

Abstract
TtgR is a multidrug binding protein in the gram negative bacteria Pseudomonas putida (DOTT1E strain) and regulates one of the key mechanisms of its antibiotic resistance by active extrusion of toxic compounds through the membranebound efflux pumps. The paper reports the molecular docking studies of Pongachalcone I, a natural pyranochalcone and reported potent inhibitor of the bacteria, on the transcriptional regulator (TtgR) enzyme (PDB Code: 2UXI) which is a key efflux pump TtgABC operon repressor. Although the bacterium is capable to expel antibiotics like Chloramphenicol, Tetracycline and Naringenin, yet Pongachalcone I has potent activity against it. The work unveils the key roles played by the residues ASN 110 and CYS 137 in the active site of the enzyme, which would be highly beneficial in providing insight into the molecular mechanism of multiple drug recognition and in designing drugs for antibiotic resistance bacteria.
Keywords: Pseudomonas putida, pyranochalcones, TtgR, docking
Background
Antibiotic resistance of bacteria is a widely spread phenomenon. One major mechanism that underlies antibiotic resistance in bacteria is the active extrusion of toxic compounds through the membranebound efflux pumps that are often regulated at the transcriptional level [1]. Resistance to antimicrobials is as a result of three main strategies namely enzymatic inactivation of the drug [2], modification of target sites [3] and extrusion by efflux [4]. The active efflux of toxic compounds is one of the common mechanisms employed by bacteria to protect themselves against the deleterious effects of toxic molecules they encounter in the environment [5]. TtgR represses the transcription of TtgABC, a key efflux pump in Pseudomonas putida, which is highly resistant to antibiotics, solvents and toxic plant secondary products [1]. The majority of the efflux systems in bacteria are nondrug specific proteins that can recognize and pump out a broad range of chemically and structurally unrelated compounds from bacteria in an energydependent manner, without drug alteration or degradation [6]. The consequence of this drug extrusion is that, it leads to a reduced intracellular concentration of the antimicrobial such that the bacterium can survive under conditions of elevated antimicrobial concentration [7].
Pyranochalcones are widely distributed naturally occurring flavonoids. Members of the pyranochalcones have been associated with a wide variety of biological activities such as antimutagenic, antimicrobial, antiulcer and antitumor activities [8]. Interestingly, Pongachalcone I, a natural pyranochalcone isolated from Tephrosia deflexa, has been reported to exhibit antibacterial activity against the bacteria Pseudomonas putida [8,9]. The present paper reports the insilico docking studies to find the key features of Pongachalcone I in superseding the efflux system of the bacteria, which will be highly beneficial in advanced drug design and in QSAR studies on antibiotic resistant bacteria.
Methodology
Substrate
The HTHtype transcriptional regulator TTGR (from PDB code: 2UXI) in Pseudomonas putida (bound with Phloretin) was taken as the substrate for docking. This substrate is particularly chosen for two reasons. Firstly, TtgR is a multidrug binding protein which represses the transcription of TtgABC. It triggers the pumping out of the toxic materials making the organism resistant to antibiotics, solvents and toxic plant secondary products. Secondly, on searching Protein Data Bank databases, only TtgR was reportedly found to bind with a plant derived flavonoid, Quercetin. Interestingly, pyranochalcones possess the structural features of Quercetin, the flavonoid and Phloretin, the plant antimicrobial.
Crystal structure of transcriptional regulator TtgR (PDB code: 2UXI) in Pseudomonas putida (bound with Phloretin) was downloaded from RCSB Protein Data Bank [10]. The structure was deposited in March 2007 with the resolution of 2.50 A and Rfactor of 0.245 (working set). Ramachandran plot generated by the PROCHECK module present in PDBsum server [11] showed about 91% residues (345 residues) falling in the most favorable region, 9% residues (34 residues) falling in the additionally allowed region while none in the disallowed region.
Ligands
Pseudomonas putida is resistant to toxic substances or antibiotics, yet the pyranochalcone Pongachalcone I (from Tephrosia deflexa), exhibited inhibitory effect on it. Keeping that in mind, this pyranochalcone was taken as ligand. TtgR efflux pump was able to ’sense‘ a number of compounds like, Phloretin, Quercetin, Chloramphenicol, Tetracycline and Naringenin (Figure 1). These compounds were reported to bind with TtgR and subsequently pumped out from the bacterial system [5]. Therefore those were also subjected to docking to examine their binding modes.
Figure 1.
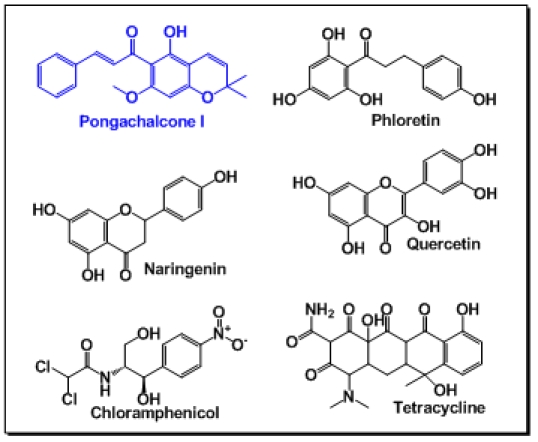
Chemical structure of the ligands used.
All the Ligands was optimized by density functional theory (DFT) at the level of B3LYP and calculations were carried out with both 6 31G* basis sets using GAUSSIAN 03 package [12,13]. The ligands were grouped into an SDF file for streaming them into simulation in sequence. The Xray crystal structures of TtgR bound with Phloretin (2UXI), Quercetin (2UXH), Chloramphenicol (2UXP) and Naringenin (2UXU) were downloaded from RCSB Database.
The Simulation Details
The structure of the protein was corrected for missing atoms or unknown units using Molegro Virtual Docker (MVD) [14]. 2UXI has a dimeric structure (A and B) with identical sequence of residues. Comparing with other TtgR crystal structures (2UXH, 2UXP, 2UXU), it was marked that one unit i.e., the unit A (sequence A) of the twin units assumes the major role in binding with a ligand. So to simplify the simulation, the unit A was taken. All other residue, water etc. were removed.
To locate the appropriate binding orientations and conformations of ligands on TtgR, docking was performed by using Molegro Virtual Docker (MVD) software package [14]. The identification of ligand binding modes is done by iteratively evaluating a number of candidate solutions (ligand conformations) and estimating the energy of their interactions with the macromolecule. MVD performs flexible ligand docking, so the optimal geometry of the ligand will be determined during the docking. The MolDock scoring function (MolDock Score) used by MVD is derived from the PLP scoring functions originally proposed by Gehlhaar et al. and later extended by Yang et al. [as cited in 15]. The MolDock scoring function further improves these scoring functions with a new hydrogen bonding term and new charge schemes. The docking scoring function is shown in supplementary material. It is a Coulomb potential with a distancedependent dielectric constant given by: D(r) = 4r. The numerical value of 332.0 fixes the units of the electrostatic energy to kilocalories per mole. To ensure that no energy contribution can be higher than the clash penalty the electrostatic energy is set to a cutoff level corresponding to a distance of 2.0 Å for distances less than 2.0 Å.
The Docking
At the beginning of docking studies, the compound Phloretin extracted from 2UXI was allowed to undergo extensive flexible docking (templeted and untempleted) on the 2UXI [A] unit truncated from 2UXI as mentioned previously. The best pose of the ligand (in terms of Molegro Rerank Score), returned by MVD was found to be in excellent agreement with the orientation of the molecule in the actual bound state. As in QSAR studies, alignment and bioactive conformation selection are two important factors for obtaining robust and meaningful models. The best docked conformation of Phloretin was taken as template for alignment and templeted docking of rest of the ligands. The substrate had only one cavity (Figure 2). The grid resolution for the binding site was kept at 0.30A.
Figure 2.
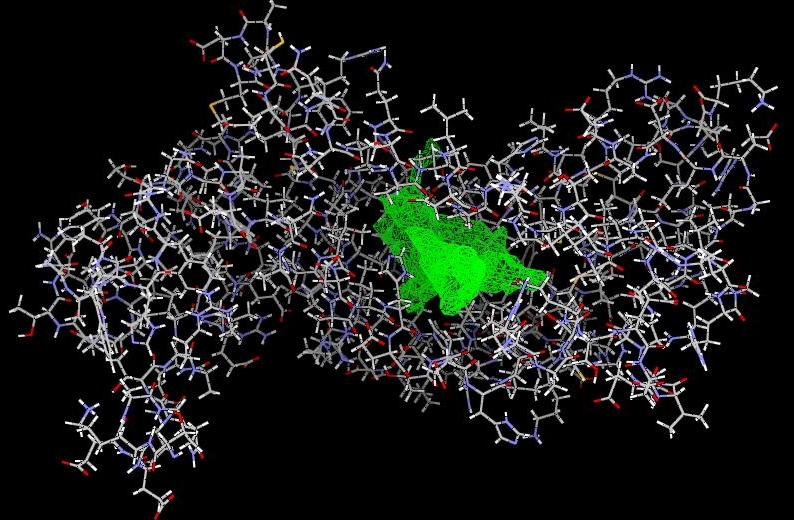
The substrate 2UXI[A] showing the cavity (green mesh) at the active site
Study of LigandSubstrate Interaction
The binding modes of the different docking poses of the ligands were investigated on Ligandscout 2.0 software package [16]. Pharmacophore corresponding to the native ligand Phloretin, and marged pharmacophore corresponding to all ligands except Pongachalcone I were also generated to pinpoint the key differences between the binding modes of the pyranochalcone and other antimicrobials studied.
Discussion
Flexible docking of Phloretin was carried out in the active site of TtgR. Ten top ranking poses for each ligand were returned in the simulation, out of which one best pose for each ligand was selected on the basis of their Rerank score. Table 1 (see supplementary material) presents the Interaction energy between the ligand and protein and Hydrogen Bond Energy for the selected alignment and conformation of each ligand. The result from the Table 1 tells that Pongachalcone I exhibited the lowest interaction with the substrate. The binding modes of the ligands were investigated on Ligandscout 2.0 software package [14]. Phermacophore was generated for the native ligand Phloretin on TtgR 2UXI [A] unit. The best poses for each ligands mentioned above, were injected into the active site of the protein. Figure 3shows the interaction of the ligands with the protein.
Figure 3.

The interaction of the ligands with the protein
From the study of interactions (Figure 3) reveals that, in the active site of the protein, ASN 110 and CYS 137 probably are the most instrumental in binding and sensing the compounds toxic to the organism. The strategic positions of these residue help on binding the ligand either from two ends (as in case of Phloretin) or from one end (as in case of Naringenin). The residues ALA74, SER77, GLU78, VAL96, ILE175 and VAL171 also play crusial role in binding, mostly by hydrophobic interactions with the ligands. Most interestingly, Pongachalcone I was not found to interact with ASN 110 or CYS 137 and probably that is why it could remain ’unnoticed‘ by the bacterial defense mechanisms.
To have a more convincing support for this mechanism, two analysis were done: Firstly, untempleted flexible docking was performed with Pongachalcone I on 2UXI [A] as substrate using Molegro suite. All of the five top ranking poses returned by Molegro were put to the Phloretin phermacophore using Ligandscout 2.0. But again no noticeable interaction of Pongachalcone I with the phermacophore region was obtained, except a weak hydrogen bond type interaction (3.3 A) with CYS137 in only one of the poses.
Secondly, in order to make the situation more stringent, phermacophores corresponding to each of Phloretin, Quercetin, Chloramphenicol, Tetracycline and Naringenin in 2UXI[A] were generated in. A combined phermacophore was formed comprising all interaction features/regions of all these compounds. Interestingly, the pyranochalcone Pongachalcone I did not exhibit any strong interaction even with this robust phermacophore also (Figure 4).
Figure 4.

A) The marged phermacophore; B) Interaction with Phloretin (shown for comparison only); C) Interaction with Pongachalcone I.
Those two studies provided an indication that, Pongachalcone I does not interact strongly with residues which are primarily responsible for binding with other antimicrobials or antibiotics.
Apart from those residues, the ligands have shown to interact with a number of other residues also. But as some of those were present in the case of Pongachalcone I and others did not have common occurrence in case of different antimicrobials studied here, it seems that those residues do not have any primary role in the efflux pump mechanism in P. putida.
Conclusion
The ProteinLigand interaction plays a significant role in structural based drug discovery/designing. In this study, molecular docking has been employed to identify the potential binding mode of a number of antimicrobials with TtgR. TtgR represses the transcription of TtgABC, a key efflux pump in Pseudomonas putida, which is highly resistant to antibiotics, solvents and toxic plant secondary products. The results were then compared with that of Pongachalcone I, a naturally occurring pyranochalcone, reported to be active against Pseudomonas putida. The study reveals that, in the active site of the protein, the residues ASN 110 and CYS 137 along with ALA74, SER77, GLU78, VAL96, ILE175 and VAL171 seem to play crucial role in binding with the ligands and triggering the efflux pump.
Although only the activity of Pongachalcone I against P. putida is reported, further work is in progress to compare the activities of a number of naturally occurring pyranochalcones. Stress is given to naturally occurring pyranochalcones to ensure the existence and availability of the compounds. That will not only help to explore the structural features for batter efficacy of this class of compounds but also will be instrumental in development of newer classes of antibacterial compounds.
Supplementary material
Acknowledgments
The authors are grateful to CDAC, India and BRAF for providing supercomputing facility with Gaussian 03 to SC. The authors are also thankful to Dr. Uday S. Chakraborty, Department of Mathematics, Assam University for valuable discussions.
Footnotes
Citation:Paul et al, Bioinformation 4(10): 473-477 (2010)
References
- 1.Alguel Y, et al. J Mol Biol. 2007;369:829. doi: 10.1016/j.jmb.2007.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies J. Science. 1994;264:375. doi: 10.1126/science.8153624. [DOI] [PubMed] [Google Scholar]
- 3.Spratt BG. Science. 1994;264:388. doi: 10.1126/science.8153626. [DOI] [PubMed] [Google Scholar]
- 4.Nakaido H. Science. 1994;264:382. [Google Scholar]
- 5.Tera W, et al. J Biol Chem. 2006;281:7102. doi: 10.1074/jbc.M511095200. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, Schweizer HP. Adv Drug Deliv Rev. 2005;57:1486. doi: 10.1016/j.addr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Marquez B. Biochimie. 2005;87:1137. doi: 10.1016/j.biochi.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Lee YR, et al. Molecules. 2007;12:1420. doi: 10.3390/12071420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kare M, et al. J Soc ouest-afr chim. 2006;11:41. [Google Scholar]
- 10. www.rcsb.org.
- 11. http://www.ebi.ac.uk/
- 12.Miller WH. Chem Phys Lett. 1990;172:62. [Google Scholar]
- 13.Perdew JP. Phys Rev Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 14. http://www.molegro.com.
- 15.Molegro Document.
- 16. http://www.inteligand.com/ligandscout/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.