

Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression at the post-transcriptional level. They play an important role in several biological processes such as cell development and differentiation. Similar to transcription factors (TFs), miRNAs regulate gene expression in a combinatorial fashion, i.e., an individual miRNA can regulate multiple genes, and an individual gene can be regulated by multiple miRNAs. The functions of TFs in biological regulatory networks have been well explored. And, recently, a few studies have explored miRNA functions in the context of gene regulation networks. However, how TFs and miRNAs function together in the gene regulatory network has not yet been examined. In this paper, we propose a new computational method to discover the gene regulatory modules that consist of miRNAs, TFs, and genes regulated by them. We analyzed the regulatory associations among the sets of predicted miRNAs and sets of TFs on the sets of genes regulated by them in the human genome. We found 182 gene regulatory modules of combinatorial regulation by miRNAs and TFs (miR-TF modules). By validating these modules with the Gene Ontology (GO) and the literature, it was found that our method allows us to detect functionally-correlated gene regulatory modules involved in specific biological processes. Moreover, our miR-TF modules provide a global view of coordinated regulation of target genes by miRNAs and TFs.
Background
MicroRNAs (miRNAs) are a class of endogenous non-coding small RNAs. They negatively regulate gene expression at the post transcriptional level through binding to complementary sites in the 3'- untranslated regions (3'-UTR) of target genes [1, 2]. The first miRNA was found by Victor Ambros and his colleagues in 1993. During the last several years, hundreds of miRNAs and their target genes have been identified in mammalian cells. Recent studies suggest that miRNAs play critical roles in multiple biological processes, including cell growth, cell differentiation, embryo development and so on [2, 3]. In cellular organisms, the regulation of gene expression is an important mechanism to control biological processes. To understand how differential gene expression is controlled at the genome-wide level, it is important to identify all factors involved, and how and when they affect gene expression. To date, differential gene expression can be regulated at the transcriptional and at the post-transcriptional levels by transcription factors (TFs) and miRNAs [4]. TFs can activate or repress transcription by binding to cis-regulatory elements located in the upstream regions of genes. Many TFs are well characterized, and corresponding binding motifs have been identified in various species. The combinatorial interactions between TFs have been well studied [5, 6]. MiRNAs repress gene expression by physically interacting with cis-regulatory elements located in the 3'-UTR of their target messenger RNAs (mRNAs). Compared with the work on transcriptional regulation by TFs, the study of biological regulatory networks mediated by miRNAs is just beginning. There are a few studies which have investigated the regulatory functions of miRNAs in the gene regulation network [7, 8].
Similar to TFs, miRNAs regulate gene expression in a combinatorial fashion, i.e., an individual miRNA can regulate multiple genes, and an individual gene can be regulated by multiple miRNAs. We also know that a gene is regulated through multiple mechanisms before producing its products. Thus, an attractive possibility is that miRNAs and TFs may cooperate in regulating the same target genes. This cooperation of miRNAs and TFs reflects the collaborative regulation at the transcriptional and post-transcriptional levels. The combined complexity of miRNAs and TFs may be relevant to creating cellular complexity in a developing organism [9]. However, how TFs and miRNAs function together in the gene regulatory network has not yet been examined. Investigating coordinated regulation of genes by miRNAs and TFs is an effort that may elucidate some of their roles in various biological processes.
Detection of microRNA regulatory modules within biological meanings has received much attention recently. Tran et al. [10] presented a rule-based learning method to find miRNA regulatory modules that contain coherent miRNAs and mRNAs. Joung and Fei [11] used an author-topic model, which is a family of probabilistic graphical models for identification of miRNA regulatory modules. Recently, Liu et al. [12] tried to identify negatively regulated patterns of miRNAs and mRNAs which are associated with specific conditions. These studies, however, only concentrated on finding miRNA functions at the post-transcriptional level. Two other recent studies also showed that miRNA mediated regulatory circuits are prevalent in the human genome [8, 13]. Thus, the question regarding to the combined roles of miRNAs and other factors in gene regulatory network still remains unanswered. Unlike previous studies as discussed above, in this paper, we propose a new computational method to discover the gene regulatory modules consisting of miRNAs, TFs, and the genes regulated by them. The method combines TF-gene binding information and miRNA target gene data for module discovery. Specifically, we analyzed the regulatory associations among the sets of predicted miRNAs and sets of TFs on the sets of regulated genes by them in the human genome. We found 182 gene regulatory modules of combinatorial regulation by miRNAs and TFs, named miR-TF regulatory modules (miR-TF modules in short). By validating these modules with the Gene Ontology (GO) and the literature, it was found that our method allows us to detect functionally-correlated gene regulatory modules involved in specific biological processes. Moreover, our miR-TF modules provide a global view of coordinated regulation of target genes by miRNAs and TFs. Results suggest that combinatorial regulation at the transcriptional and post-transcriptional levels is more complicated than previously thought. Taken together, our results may provide insights into how miRNAs and TFs function in the complex regulatory network of the human genome.
Methodology
Datasets
We obtained the miRNA regulatory signatures and TF regulatory signatures from CRSD database [14]. CRSD utilizes and integrates six well-known, large-scale databases, including the human UniGene, TRANSFAC, mature miRNAs, putative promoter, pathway and GO databases. The miRNA regulatory signature database was constructed using mature human miRNA from miRBase [15] and 3'-UTR sequences, and TF regulatory signature database was constructed using TRANSFAC [16] and the promoter database [17]. From these databases, we build a data set that contains three components, miRNAs, TFs, and genes regulated by them. To avoid trivial cases of binding associations, each gene in our data set is regulated by at least two miRNAs and two TFs with a significant binding score (p-value <5 x 10−2 for miRNA-target gene binding, and p-value < 5 x 10−2 for TF-target gene binding). Specifically, our data set contains 267 mature miRNAs, 483 TFs, and 1253 genes regulated by miRNAs and TFs. On average, each miRNA binds to 22 genes and each TF binds to 17 genes. In addition, each gene was regulated by 5 miRNAs and 6 TFs on average. A schematic description of the data set construction is illustrated in Figure 1.
Figure 1.
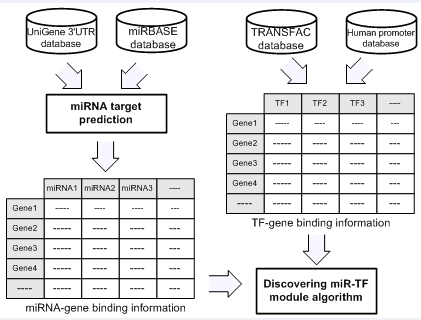
Schematic description of the data set construction. The miRNA-gene binding information is constructed from miRNA and 3’-UTR databases by using a miRNA target prediction, and the TF-gene binding information is constructed by TF binding site prediction integrating promoter and TRANSFAC databases.
Algorithm
In this section we present our algorithm for discovering miR-TF regulatory modules. As mentioned above, each module consists of genes, miRNAs, and TFs that all these miRNAs and TFs regulate these genes with a stringent p-value. Our method exhaustively and efficiently searches on the entire combinatorial space of subsets of miRNAs and subsets of TFs to discover what factors control what genes.Algorithm: Let A be a matrix of miRNA-gene binding p-values, where rows correspond to genes and columns correspond to miRNAs, so that Aij denotes the binding p-value of gene i with miRNAj. And, let B be a matrix of TF-gene binding p-values, where rows correspond to genes and columns correspond to TFs, so that Bij denotes the binding p-value of gene i with TF j. Let M(i, p) denotes the set of all miRNAs that bind to gene i with a p-value less than p. Let T(i, p) denotes the set of all TFs that bind to gene i with a p-value less than p. Let G(X, p) be a set of all genes to which all the factors (i.e. miRNAs or TFs) in X bind with a given significance threshold p. Our algorithm begins by going over all genes i. Using a strict p-value p1, we search all subsets of M’⊆ M(i, p1) that not have been explored in previous steps (i.e. the function IsAvailable(M’) is true). For each M’, we search all subsets of T’⊆ T(i, p2) that not have been explored in previous steps (i.e. the function IsAvailable(T’) is true). Then, we find the set of all genes G that are bound by all miRNAs in M’ and all TFs in T’ with given significance thresholds p1 and p2. Finally, if the number of genes in G is greater than 1, we obtain a module and mark all the subsets of M’ and T’, so that they are not considered next time. All steps of the algorithm are presented in Algorithm 1. Basically, our algorithm works with a dataset containing more than one hundred miRNAs and TFs. Thus, there are potentially exponential numbers of larger-size subsets of miRNAs (or TFs). However, as we showed in the previous section, each gene is bound by only 5 miRNAs and 6 TFs on average. In addition, we mark all the subsets of M’ and T’ when they are involved in the same module, so that the number of subsets of T and M to be considered is reduced in the algorithm. Therefore, the number of actual subsets we are searching is much smaller.
Results and Discussion
miRNA-TF regulatory module detection
We have applied our module discovery algorithm to genome-wide binding data for 267 mature miRNAs and 483 TFs. In order to determine the appropriate values for miRNA-gene interaction threshold p1 and TF-gene interaction threshold p2, we have tested binding data at a number of different confidence levels (0.05, 0.01, 0.005, and 0.001). Table 1 ( supplementary material) shows the number of modules found by our algorithm with different p-value thresholds. As can be seen, when we relax the binding thresholds, the number of modules found increases. For further analysis, we selected the values of p1 and p2 equal to 0.01. Consequently, we obtained 182 miR-TF modules. These modules contain the relationships between 107 miRNAs, 176 TFs, and 261 target genes regulated by them (Table 1 - supplementary material). Among 182 miR-TF modules found, we take fifteen modules as examples (Table 2 - supplementary material), the remaining module data can be found in the supplement files.
As mentioned above, each module consists of three components, miRNAs, TFs, and genes regulated by all miRNAs and TFs in the module. And the relationship between one gene and one miRNA or TF represents the strength of binding score. We found that some of our modules share a subset of miRNAs or TFs on regulation of the target genes. This is because of the complexity of the miRNA regulation network. Consequently, our results provide a global view of the combinatorial coregulation of two main factors on gene regulation network. A part of the network constructs from miR-TF modules is shown in Figure 2. Three modules (68, 66, and 67) share a common miRNA hsa-miR-125b; two modules (66 and 67) share two miRNAs: hsa-miR-125a and hsa-miR-125b. Also, modules 68 and 66 have the same TF Zic3. This suggests that the coordinated regulation of target genes by miRNAs and TFs is more complicated than previously thought.
Figure 2.
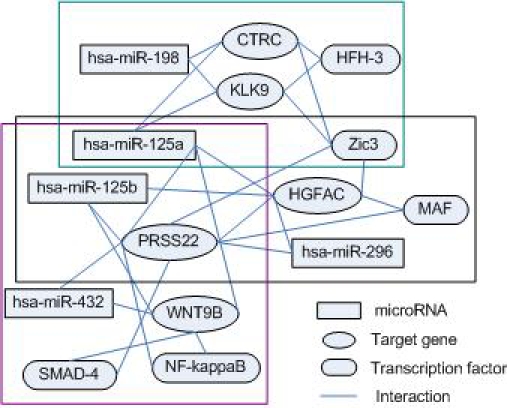
An illustration of three miR-TF modules discovered by our algorithm Inside green rectangle, black rectangle, and cyan rectangle are modules 68, 66, and 67, respectively. The line from a miRNA/TF to a gene indicates that miRNA/TF regulates that target gene. MiRNAs repress target genes, while TFs may activate or repress target genes.
Validation using Gene Ontology annotations
To test if the regulated target genes and the host genes of TFs for each miR-TF module might be enriched functionally based on arbitrary GO terms, we performed GO annotation and significance analysis using GOstat [18]. We observed terms associated significantly with the target genes included in the GO gene-association database. To find significantly overrepresented GO terms associated with a given gene set, GOstat counts the number of appearances of each GO term for the genes in this group, as well as in the reference group. Then, for each GO term, a p-value is calculated, representing the probability that the observed numbers of counts could have resulted from randomly distributing this GO term between the tested group and the reference group. The GO terms most specific to the analyzed list of genes will have the lowest p-values.
Table 3 ( supplementary material) shows fifteen significant GO terms directly related to the set of regulated target genes and host genes of TFs in miR-TF modules. Some of them are annotated as regulating specific biological processes (GO:0019219, GO:0019222, GO:0045449, GO:0031323). Other terms have function in organ morphogenesis (GO:000987), gland development (GO:0048732), and organ development (GO:0048513). In addition, GO terms associated with the gene set of miR-TF modules are not so general, for example: GO:000987, GO:0019222, and GO:0019219 are located at levels 8, 6, and 7 in the hierarchical structure of Gene Ontology. Thus we speculate that our miRNA-TF modules may be functional biological modules in gene regulation and molecular development.
Functional validation from the literature
We did a literature review and found that some miRNAs and TFs in miR-TF modules were discovered to have associations with cancer development. Interestingly, several miRNAs have been confirmed to be related to lung and other human cancers. For example, hsa-let-7d acted as a tumor suppressor in human lung cancer; hsa-miR-15a was downregulated in B-cell chronic and lymphocytic leukemia [19]. HsamiR- 125b was downregulated and hsa-miR-25 was upregulated in human breast cancer. These facts suggest that these miRNAs may potentially act as tumor suppressors. Several transcription factors in miR-TF modules have roles in the development of several types of cancers. NF-κВ kappa prevents apoptosis in several untransformed and tumor cell types. AP transcription factor family (AP-1 and AP-2) is encoded from a gene which is known to regulate a variety of cellular processes including cellular proliferation, differentiation, and oncogenic transformation [20]. In addition, ETS family regulates numerous genes, and is involved in stem cell development, and tumorigenesis [21]. Taken together with evidence shown in Table 4, it is reasonable for us to think that the miR-TF modules we have discovered may be related to human cancers.
Conclusion
Transcription factors and microRNAs are two main and important factors regulating gene expression. TFs play the functions at the transcriptional level by binding to promoter regions of the target genes. In other way, miRNAs regulate the messenger RNAs at the post-transcriptional level by binding to their 3’-UTR. The functions of miRNAs and TFs in biological regulatory networks recently have been quite well explored. However, the combinatorial regulation by miRNAs and TFs on the common target genes is less understood. In this paper, we introduced a computational method for discovering functional miR-TF modules and applied this method to the data set in the human genome.
Using GO annotations and the literature review, we found that our algorithm allows us to detect functionally correlated gene regulatory modules involved in specific biological processes. Specifically, miRNAs and the host gene of TFs in our modules may be involved in some development processes, and may be involved in several types of cancer diseases. Moreover, our miR-TF modules provide a global view of coordinated regulation of target genes by miRNAs and TFs.
Supplementary material
Acknowledgments
The first author has been supported by Japanese government scholarship (Monbukagakusho) to study in Japan. This work is also supported in part by NAFOSTED project No. 102.03.21.09 from the Ministry of Science and Technology, Vietnam. The author would like to thank Dr. Chun-Chi Liu and his colleagues for sharing the CRSD database.
Footnotes
Citation:Tran et al, Bioinformation 4(8): 371-377 (2010)
References
- 1.Ambros A. Nature. 2004;431:350. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. Cell. 2004;116:281. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.He L, Hannon GJ. Nature Review. 2004;5:522. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 4.Chen K, Rajewsky N. Nature Reviews. 2007;8:93. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 5.Segal E, et al. Nature Genetics. 2003;34:166. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- 6.Yu H, Gerstein M. Proc Natl Acad Sci. 2006;103:14724. doi: 10.1073/pnas.0508637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui Q, et al. Mol Syst Biol. 2006;2:46. doi: 10.1038/msb4100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsang J, et al. Mol Cell. 2007;26:753. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hobert O. TRENDS in Biochemical Sciences. 2004;29:462. doi: 10.1016/j.tibs.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Tran DH, et al. BMC Bioinformatics. 2008;9:S5. doi: 10.1186/1471-2105-9-S12-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joung JG, Fei Z. Bioinformatics. 2009;25:387. doi: 10.1093/bioinformatics/btn626. [DOI] [PubMed] [Google Scholar]
- 12.Liu B, et al. J Biomedical Infor. 2009;42:685. doi: 10.1016/j.jbi.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Shalgi R, et al. PLoS Comput Biol. 2007;3:42. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu CC, et al. Nucleic Acids Res. 2006;34:W571. doi: 10.1093/nar/gkl279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffiths-Jones S, et al. Nucleic Acids Res. 2006;34:D140 . doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matys V, et al. Nucleic Acids Res. 2003;31:374. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halees AS, Weng Z. Nucleic Acids Res. 2004;32:W191. doi: 10.1093/nar/gkh433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beissbarth T, Speed TP. Bioinformatics. 2004;20:1464. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 19.Calin GA, et al. PNAS. 2002;99:15524. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen Q, et al. Oncogene. 2008;27:366. doi: 10.1038/sj.onc.1210643. [DOI] [PubMed] [Google Scholar]
- 21.Dwyer J, et al. Ann N Y Acad Sci. 2007;1114:36. doi: 10.1196/annals.1396.022. [DOI] [PubMed] [Google Scholar]
- 22.Johnson CD. Cancer Res. 2007;67:7713. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 23.Iorio MV, et al. Cancer Res. 2005;65:7065. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 24.He L, et al. Nature. 2005;435:828. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Volinia S, et al. PNAS. 2006;103:2257. [Google Scholar]
- 26.Vincent B, et al. Bio Phar. 2000;60:1085. [Google Scholar]
- 27.Donnell KA. Nature. 2005;435:839. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.