking, self-renewal, and maintenance of HSCs. For example, selective ablation of the vascular niche abrogates megakaryocytopoiesis, since interaction of megakaryocytic progenitors with the BM’s sinusoidal vessels is essential for thrombopoiesis (Avecilla et al., 2004). However, targeting the osteoblastic niche without affecting the vascular niche is cumbersome, since osteoblast-derived angiogenic factors may modulate establishment of the vascular niche (Kopp et al., 2005). In addition, identification of molecular markers to track the itinerary of a true HSC within the BM in real time is necessary in order to eavesdrop on the subtle molecular conversations between HSCs and their dynamic niches.
References
- Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, Poznansky MC, Kos CH, Pollak MR, Brown EM, Scadden DT. Nature. 2006;439:599–603. doi: 10.1038/nature04247. [DOI] [PubMed] [Google Scholar]
- Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, Shido K, Jin DK, Dias S, Zhang F, Hartman TE, et al. Nat Med. 2004;10:64–71. doi: 10.1038/nm973. [DOI] [PubMed] [Google Scholar]
- Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, et al. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, et al. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Radice GL, Morrison SJ. Cell Stem Cell. 2007;1(this issue):204–217. doi: 10.1016/j.stem.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Kopp HG, Avecilla ST, Hooper AT, Shmelkov SV, Ramos CA, Zhang F, Rafii S. Blood. 2005;106:505–513. doi: 10.1182/blood-2004-11-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Kostetskii I, Radice GL. Dev Dyn. 2005;232:336–344. doi: 10.1002/dvdy.20241. [DOI] [PubMed] [Google Scholar]
- Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL. Blood. 2004;103:3258–3264. doi: 10.1182/blood-2003-11-4011. [DOI] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, et al. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]